

Кинетика и катализ, 2022, T. 63, № 1, стр. 16-32
Никелевые катализаторы для электроокисления водорода и борогидрида: современное состояние и перспективы
А. Г. Ощепков a, *, Е. Р. Савинова b, **
a ФГБУН ФИЦ Институт катализа им. Г.К. Борескова СО РАН
630090 Новосибирск, просп. Акад. Лавреньева, 5, Россия
b Institut de Chimie et Procédés pour l’Energie, l’Environnement et la Santé, UMR 7515 CNRS-University of Strasbourg
67087 Strasbourg Cedex, 25 rue Becquerel, France
* E-mail: oshchepkov@catalysis.ru
** E-mail: Elena.Savinova@unistra.fr
Поступила в редакцию 02.06.2021
После доработки 27.07.2021
Принята к публикации 25.08.2021
- EDN: POGTYI
- DOI: 10.31857/S0453881122010130
Аннотация
Никель широко применяется в гетерогенном катализе в процессах гидрирования, паровой конверсии метана, метанирования и других. В гетерогенном электрокатализе никель менее распространен, но в последние десятилетия интерес к нему растет. Настоящая работа представляет собой краткий обзор литературы, посвященной никелевым электрокатализаторам для реакций электроокисления водорода и борогидрида, в котором особое внимание уделено материалам, содержащим гетероструктуры на основе металлического никеля и оксида (гидроксида) никеля.
1. ВВЕДЕНИЕ
По сравнению с гетерогенным катализом, гетерогенный электрокатализ – относительно молодая дисциплина. Термин “электрокатализ” был введен Кобозевым и Монблановой [1], которые исследовали реакцию катодного выделения водорода на металлических электродах в водных электролитах (уравнение I в кислоте или уравнение II в щелочи):
(I)
${\text{2}}{{{\text{H}}}^{ + }} + {\text{ }}2{{{\text{e}}}^{--}} \leftrightarrow {\text{ }}{{{\text{H}}}_{2}},$(II)
${\text{2}}{{{\text{H}}}_{{\text{2}}}}{\text{O + 2}}{{{\text{e}}}^{--}} \leftrightarrow {\text{ }}{{{\text{H}}}_{{\text{2}}}}{\text{ + 2O}}{{{\text{H}}}^{--}}.$На основании зависимости кинетики катодного выделения водорода от материала электрода [2] был сделан вывод о том, что последний является катализатором электродной реакции. Гетерогенный электрокатализ (который в этом обзоре мы будем называть просто электрокатализом) имеет значительное сходство с гетерогенным катализом. В частности, кинетика протекающих реакций зависит от прочности адсорбции промежуточных продуктов реакции (Had в случае катодного выделения водорода) на поверхности электрокатализатора. Основные особенности, отличающие электрокатализ от традиционного гетерогенного катализа, обусловлены тем, что в первом случае процесс происходит на границе раздела электрод/электролит, и, по крайней мере, одна из стадий реакции включает перенос заряда. Таким образом, наряду с температурой, электродный потенциал в электрокатализе является одним из основных параметров, определяющих кинетику и механизм реакции. Электрокатализатор ускоряет кинетику, позволяя тем самым осуществлять процесс при потенциалах, близких к равновесным. В электрокатализе используются различные показатели активности, из которых наиболее распространены перенапряжение (представляющее собой разницу между потенциалом электрода при определенном значении тока и равновесным потенциалом), необходимое для достижения определенной плотности тока, или плотность тока обмена при равновесном потенциале. В обоих случаях для корректного сравнения различных каталитических материалов требуется, чтобы токи были нормированы либо на их площадь поверхности (удельная активность), либо на их массу (активность на единицу массы).
Первые достижения в области электрокатализа были связаны с исследованием кинетики реакции выделения водорода. Этот процесс имеет важное практическое значение, поскольку протекает на катоде при электролизе воды. Было установлено, что в кислой среде скорость реакции для “активных” (Pt) и “неактивных” (Hg) электродных материалов отличается на 12 порядков. Дальнейший прогресс в области электрокатализа был тесно связан с разработкой систем преобразования и хранения энергии, таких как электролизеры и топливные элементы. Первые электрокатализаторы топливных элементов были основаны на платине. Однако зависимость от дефицитных и дорогих благородных металлов отрицательно сказывается на практическом использовании таких устройств производства и преобразования энергии, как топливные элементы и электролизеры. Таким образом, в настоящее время все большее внимание привлекают катализаторы на основе переходных металлов, не содержащие благородных металлов. Достигнутый в последние десятилетия прогресс в области анионообменных мембран способстует развитию электрокатализа на переходных металлах (таких как Ni), которые в щелочной среде (в зависимости от потенциала электрода) стабильны либо в металлической, либо в окисленной (оксид, гидроксид, оксигидроксид) форме.
Никель относится к наиболее распространенным переходным металлам на Земле. Его предполагаемое количество составляет от 17 000 до 19 000 миллионных долей, при этом бóльшая часть никеля сосредоточена в ядре Земли и около 47 миллионных долей – в континентальной коре [3]. Никель широко используется в гетерогенном катализе, в частности, в процессах паровой конверсии метана и риформинга с углекислым газом, селективного и полного гидрирования, гидрогенолиза, метанирования и ряде других процессов [4, 5]. Было показано, что в процессах, катализируемых Ni, участвуют как поверхностные, так и приповерхностные интермедиаты. Так, паровая конверсия метана на никелевых катализаторах включает адсорбированные формы H, O, OH и CHx [6], тогда как в гидрировании этилена возможно участие абсорбированного водорода, который в ходе реакции выходит из объема на поверхность катализатора [7]. Поскольку миграция H в приповерхностные слои Ni является медленной стадией в связи с ее высоким активационным барьером, для проведения таких реакций гидрирования требуется высокое давление водорода (что отличает Ni от Pd) [8]. В некоторых публикациях сообщается о протекании реакций гидрирования на композитных материалах, содержащих гетероструктуры Ni/NiO. Например, селективное гидрирование 2(5H)-фуранона до γ-бутиролактона [9] или левулиновой кислоты до γ-валеролактона [10] протекает на гетероструктурах Ni/NiO по механизму Лэнгмюра–Хиншельвуда между адсорбированным водородом и адсорбированными органическими молекулами [10]. Было показано, что NiO способствует адсорбции органических молекул и снижает энергетический барьер диссоциативной адсорбции водорода на Ni. Кроме того, использование нанокомпозитов Ni/NiO позволяет проводить различные реакции в мягких условиях (низкое давление и температура) с высокой селективностью и активностью [10, 11].
В электрокатализе никель используется менее широко, но в последние десятилетия интерес к этому металлу существенно вырос. Материалы на основе никеля активно изучаются в реакции катодного выделении водорода в течение многих десятилетий в связи с промышленным применением систем щелочного электролиза воды. Начиная с 2000-х гг. катализаторы на основе Ni активно исследуются для реакции анодного окисления водорода, для окисления некоторых неорганических (таких как борогидрид, гидразин и т.д.) или органических (таких как спирты, мочевина и т.д.) топлив, для окисления воды до кислорода (реакция выделения кислорода) и некоторых других процессов.
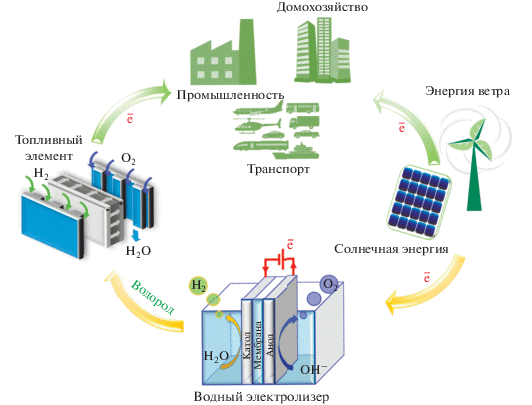
Представленная обзорная статья посвящена электрокатализу реакций окисления водорода и борогидрида, которые требуют присутствия металлических центров на поверхности никеля и протекают вблизи равновесного потенциала водородного электрода (0 В по шкале обратимого водородного электрода, ОВЭ). Практический интерес к этим реакциям связан с их протеканием в топливных элементах, в которых электричество генерируется за счет электроокисления топлива на аноде и электровосстановления кислорода (обычно поступающего из воздуха) на катоде (рис. 1). Если топливо производится путем электролиза (например, водород может быть получен посредством электролиза воды, рис. 1), то создается замкнутый цикл, который позволяет запасать возобновляемую электроэнергию в виде топлива и в дальнейшем использовать ее по назначению.
Рис. 1.
Схематическое изображение цикла конверсии водорода, включая производство “зеленого” водорода электролизом воды с его дальнейшим использованием в топливном элементе для производства электроэнергии в местах потребления.
Обзор построен следующим образом. В разделе 2 мы кратко рассмотрим свойства межфазной границы никель/водный электролит в интервале потенциалов, соответствующем протеканию реакций окисления водорода и водородсодержащих топлив. В разделе 3 с привлечением экспериментальных данных, методов исследования поверхности и вычислительных методов будет показано, что водород очень прочно адсорбируется на металлическом никеле, что является наиболее вероятной причиной его низкой активности в анодном окислении водорода. Интересно, что частичное окисление поверхности Ni позволяет значительно увеличить скорость реакции. Согласно данным кинетического моделирования это связано с уменьшением абсолютного значения энергии адсорбции атомарного водорода на Ni. Раздел 4 обзора посвящен использованию никеля в качестве перспективного электрокаталитического материала для “прямых” топливных элементов, работающих на водородсодержащих жидких топливах. После краткого рассмотрения различных типов перспективных топлив мы сосредоточимся на обсуждении реакции окисления борогидрида. В данном процессе нахождение никеля в металлическом, а не в частично окисленном, состоянии является очевидным преимуществом. В конце обзора представлены выводы и перспективы.
2. ЭЛЕКТРОХИМИЧЕСКИЕ СВОЙСТВА НИКЕЛЯ
В связи с важной ролью Ni в гетерогенном катализе адсорбция различных молекул (H2, O2, H2O, CO, углеводородов и т.д.) на его поверхности широко изучалась с использованием как экспериментальных, так и вычислительных методов (см. источник [12] и ссылки в нем). В табл. 1 представлены рассчитанные с помощью теории функционала плотности значения энергии адсорбции для некоторых адсорбатов на грани (111) Ni и, в качестве сравнения, на гранях (111) Pt, Au и Pd. Заметим, что эти значения хорошо согласуются с экспериментальными данными (см. источник [13]). Можно видеть, что Н, OH и особенно O прочно адсорбируются на поверхности Ni. Энергии адсорбции H и OH близки друг к другу, но по-разному зависят от степени заполнения поверхности. Действительно, прочность адсорбции H на Ni(111) незначительно уменьшается с увеличением степени заполнения [14], тогда как сильное отталкивающее взаимодействие между адсорбатами OHad приводит к значительному снижению абсолютного значения энергии адсорбции ОНad при высокой степени заполнения [15].
Таблица 1.
Энергии адсорбции на гранях (111) Ni, Pt, Au и Pd, рассчитанные методом функционала плотности при нулевой степени заполнения поверхности
| Форма | Энергия адсорбции, эВ [ссылка] | |||
|---|---|---|---|---|
| Ni(111) | Pt(111) | Au(111) | Pd(111) | |
| H* | –2.81 [16] –2.81 [17] –2.86 [18] –2.89 [12] –2.94 [8] |
–2.70 [16] –2.72 [19] –2.72 [8] |
–2.04 [16] –2.12 [20] –2.18 [8] |
–2.83 [16] –2.88 [8] |
| OH* | –2.98 [16] –3.10 [21] –3.37 [18] –3.42 [22] –3.42 [17] |
–2.11 [16] –2.26 [19] –2.34 [22] |
–1.56 [16] –1.79 [20] –2.21 [22] |
–2.22 [16] –2.62 [22] |
| O* | –4.94 [16] –5.13 [12] –5.67 [17] |
–3.73 [16] | –2.47 [16] | –3.73 [16] |
| H2O* | –0.29 [17] –0.30 [18] –0.33 [22] |
–0.25 [19] –0.29 [22] |
–0.11 [20] –0.12 [22] |
–0.30 [22] |
| BH4* | –3.68 [18] –3.50 [23] |
– | –1.73 [20] –1.96 [23] |
– |
| BH + 3H* | – | –4.73 [19] –4.56 [23] |
– | –3.63 [23] |
Значения, представленные в табл. 1, были рассчитаны для границы раздела твердое тело/вакуум. При исследовании адсорбционных процессов в электрохимических системах удобным методом анализа является циклическая вольтамперометрия (ЦВА). В данном методе на рабочий электрод подается линейная развертка потенциала таким образом, что потенциал в конце цикла измерения возращается к первоначальному значению, а ток измеряется в зависимости от приложенного потенциала. Следует отметить, что этот метод дает информацию только о процессах, в которых происходит перенос заряда через границу раздела электрод/электролит. Учитывая нестабильность никелевых электродов в кислых средах, его электрохимические свойства преимущественно изучались в щелочных или нейтральных электролитах. Электрохимическое поведение никеля в щелочной среде исследуется более века и недавно было обобщено в ряде работ [24–29]. Наиболее важные характерные особенности ЦВА поликристаллического Ni-электрода в водном щелочном электролите представлены на рис. 2а. В зависимости от потенциала электрода можно наблюдать катодное выделение водорода или адсорбцию/десорбцию OH и H на поверхности Ni.
Рис. 2.
a – ЦВА поликристаллического Ni-электрода в деаэрированном 0.1 моль/л NaOH электролите при скорости развертки (v) 50 мВ/с и температуре (T) 25°C. Первая кривая ЦВА на рис. 2a (обозначена штрихпунктирной синей линией) зарегистрирована в диапазоне потенциалов от –0.2 до 0.5 В отн. ОВЭ, в то время как две другие кривые (первый цикл – сплошная черная линия, и второй цикл – пунктирная красная линия) получены в диапазоне потенциалов от –0.3 до 1.6 В отн. ОВЭ. б – Схема Боде для окислительно-восстановительного превращения Ni(OH)2/NiOOH. в – Заполнение поверхности электрода различными адсорбатами, смоделированное для анодного хода ЦВА, полученной на Ni- и Ni/NiOx-электродах в деаэрированном 0.1 моль/л NaOH электролите при v = 5 мВ/с и T = 25°C. г и д – In situ cпектры комбинационного рассеяния, полученные с использованием Ni-электрода при низких (г) и высоких (д) потенциалах в щелочном электролите. Использовано с разрешения: а – источник [29]; б – источник [38]; в – источник [36]; г – источник [39]; д – источник [40]. Copyright: a – 2020, American Chemical Society; б – 2015, American Chemical Society; в – 2018, Elsevier; г и д – по лицензии Creative Commons.

При потенциалах меньше 0 В отн. ОВЭ основной электрохимической реакцией на поверхности Ni-электрода является катодное выделение водорода (уравнение (II)). Реакция протекает через образование адсорбированного водорода (Ni–Had) в соответствии со стадией Фольмера (уравнение (III)) с последующим выделением молекулы H2 в результате либо стадии Гейровского (уравнение (IV)), либо стадии Тафеля (уравнение (V)) (в зависимости от условий).
(III)
${{{\text{H}}}_{{\text{2}}}}{\text{O + Ni + }}{{{\text{e}}}^{--}} \rightleftarrows {\text{Ni}}--{{{\text{H}}}_{{{\text{ad}}}}} + {\text{ O}}{{{\text{H}}}^{--}},{\text{ стадия Фольмера}},$(IV)
${\text{Ni--}}{{{\text{H}}}_{{{\text{ad}}}}}{\text{ + }}{{{\text{H}}}_{{\text{2}}}}{\text{O + }}{{{\text{e}}}^{--}} \rightleftarrows {{{\text{H}}}_{{\text{2}}}}{\text{ + O}}{{{\text{H}}}^{--}} + {\text{ Ni, стадия Гейровского}},$(V)
${\text{2Ni--}}{{{\text{H}}}_{{{\text{ad}}}}} \rightleftarrows {{{\text{H}}}_{{\text{2}}}}{\text{ + 2Ni, стадия Тафеля}}{\text{.}}$На анодном ходе ЦВА, при потенциалах выше 0 В отн. ОВЭ, поверхность металлического Ni окисляется с образованием сначала поверхностного слоя Ni(OH)2 (возможно, с некоторым вкладом NiO), который при потенциалах выше 1.2 В отн. ОВЭ окисляется до NiOOH. Структура различных оксидов Ni схематично представлена на рис. 2б. Ранее пики на ЦВА Ni-электрода в интервале потенциалов ниже 0.5 В отн. ОВЭ связывали исключительно с процессами образования/восстановления α-Ni(OH)2, которые включают перенос 2 электронов и протекают через образование промежуточного адсорбата Ni–OHad (уравнения (VI) и (VII)). Однако, согласно современным представлениям [29], основанным на экспериментальных и теоретических данных, поверхность металлического Ni при потенциалах меньше примерно 0.1 В отн. ОВЭ покрыта адсорбированным водородом (рис. 2в), окисление которого в обратной стадии Фольмера (уравнение (III)) предшествует образованию обратимого α-Ni(OH)2. Таким образом, в анодный пик, наблюдающийся в интервале потенциалов между 0 и 0.5 В отн. ОВЭ, дают вклад три процесса, последовательно протекающих на поверхности: обратная стадия Фольмера, электрохимическая адсорбция OH (уравнение (VI)) и окисление OH с образованием монослоя Ni(OH)2 (уравнение (VII)). В результате суммарное число перенесенных электронов равняется 3. При более высоких потенциалах α-Ni(OH)2 необратимо превращается в β-Ni(OH)2, который имеет плотноупакованную структуру вследствие отсутствия молекул воды, интеркалированных между слоями гидроксида Ni, как показано на рис. 2б. Это объясняет гораздо бóльшую устойчивость β-Ni(OH)2 к электрохимическому восстановлению до металлического Ni [25]. Кроме того, при этих потенциалах образуется некоторое количество приповерхностного NiO [24, 27, 30, 31]. Дальнейшее увеличение потенциала приводит к обратимому переходу Ni(OH)2 в β-NiOOH или γ-NiOOH (рис. 2б). В γ-NiOOH средняя степень окисления никеля превышает 3 (3.22) [32]. Формирование поверхностных оксидов никеля было подтверждено методом спектроскопии комбинационного рассеяния in situ (рис. 2г и 2д).
(VI)
${\text{O}}{{{\text{H}}}^{--}}{\text{ + Ni }} \rightleftarrows {\text{ Ni--O}}{{{\text{H}}}_{{{\text{ad}}}}}{\text{ + }}{{{\text{e}}}^{--}},$(VII)
${\text{Ni--O}}{{{\text{H}}}_{{{\text{ad}}}}} + {\text{ O}}{{{\text{H}}}^{--}} \rightleftarrows {\text{ Ni}}{{\left( {{\text{OH}}} \right)}_{{\text{2}}}}{\text{ + }}{{{\text{e}}}^{--}},$(VIII)
${\text{Ni}}{{\left( {{\text{OH}}} \right)}_{{\text{2}}}}{\text{ + O}}{{{\text{H}}}^{--}} \rightleftarrows {\text{ NiOOH + }}{{{\text{H}}}_{{\text{2}}}}{\text{O + }}{{{\text{e}}}^{--}}.$В присутствии на поверхности электрода “стабильных” оксидов форма ЦВА Ni значительно изменяется, как видно из сравнения сплошной и пунктирной кривых на рис. 2а. Эти изменения сопровождаются изменением поверхностных свойств Ni-электрода. Действительно, по данным микрокинетического моделирования при потенциалах до 0.1 В отн. ОВЭ поверхность металлического Ni полностью покрыта адсорбированным водородом, тогда как степень заполнения Нad снижается при наличии на поверхности оксидов (рис. 2в). Согласно результатам расчетов методами функционала плотности [33, 34] и микрокинетического моделирования [35–37] снижение степени заполнения поверхности адсорбированным водородом связано с уменьшением абсолютного значения энергии Гиббса адсорбции H на металлических центрах, расположенных вблизи поверхностных оксидных форм. Между тем адсорбция ОН в присутствии поверхностных оксидов становится более прочной (рис. 2в).
3. РЕАКЦИЯ ОКИСЛЕНИЯ ВОДОРОДА НА НИКЕЛЕ
Реакция анодного окисления водорода (которая протекает при потенциалах выше 0 В отн. ОВЭ) на металлических катализаторах происходит с образованием адсорбированного водорода (в качестве промежуточного соединения) и включает стадии Фольмера, Гейровского и Тафеля (уравнения (III)–(V)). Следовательно, величина энергии связи M–Had становится одним из наиболее важных параметров, влияющих на скорость реакции. Действительно, было показано, что наиболее каталитически активные металлы, такие как Pt и Pd, в соответствии с принципом Сабатье, связывают водород не слишком сильно, но и не слишком слабо [41]. На никеле адсорбция водорода прочнее, чем на Pt, что приводит к его значительно более низкой активности в реакциях водородного электрода (катодного выделения и анодного окисления водорода). Еще один недостаток никелевых электродов – их склонность к окислению при потенциалах выше 0.4 В отн. ОВЭ. Поскольку металлические центры необходимы для адсорбции водорода, полное заполнение поверхности никеля оксидами подавляет его каталитическую активность в водородных реакциях. Ni является очень реакционноспособным металлом, и образование оксидов на его поверхности может происходить либо во время приготовления электрокатализаторов (синтез высокодисперсных никелевых катализаторов часто заканчивается стадией пассивации, чтобы избежать их воспламенения при контакте с воздухом), либо при их хранении в условиях окружающей среды, либо при проведении электрохимических измерений (образование NiOOH при высоких анодных потенциалах, которое обсуждалось в разделе 2 настоящего обзора). Присутствие поверхностных оксидов оказывает значительное влияние на электрокаталитическую активность Ni в водородных реакциях и может либо повысить (см. обсуждение ниже), либо полностью подавить ее.
В литературе предлагаются различные способы повышения активности никелевых электродов в водородных реакциях, среди которых (i) добавление второго металла M (Cu, Mo, Co, и т.д.), (ii) введение гетероатомов (таких как N, P, S и B) в решетку Ni или в структуру углеродного носителя и (iii) модификация состояния поверхности Ni электрода. В табл. 2 на примере некоторых литературных данных показано, как различные факторы влияют на активность Ni-электрода в реакциях водородного электрода.
Таблица 2.
Сравнение активности никельсодержащих и коммерческих платиновых и палладиевых катализаторов в анодном окислении водорода в щелочной среде
| Электрокатализатор | T, °C | j0, мкA ${\text{см}}_{{{\text{Ni}}}}^{{ - 2}}$ | i0, A ${\text{г}}_{{{\text{Ni}}}}^{{ - 1}}$ | $i_{{\text{к}}}^{{50\,\,{\text{мВ}}}}$, A ${\text{г}}_{{{\text{Ni}}}}^{{ - 1}}$ | $j_{{\text{к}}}^{{50\,\,{\text{мВ}}}}$, мкA ${\text{см}}_{{{\text{Ni}}}}^{{ - 2}}$ | Источник |
|---|---|---|---|---|---|---|
| 46% Pt/C (TKK) | 21 | 570 ± 70 мкA ${\text{см}}_{{{\text{Pt}}}}^{{ - 2}}$ | 350 ± 50 A ${\text{г}}_{{{\text{Pt}}}}^{{ - 1}}$ |
371 A ${\text{г}}_{{{\text{Pt}}}}^{{ - 1}}$ |
600 мкА ${\text{см}}_{{{\text{Pt}}}}^{{ - 2}}$ |
[67] |
| 20% Pd/C (PremetekCo.) | 20 | 52 ± 2 мкA ${\text{см}}_{{{\text{Pd}}}}^{{ - 2}}$ |
38 ± 4 A ${\text{г}}_{{{\text{Pd}}}}^{{ - 1}}$ |
95 A ${\text{г}}_{{{\text{Pd}}}}^{{ - 1}}$ |
127 мкА ${\text{см}}_{{{\text{Pd}}}}^{{ - 2}}$ |
[68] |
| 60% Ni/KB | 25 | 28.6 ± 0.3 | 7.0 ± 0.1 | – | – | [69] |
| 42% Ni/XC-72 | 25 | 6.2 ± 0.6 | 5.7 ± 0.2 | – | – | [64] |
| 42% Ni-NiOx/XC-72 | 25 | 56 ± 10 | 22.4 ± 4.3 | 32.1 ± 4.8 | 83 ± 12 | |
| 63.2% Ni/NiO/C | – | 26 | – | 5 | – | [63] |
| 51.4% Ni3N/XC-72R | – | 14 | 12 | 24.38 | – | [70] |
| 71.7% np-Ni3N/C | КТ | – | 10.3 | 29.75 | – | [60] |
| 70% Ni/N-CNT | КТ | 28 | 3.5 | 9.3 | 75 | [71] |
| 32.5% Ni/h-BN/KB | 25 | 23 | 3.5 | – | – | [59] |
| 45% Ni/C | 30 | 14 ± 5 | 1.0 ± 0.2 | – | – | [58] |
| 54% Ni/BC | 30 | 13 ± 5 | 2.0 ± 0.7 | – | – | |
| 55% Ni/NC | 30 | 30 ± 5 | 4.8 ± 0.8 | – | – | |
| 55% Ni/SC | 30 | 40 ± 3 | 7.4 ± 0.9 | 11 | – | |
| 46.1% CeO2(r)-Ni/XC-72 | 30 | 38 ± 2 | – | 12.8 ± 0.3 | – | [55] |
| Ni4Mo + KB | – | – | 26.1 | 78.7 | – | [53] |
| 50% Ni9Mo1/KB | 25 | 27 ± 2 | 4.5 ± 0.2 | – | – | [49] |
| Co0.17Ni4.49Mo1 | 20 | 15 | – | – | 44 ± 5 | [50] |
| 25% Ni0.95Cu0.05/XC-72 | 25 | 14 ± 1 | 2.5 ± 0.2 | – | – | [43] |
| 50% Ni0.95Cu0.05/KB | КТ | 25 ± 1 | 0.9 ± 0.1 | – | – | [44] |
| 50% Ni0.95Cu0.05/ Denka-AApyr | 25 | – | 10 | – | – | [46] |
| Ni5.2WCu2.2 | 25 | 14 | 2.54 | – | – | [48] |
Обозначения: T – температура (КТ означает комнатную температуру); j0 и i0 –плотности тока обмена, нормированные на площадь поверхности и на массу соответственно; $j_{{\text{к}}}^{{50\,{\text{мВ}}}}$ и $i_{{\text{к}}}^{{50\,{\text{мВ}}}}$ – плотности кинетического тока при E = 50 мВ отн. ОВЭ, нормированные на площадь поверхности и на массу соответственно. Прочерки означают отсутствие данных.
Легирование Ni другим металлом – один из самых распространенных подходов к изменению его электрокаталитических свойств. Среди неблагородных металлов, приводящих к повышению активности Ni в анодном окислении водорода, наиболее широко используются медь [42–47] (в сочетании с вольфрамом [48]), молибден [49–53] (в сочетании с кобальтом [50]), кобальт [47], железо [47, 54] и церий [55]. Обычно наблюдаемое увеличение электрокаталитической активности Ni связывают с уменьшением прочности связи адсорбированного водорода с поверхностью в результате электронного эффекта, вызванного вторым металлом.
Следует, однако, отметить, что в некоторых исследованиях предлагается альтернативное объяснение. В частности, улучшенные характеристики CeO2(r)-Ni/XC-72 (где CeO2(r) обозначает CeO2, подвергнутый восстановительной обработке) авторы приписывают “бифункциональному” механизму, согласно которому адсорбция ОН происходит на поверхности частично восстановленного CeO2, содержащего кислородные вакансии, в то время как Н адсорбируется на близлежащих центрах Ni. Реакция завершается рекомбинацией Had и OHad с образованием H2O в соответствии с уравнением (IX):
(IX)
${{{\text{H}}}_{{{\text{ad}}}}} + {\text{O}}{{{\text{H}}}_{{{\text{ad}}}}} \rightleftarrows {{{\text{H}}}_{{\text{2}}}}{\text{O}}.$Указанный механизм имеет некоторое сходство с механизмом парциального окисления метана на никеле, нанесенном на допированный гадолинием оксид церия, который был предложен в гетерогенном катализе на основе in situ исследований методом спектроскопии комбинационного рассеяния [56]. Этот механизм включает стадию окисления углерода, образующегося в результате диссоциативной адсорбции метана на поверхности Ni, решеточным кислородом допированного оксида церия. Что касается электрокаталитического окисления водорода на монометаллических Ni электродах, то даже если адсорбаты Had и OHad могут сосуществовать на поверхности (рис. 2в), их прочная адсорбция делает стадию рекомбинации (IX) маловероятной [57].
Модификация углеродных носителей гетероатомами обычно проводится путем пиролиза органических молекул (таких как дибензилдисульфид, меламин, фенилборная кислота и т.д.) на их поверхности. Учитывая взаимодействие металла с носителем, было высказано предположение, что введение гетероатомов в состав носителя влияет на электронную структуру нанесенных наночастиц Ni, способствуя тем самым ослаблению прочности адсорбции водорода на никеле [58]. Кроме того, было показано, что покрытие поверхности наночастиц Ni слоем гексагонального нитрида бора снижает их склонность к окислению [59]. Однако наиболее интересные результаты были получены при введении гетероатомов, например азота, в структуру наночастиц Ni. Так, предполагается, что энергия адсорбции водорода на образующейся фазе Ni3N близка к оптимальной, что, наряду с более низкой энергией активации стадии образования воды, объясняет ее высокую активность в анодном окислении водорода по сравнению с монометаллическим никелем [60, 61].
Как показано в разделе 2, образование стабильных оксидов Ni (а именно β-Ni(OH)2 и в некоторой степени NiO) на поверхности электрода может происходить уже в процессе приготовления катализатора, его электрохимической обработки или при контакте с воздухом и существенно влияет на его межфазные свойства. Сосуществование оксидов Ni (предположительно являющихся центрами для адсорбции H2O) с металлическим Ni (центрами для адсорбции водорода) приводит к формированияю гетероструктур Ni/NiOx, которые проявляют значительно более высокую удельную активность в анодном окислении водорода по сравнению с металлическим Ni. Напомним, что металлический Ni обладает очень низкой активностью в анодном окислении водорода, в то время как никелевый электрод, полностью покрытый оксидами, вообще неактивен (из-за отсутствия на поверхности центров, необходимых для адсорбции водорода). Поэтому неудивительно, что активность никеля в водородных реакциях зависит от степени заполнения его поверхности оксидами (рис. 3а), при этом оптимум наблюдается примерно при 30–50% заполнении (в зависимости от морфологии Ni-электрода) [37, 62]. Результаты расчетов методом функционала плотности [33, 34] и микрокинетического моделирования [35–37] указывают на ослабление прочности адсорбции H на Ni-центрах, расположенных вблизи кислородсодержащих поверхностных участков. Кроме того, присутствие поверхностных оксидов, вероятно, стабилизирует молекулы воды, предположительно, за счет водородных связей. Оба фактора приводят к увеличению константы скорости стадии Фольмера, которая считается скоростьопределяющей в реакции окисления водорода на Ni [29]. Кроме того, присутствие оксидов Ni на поверхности электрода может оказывать воздействие на структуру двойного электрического слоя, который в свою очередь также влияет на скорость реакции. Следует заметить, что “бифункциональный” механизм, описанный выше для электрокатализатора CeO2(r)-Ni/XC-72, может иметь место и в случае гетероструктур Ni/NiOx [63]. Однако в настоящее время невозможно сказать, какой из факторов – вклад стадии рекомбинации (уравнение IX) или повышение константы скорости стадии Фольмера в присутствии поверхностных оксидов – является определяющим в механизме анодного окисления водорода на Ni/NiOx [37]. Эксперименты, проведенные при различных температурах (рис. 3б), показали небольшое снижение кажущейся энергии активации с 30 кДж/моль на металлическом Ni до 26 кДж/моль на электроде Ni/NiOx, что согласуется с уменьшением прочности адсорбции Had на Ni/NiOx по сравнению с металлическим Ni-электродом [36].
Рис. 3.
a – Зависимость плотности тока обмена водородного электрода, нормированного либо на электрохимически активную (ECSA), либо на геометрическую площадь поверхности массивных электродов Ni/NiOx от степени заполнения поверхности оксидами никеля ${{\Theta }_{{{\text{Ni}}{{{\text{O}}}_{x}}}}}.$ б – Зависимость плотности тока обмена водородного электрода от температуры в координатах Аррениуса, полученная для массивного никелевого электрода с металлической поверхностью (Ni) и для частично окисленного электрода (Ni/NiOx) со степенью заполнения NiOx ~ 15%. в – Изображение катализатора Ni/NiOx/XC-72, полученное методом просвечивающей электронной микроскопии, г – сравнение удельной активности для массивных Ni и Ni/NiOx электродов и соответствующих наночастиц. Использовано с разрешения: а – источник [37], б – источник [36], в – источник [64]. Copyright: a – 2020, Springer publisher; б и в – 2018, Elsevier.

Наряду с контролем состояния поверхности Ni, определяющего его удельную активность в анодном окислении водорода, еще одной важной задачей является получение электрокаталитических материалов с высокой удельной поверхностью для достижения высокой активности на единицу массы. Электроосаждение – удобный способ приготовления нанесенных наночастиц металла. Оно не требует ни органических лигандов (которые в случае неполного удаления с поверхности могут оказывать влияние на каталитическую активность вследствие блокирования активных центров), ни стадии пассивации (которая необходима при использовании химических методов синтеза и приводит к образованию поверхностного слоя из оксидов Ni). Кроме того, контроль потенциала электрода позволяет управлять процессами нуклеации и роста частиц при электроосаждении. Mетодом электроосаждения был приготовлен электрокатализатор Ni/XC-72, содержащий равномерно распределенные по поверхности углеродного носителя наночастицы Ni размером около 10 нм (рис. 3в). Целенаправленное частичное окисление их поверхности позволило получить высокоактивный электрокатализатор Ni/NiOx/XC-72 с оптимальным соотношением Ni и NiOx на поверхности. Он показал одно из самых высоких (среди катализаторов, не содержащих благородных металлов) значений удельной активности в анодном окислении водорода, которое сопоставимо с активностью коммерческого электрокатализатора Pd/C (табл. 2) [64]. Отметим, что удельная активность наночастиц Ni (как в металлическом, так и частично окисленном состоянии) выше активности массивного никеля (рис. 3г), что может указывать на размерный эффект и согласуется с данными Давыдовой и соавт. [65]. Однако детальных исследований размерного эффекта для водородных реакций на никеле не проводилось. Кроме того, было показано, что гетероструктуры Ni/NiOx гораздо устойчивее по отношению к отравлению монооксидом углерода по сравнению с платиновыми катализаторами [63]. Учитывая то, что в настоящее время водород преимущественно производится, в основном, путем паровой конверсии природного газа и обычно содержит примесь монооксида углерода, устойчивость к нему катализаторов окисления водорода является серьезным достоинством.
Наконец, мы полагаем, что влияние поверхностных оксидов необходимо учитывать, в том числе, и для биметаллических катализаторов окисления водорода. Действительно, было показано, что добавление второго металла может стабилизировать либо металлические (медь [42]), либо окисленные (хром [66]) центры на поверхности Ni и, таким образом, косвенно влиять на активность таких никельсодержащих электродов.
4. ОКИСЛЕНИЕ ВОДОРОДСОДЕРЖАЩИХ БЕЗУГЛЕРОДНЫХ ЖИДКИХ ТОПЛИВ НА НИКЕЛЕ
Интерес к водородсодержащим безуглеродным жидким топливам обусловлен их высокой (по сравнению с водородом) плотностью энергии на единицу объема, удобством хранения и отсутствием выбросов углекислого газа при их окислении (по сравнению с органическими топливами, такими как метанол, этанол и т.д.). В табл. 3 приведены значения плотности энергии (на единицу объема) некоторых видов безуглеродных топлив в сравнении с водородом. Можно отметить, что жидкие топлива довольно привлекательны по сравнению со сжатым или сжиженным водородом, особенно если учесть энергию, которую необходимо затратить для сжижения водорода, а также потери вследствие его испарения (в частности, из-за самопроизвольной орто-пара конверсии водорода [72]) и проблемы, связанные с транспортировкой водорода. Что касается хранения водорода в обратимо связанном состоянии (например, в виде твердых гидридов), то ни один из доступных в настоящее время материалов не удовлетворяет одновременно совокупности всех требований, предъявляемых к его эффективному хранению [73].
Таблица 3.
Стандартные электродные потенциалы и теоретические* значения плотности энергии для безуглеродных топлив [74]
| Топливо | Анодная реакция | Стандартный электродный потенциал (В отн. ОВЭ) |
Теоретическая плотность энергии на единицу объема, Вт ч л–1 |
|---|---|---|---|
| Жидкий водород | 2H2O + 2e– = H2 + 2OH– | 0 | 2350 |
| Сжатый водород (70 МПа) |
2H2O + 2e– = H2 + 2OH– | 0 | 1300 |
| Аммиак | 2NH3 + 6OH– = 2N2 + 6H2O + 6e– | –0.06 | 1704 (35 мас. %) |
| Гидразин | N2H4 + 4OH– = N2 + 4H2O + 4e– | –0.33 | 4269 (чистый) |
| Аминборан | NH3BH3 + 6OH– = ${\text{BO}}_{2}^{ - }$ + ${\text{NH}}_{4}^{ + }$ + 4H2O + 6e– | –0.39 | 655 (2 M) |
| Борогидрид | ${\text{BH}}_{4}^{ - }$ + 8OH– = ${\text{BO}}_{2}^{ - }$ + 6H2O + 8e– | –0.41 | 2940 (30 мас. %) |
Для окисления жидких топлив (аммиака [75], гидразина [76, 77], амминборана и борогидрида [78–80]) применялись различные материалы, среди которых наиболее широко исследована Pt. Платина и материалы на ее основе показывают высокие токи в реакции окисления гидразина, амминборана и борогидрида при потенциалах выше 0 В отн. ОВЭ. Однако высокая активность платины в катодном выделении водорода препятствует окислению топлив при потенциалах ниже 0 В отн. ОВЭ и не позволяет в полной мере использовать преимущество их низких стандартных потенциалов (табл. 3). Катализаторы на основе Pt имеют еще один недостаток – быстрое “самоотравление” прочно адсорбированными промежуточными продуктами реакции, такими как BHad в случае окисления борогидрида, образующимися в результате диссоциативной адсорбции ${\text{BH}}_{4}^{ - }$ на поверхности электрода.
В настоящем обзоре мы сосредоточимся на обсуждении реакции окисления борогидрида на никелевых электродах. Однако вначале необходимо кратко обсудить показатели активности для этой реакции. В отличие от водородных реакций, определение плотности тока обмена для окисления борогидрида представляет собой нетривиальную задачу, поскольку реакция необратима, и при нулевом токе на границе раздела электрокатализатор/электролит устанавливается так называемый потенциал разомкнутой цепи (ПРЦ), а не равновесный потенциал. Последнее связано с одновременным протеканием выделения водорода и окисления борогидрида в области отрицательных потенциалов в шкале ОВЭ. Следовательно, подходящими показателями активности в окислении борогидрида являются значения ПРЦ и плотности тока при заданном потенциале (j(E)), положительном по отношению к ПРЦ. Чем ниже ПРЦ и выше j(E), тем выше эффективность прямого борогидридного топливного элемента.
Начиная с 2000 г. в окислении борогидрида были исследованы многие Ni-содержащие материалы, включая сплавы Ni с такими металлами, как Co [81–83], Cu [84–86], Ru [87, 88], Pd [89–92], Au [93–96], Pt [90, 97–103] и редкоземельными металлами [104–107]. Однако большинство этих материалов имело в своем составе благородные металлы, которые и определяли активность катализатора в окислении борогидрида. Катализаторы, не содержащие благородных металлов, немногочисленны и зачастую не проявляют заметной активности в окислении борогидрида при потенциалах, близких или меньших 0 В отн. ОВЭ. Следует отметить, что, как и в случае с анодным окислением водорода, до недавнего времени редко уделялось должное внимание влиянию состояния поверхности электродов на их активность в окислении борогидрида. Отсутствие контроля за состоянием поверхности Ni привело к значительным расхождениям в показателях активности материалов на основе Ni в данном процессе. Анализ литературы показывает, что низкая активность и высокие значения ПРЦ наблюдались для никелевых электродов, поверхность которых была в значительной степени окислена [94, 108]. Об этом говорит отсутствие анодных пиков при потенциалах ниже 0.5 В отн. ОВЭ на ЦВА в фоновом электролите в этих исследованиях. Напротив, в работах, где авторы избегали окисления поверхности электрода при высоких потенциалах, отмечалась заметная активность никеля в окислении борогидрида при достаточно низких значениях потенциала [106, 109, 110]. Таким образом, анализ литературы свидетельствует о том, что аналогично рассмотренному в разделе 3 примеру с окислением водорода для достижения высокой электрокаталитической активности никеля в окислении борогидрида необходим тщательный контроль состояния его поверхности. Однако, как показано ниже, в отличие от окисления водорода, для окисления борогидрида следует избегать присутствия оксидов Ni на поверхности электрода.
На рис. 4а приведены ЦВА наночастиц никеля, полученных методом электроосаждения на поверхность углеродного носителя (материал, сходный с представленным на рис. 3в), в присутствии NaBH4. Можно видеть, что “частично окисленный” (красная кривая 2) Ni-электрод, содержащий как металлические, так и NiOx-центры на поверхности, обладая чрезвычайно высокой активностью в водородных реакциях (см. раздел 3 этого обзора), демонстрирует меньшие токи в окислении борогидрида и более положительный ПРЦ по сравнению с металлическим Ni-электродом (синяя кривая 3). При этом активность в окислении борогидрида электрода, полностью покрытого оксидами Ni, еще ниже (черная кривая 1). Низкая скорость катодного выделения водорода, которая, как указано выше, связана с прочной адсорбцией атомов водорода на металлическом Ni, объясняет существенно меньшее значение ПРЦ для Ni, что является его значительным преимуществом по сравнению с Pt в реакции окисления борогидрида (рис. 4б). Измерения in situ с помощью метода ИК-Фурье спектроскопии (инфракрасная спектроскопия с преобразованием Фурье) (рис. 4в) показали образование бората $\left( {{\text{B(OH)}}_{4}^{ - }} \right),$ конечного продукта окисления борогидрида, уже при ПРЦ [111]. По мере увеличения потенциала электрода скорость окисления борогидрида начинает превышать скорость выделения водорода, что приводит к изменению рН вблизи поверхности электрода и частичному превращению ${\text{B(OH)}}_{4}^{ - }$ в B(OH)3. Стоит отметить, что измерения с использованием метода ИК-Фурье спектроскопии не позволили обнаружить каких-либо растворимых промежуточных соединений (например, BH3OH–), образование которых обсуждалось для Pt и Au в более ранних публикациях [112–114]. Отсутствие в растворе продуктов частичного окисления согласуется с данными расчетов методом функционала плотности, которые свидетельствуют о нестабильности продуктов парциального окисления борогидрида на поверхности никеля и их преимущественном окислении до B(OH)3. Согласно данным теоретических расчетов [18, 111] высокая скорость окисления борогидрида на Ni при низких электродных потенциалах обусловлена не только медленной скоростью выделения водорода, но также и более благоприятными энергиями адсорбции Had, OHad и борсодержащих реакционноспособных промежуточных соединений на Ni по сравнению с Pt, Pd или Au. В отличие от Pt, на поверхности которой BH4 диссоциирует с образованием адсорбированного BHad (табл. 1), что ведет к сильному отравлению поверхности катализатора, на Ni(111) происходит молекулярная адсорбция BH4 (табл. 1), что приводит к гораздо меньшему отравлению поверхности [18]. За стадией молекулярной адсорбции на Ni следует последовательная диссоциация связей B–H и образование связей B–OH.
Рис. 4.
ЦВА, полученные на вращающемся дисковом электроде для катализаторов на основе никеля, электроосажденного на углерод (NiED/C) с одинаковым содержанием никеля, но различным состоянием поверхности (а) и металлического никеля электроосажденного на углерод в сравнении с Pt/C, Pd/C, и Au/C в деаэрированном 1.0 моль/л NaOH + + 5 ммоль/л NaBH4 электролите, v = 20 мВ/с, ω = 1600 об./мин (б); in situ ИК-Фурье спектры массивного Ni электрода в 0.2 моль/л NaBH4 + 0.1 моль/л NaOH электролите при различных потенциалах электрода, указанных на графике (в); измерения методом дифференциальной электрохимической масс-спектрометрии на протравленном войлочном никелевом электроде в деаэрированном 1 моль/л NaOH + 50 ммоль/л NaBD4 электролите, v = 10 мВ/с (г). Фарадеевский ток (Iф – черная кривая), ионные токи (H2 – красная, HD – зеленая, D2 – синяя кривая), пересчитанные в фарадеевские с использованием независимо определенной калибровочной постоянной (K = 2.197 × 1010). Использовано с разрешения: а, б – источник [18]; в, г – источник [111]. Copyright: a, б – 2019, American Chemical Society; в, г – 2021, Elsevier.

Примечательно, что Ni превосходит Pt не только в трехэлектродной ячейке с жидким электролитом, но и в топливном элементе с полимерной мембраной (рис. 5). При этом наилучшие характеристики показал макропористый электрод на основе никелевого “войлока”, на поверхность которого электроосаждены наночастицы никеля (рис. 5).
Рис. 5.
Характеристики (напряжение на элементе и мощность в зависимости от плотности тока) прямого борогидридного топливного элемента (а) и ток при постоянном напряжении 1 В (без учета омических потерь) в зависимости от времени (б) для электродов NiED/eNFT (травленный Ni-войлок, на поверхность которого нанесены наночастицы Ni методом электроосаждения), NiED/GDL (Ni, электроосажденный на поверхность газодиффузионного электрода), Pt black/GDL (платиновая чернь, нанесенная на поверхность газодиффузионного электрода) и Pt/GDL (наночастицы Pt, нанесенные на поверхность газодиффузионного электрода) при T = 60°C, скорости потока электролита 50 мл/мин; фотографии, полученные с помощью растрового электронного микроскопа, для чистой углеродной бумаги (Sigracet 38 AA, SGL carbon), использованной в качестве газодиффузионного слоя (GDL) (в), Ni-войлока (д) и Ni-частиц, электроосажденных на углеродную бумагу SGL (г) или травленный Ni-войлок (е). Использовано с разрешения: а, б, д, е – источник [115]; в, г – источник [18]. Copyright: a, б, д, е – 2020, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim; в, г – 2019, American Chemical Society.

Следует отметить, что наряду с достоинствами (чрезвычайно низкий ПРЦ и высокие токи окисления при низких потенциалах), Ni-электроды обладают и недостатком по сравнению с Pt. Действительно, из рис. 4б видно, что предельный ток окисления борогидрида на Ni-электроде в два раза меньше, чем на Pt. Это позволяет предположить, что реакция на нем протекает с переносом только 4 электронов вместо максимально возможных 8. Данное предположение подтверждается измерениями методом дифференциальной электрохимической масс-спектрометрии (рис. 4г), которые свидетельствуют об образовании двух молекул водорода на один анион ${\text{BH}}_{4}^{ - }$ в процессе анодного окисления борогидрида на Ni. По результатам исследования с использованием ${\text{BD}}_{4}^{ - }$ было установлено, что водород образуется из борогидрида, а не из молекул воды.
В будущем представляет интерес разработка электрокаталитических материалов, которые, с одной стороны, будут обладать высокими токами окисления борогидрида при низких (ниже 0 В отн. ОВЭ) электродных потенциалах (что характерно для металлического Ni), а с другой стороны, позволят достичь максимально возможного (8) количества переносимых электронов (что характерно для Pt).
5. ВЫВОДЫ И ПЕРСПЕКТИВЫ
Никель широко распространен в гетерогенном катализе, однако реже применяется для “низкотемпературных” (с температурой ниже 100°C) электрокаталитических процессов. Это связано не с низкой электрокаталитической активностью Ni, а скорее с реакционной способностью этого металла, приводящей к его взаимодействию с кислородом (и, в конечном счете, с другими кислородсодержащими соединениями) при приготовлении катализатора, его исследовании физико-химическими методами, при хранении в условиях окружающей среды и использовании в электрохимической ячейке. Высокотемпературная обработка поверхности, которая необходима для восстановления поверхностных оксидов и обычно применяется в случае гетерогенных каталитических процессов в газовой фазе, вряд ли совместима с низкотемпературными электрокаталитическими процессами. Электрокатализаторы на основе Ni, как правило, хранятся в условиях окружающей среды, что ведет к окислению их поверхности и требует последующего целенаправленного электрохимического восстановления, которое обычно проводится при комнатной температуре. К сожалению, оксиды Ni трудно восстанавливаются при низких температурах. Это приводит к невоспроизводимому состоянию поверхности и, как следствие, большому разбросу в величинах активности никелевых электрокатализаторов.
В представленном обзоре показано, что тщательный контроль состояния поверхности Ni-электрокатализаторов позволяет получить рекордные показатели активности в низкотемпературных реакциях окисления различных водородсодержащих топлив, например H2 и NaBH4. Показано, что для окисления борогидрида необходим металлический никель, который превосходит Pt при низких потенциалах (что предпочтительно для топливных элементов) и позволяет достичь выдающихся показателей при использовании на аноде прямых борогидридных топливных элементов. Тем не менее, недостатком окисления борогидрида на никеле является протекание реакции с переносом не восьми (соответствующих полному окислению борогидрида), а только четырех электронов. Еще четыре электрона расходуются на образование молекулярного водорода. Таким образом, дальнейшие исследования должны быть направлены на разработку биметаллических или композитных электродов на основе Ni, которые могли бы сочетать высокую активность окисления борогидрида при низких потенциалах с высокой селективностью по отношению к восьмиэлектронному процессу.
В отличие от окисления борогидрида окисление молекулярного водорода на металлическом Ni является медленным процессом. Реакцию можно значительно ускорить, если наряду с металлическими центрами на поверхности будут присутствовать (гидр)оксиды Ni, роль которых двояка. С одной стороны, они оказывают электронное воздействие на близлежащие центры металлического Ni, снижая прочность адсорбции на них водорода, которая в противном случае слишком высока, что приводит к блокированию поверхности металлического Ni адсорбированным водородом. С другой стороны, оксиды Ni представляют собой центры для адсорбции воды (бифункциональный эффект), увеличивая константу скорости скоростьопределяющей стадии Фольмера. Кроме того, оксиды Ni, вероятно, влияют на структуру двойного электрического слоя на поверхности электрода, что также повышает скорость реакции окисления водорода. В случае композитных электродов на основе Ni/NiOx наибольшие значения скорости реакции окисления водорода были зафиксированы при степени заполнения поверхности NiOx от 30 до 50%. Полностью окисленный (пассивированный) Ni-электрод неактивен из-за отсутствия поверхностных центров для адсорбции H2, в то время как поверхность металлического Ni в отсутствие поверхностных оксидов блокирована прочно адсорбированным водородом, что делает его неактивным в области низких потенциалов. Необходимость оптимального соотношения Ni- и NiOx-центров на поверхности Ni-электрокатализаторов затрудняет обеспечение высокой активности в реакции окисления водорода в течение длительного времени. Действительно, длительное функционирование гетероструктур Ni/NiOx вблизи равновесного электродного потенциала (что желательно для применения в топливных элементах) приводит к уменьшению содержания метастабильного NiOx и сопутствующему снижению каталитической активности. С другой стороны, воздействие высоких анодных потенциалов приводит к пассивации поверхности, что также сопровождается снижением активности. Таким образом, для применения никелевых катализаторов в топливных элементах с анионообменной мембраной необходимо искать подходы для стабилизации активных (но метастабильных) гетероструктур Ni/NiOx. Среди них перспективными представляются подходы, основанные на взаимодействии металл–носитель, на использовании биметаллических наноматериалов или введении гетероатомов.
В будущем особое внимание следует уделить исследованию стабильности катализаторов в условиях реакции и разработке подходов к созданию электрокаталитических материалов на основе Ni, способных работать в течение длительного времени. Возможные процессы деградации включают вышеупомянутое чрезмерное окисление или восстановление поверхности (в зависимости от сферы применения), отравление примесями (CO и т.д.), агломерацию наночастиц Ni, растворение Ni и коррозию углеродных носителей. Хочется обратить внимание на следующее: имеющиеся к настоящему времени результаты дают основания полагать, что по сравнению с Pt наноматериалы на основе Ni обладают более высокой стабильностью в щелочных электролитах при умеренных электродных потенциалах [29].
Методы приготовления наноматериалов на основе Ni для низкотемпературных электрокаталитических процессов должны удовлетворять ряду требований: (i) полученные катализаторы должны обладать высокой удельной площадью поверхности (следовательно, необходимо синтезировать никель в форме наночастиц); (ii) носитель должен иметь достаточную электронную проводимость (этому требованию удовлетворяют, например, углеродные носители); (iii) способы приготовления должны обеспечивать возможность контролируемой “настройки” состояния поверхности никелевых наночастиц с целью оптимизации содержания металлических и оксидных центров на поверхности (в зависимости от сферы применения). Электрохимическое осаждение Ni из водных растворов солей является удобным способом приготовления высокодисперсных наночастиц Ni, который позволяет избежать стадии пассивации. Последняя, как правило, используется в химических методах приготовления и пагубно влияет на электрокаталитические свойства никеля.
Прогресс, достигнутый в последние годы в понимании реакций окисления водорода и борогидрида на никелевых и никельсодержащих электродах, разрабатываемых для низкотемпературных топливных элементов, открывает перспективы для создания материалов, не содержащих благородных металлов, для других практически значимых применений, таких как электросинтез химических соединений, например, с использованием компонентов биомассы.
Список литературы
Кобозев Н.И., Монбланова В.В. // Журн. Физ. Химии. 1936. Т. 7. С. 645.
Кобозев Н.И., Некрасов Н.И. // Электрохимия. 1939. Т. 36. № 68. С. 529.
Griffin W.L., Chassé M. / Encycl. Geochemistry. Ed. White W.M., Springer International Publishing Switzerland, 2016. P. 1.
De S., Zhang J., Luque R., Yan N. // Energy Environ. Sci. 2016. V. 9. № 11. P. 3314.
Stoffels M.A., Klauck F.J.R., Hamadi T., Glorius F., Leker J. // Adv. Synth. Catal. 2020. V. 362. № 6. P. 1258.
Maier L., Schädel B., Delgado K.H., Tischer S., Deutschmann O. // Top. Catal. 2011. V. 54. № 13–15. P. 845.
Ceyer S.T. // Acc. Chem. Res. 2001. V. 34. № 9. P. 737.
Ferrin P., Kandoi S., Nilekar A.U., Mavrikakis M. // Surf. Sci. 2012. V. 606. № 7–8. P. 679.
Wang Y., Cao F., Lin W., Zhao F., Zhou J., Li S., Qin G. // J. Mater. Chem. 2019. V. 7. № 30. P. 17834.
Song S., Yao S., Cao J., Di L., Wu G., Guan N., Li L. // Appl. Catal. B: Environ. 2017. V. 217. P. 115.
Chen S., Qian T.T., Ling L.L., Zhang W., Gong B.B., Jiang H. // ChemSusChem. 2020. V. 13. № 20. P. 5507.
Bai Y., Kirvassilis D., Xu L., Mavrikakis M. // Surf. Sci. 2019. V. 679. P. 240.
Christmann K., Schober O., Ertl G., Neumann M. // J. Chem. Phys. 1974. V. 4528. № 1974. P. 4528.
Greeley J., Mavrikakis M. // Surf. Sci. 2003. V. 540. № 2–3. P. 215.
Juarez F., Salmazo D., Savinova E.R., Quaino P., Belletti G., Santos E., Schmickler W. // J. Electroanal. Chem. 2019. V. 832. P. 137.
Herron J.A., Scaranto J., Ferrin P., Li S., Mavrikakis M. // ACS Catal. 2014. V. 4. № 12. P. 4434.
Zhu Y.A., Chen D., Zhou X.G., Yuan W.K. // Catal. Today. 2009. V. 148. № 3–4. P. 260.
Oshchepkov A.G., Braesch G., Ould-Amara S., Rostamikia G., Maranzana G., Bonnefont A., Papaefthimiou V., Janik M.J., Chatenet M., Savinova E.R. // ACS Catal. 2019. V. 9. № 9. P. 8520.
Rostamikia G., Janik M.J. // Electrochim. Acta. 2010. V. 55. № 3. P. 1175.
Rostamikia G., Janik M.J. // J. Electrochem. Soc. 2009. V. 156. № 1. P. B86.
Mohsenzadeh A., Richards T., Bolton K. // Surf. Sci. 2016. V. 644. P. 53.
Huang S.-C., Lin C.-H., Wang J.-H. // J. Phys. Chem. C. 2010. V. 114. № 21. P. 9826.
Escaño M.C.S. // SPR Electrochem. 2017. V. 14. № 3. P. 1.
Hall D.S., Bock C., MacDougall B.R. // J. Electrochem. Soc. 2013. V. 160. № 3. P. F235.
Hall D.S., Lockwood D.J., Bock C., MacDougall B.R. // Proc. R. Soc. A Math. Phys. Eng. Sci. 2015. V. 471. № 2174. P. 20140792.
Alsabet M., Grden M., Jerkiewicz G. // Electrocatal. 2011. V. 2. № 4. P. 317.
Alsabet M., Grden M., Jerkiewicz G. // Electrocatal. 2014. V. 5. № 2. P. 136.
Alsabet M., Grdeń M., Jerkiewicz G. // Electrocatal. 2015. V. 6. № 1. P. 60.
Oshchepkov A.G., Braesch G., Bonnefont A., Savinova E.R., Chatenet M. // ACS Catal. 2020. V. 10. № 13. P. 7043.
Medway S.L.L., Lucas C.A.A., Kowal A., Nichols R.J.J., Johnson D. // J. Electroanal. Chem. 2006. V. 587. № 1. P. 172.
Melendres C.A., Pankuch M. // J. Electroanal. Chem. 1992. V. 333. № 1–2. P. 103.
Morishita M., Ochiai S., Kakeya T., Ozaki T., Kwawabe Y., Watada M., Tanase S., Sakai T. // Electrochem. 2008. V. 76. № 11. P. 802.
Dong Y., Dang J., Wang W., Yin S., Wang Y. // ACS Appl. Mater. Interfaces. 2018. V. 10. № 46. P. 39624.
Liu S., Ishimoto T., Koyama M. // Appl. Surf. Sci. 2015. V. 333. P. 86.
Oshchepkov A.G., Bonnefont A., Saveleva V.A., Papaefthimiou V., Zafeiratos S., Pronkin S.N., Parmon V.N., Savinova E.R. // Top. Catal. 2016. V. 59. № 15–16. P. 1319.
Oshchepkov A.G., Bonnefont A., Parmon V.N., Savinova E.R. // Electrochim. Acta. 2018. V. 269. P. 111.
Oshchepkov A.G., Bonnefont A., Savinova E.R. // Electrocatal. 2020. V. 11. № 2. P. 133.
Klaus S., Cai Y., Louie M.W., Trotochaud L., Bell A.T. // J. Phys. Chem. C. 2015. V. 53. № 9. P. 1689.
Faid A.Y., Barnett A.O., Seland F., Sunde S. // Electrochim. Acta. 2020. V. 361. P. 137040.
Diaz-Morales O., Ferrus-Suspedra D., Koper M.T.M.M. // Chem. Sci. 2016. V. 7. № 4. P. 2639.
Sheng W., Myint M., Chen J.G., Yan Y. // Energy Environ. Sci. 2013. V. 6. № 5. P. 1509.
Oshchepkov A.G., Simonov P.A., Cherstiouk O.V., Nazmutdinov R.R., Glukhov D.V., Zaikovskii V.I., Kardash T.Y., Kvon R.I., Bonnefont A., Simonov A.N., Parmon V.N., Savinova E.R. // Top. Catal. 2015. V. 58. № 18–20. P. 1181.
Cherstiouk O.V., Simonov P.A., Oshchepkov A.G., Zaikovskii V.I., Kardash T.Y., Bonnefont A., Parmon V.N., Savinova E.R. // J. Electroanal. Chem. 2016. V. 783. P. 146.
Roy A., Talarposhti M.R., Normile S.J., Zenyuk I.V., De Andrade V., Artyushkova K., Serov A., Atanassov P. // Sustain. Energy Fuels. 2018. V. 2. № 10. P. 2268.
Wang G., Li W., Huang B., Xiao L., Lu J., Zhuang L. // ACS Appl. Energy Mater. 2019. V. 2. № 5. P. 3160.
Rezaei Talarposhti M., Asset T., Roy A.J., Artyushkova K., Tsui L. kun, Garzon F.H., Serov A., Atanassov P. // Electrochem. Commun. 2021. V. 125. P. 106999.
Davydova E., Zaffran J., Dhaka K., Toroker M., Dekel D. // Catalysts. 2018. V. 8. № 10. P. 454.
Qin S., Duan Y., Zhang X.L., Zheng L.R., Gao F.Y., Yang P.P., Niu Z.Z., Liu R., Yang Y., Zheng X.S., Zhu J.F., Gao M.R. // Nat. Commun. 2021. V. 12. № 1. P. 1.
Kabir S.A., Lemire K., Artyushkova K., Roy A., Odgaard M., Schlueter D., Oshchepkov A., Bonnefont A., Savinova E., Sabarirajan D., Mandal P., Crumlin E., Zenyuk I.V., Atanassov P., Serov A. // J. Mater. Chem. A. 2017. V. 5. P. 24433.
Sheng W., Bivens A.P., Myint M., Zhuang Z., Forest R.V., Fang Q., Chen J.G., Yan Y. // Energy Environ. Sci. 2014. V. 7. № 5. P. 1719.
Deng S., Liu X., Huang T., Zhao T., Lu Y., Cheng J., Shen T., Liang J., Wang D. // Electrochim. Acta. 2019. V. 324. P. 134892.
Kim H., Park H., Tran D.S., Kim S.K. // J. Ind. Eng. Chem. 2021. V. 94. P. 309.
Wang M., Yang H., Shi J., Chen Y., Zhou Y., Wang L., Di S., Zhao X., Zhong J., Cheng T., Zhou W., Li Y. // Angew. Chemie – Int. Ed. 2021. V. 60. № 11. P. 5771.
Biemolt J., Douglin J.C., Singh R.K., Davydova E.S., Yan N., Rothenberg G., Dekel D.R. // Energy Technol. 2021. V. 9. № 4. P. 5.
Yang F., Bao X., Li P., Wang X., Cheng G., Chen S., Luo W. // Angew. Chemie–Int. Ed. 2019. V. 58. № 40. P. 14179.
Ouyang M., Boldrin P., Maher R.C., Chen X., Liu X., Cohen L.F., Brandon N.P. // Appl. Catal. B: Environ. 2019. V. 248. P. 332.
Salmazo D., Juarez M.F., Oshchepkov A.G., Cherstiouk O.V., Bonnefont A., Shermukhamedov S.A., Nazmutdinov R.R., Schmickler W., Savinova E.R. // Electrochim. Acta. 2019. V. 305. P. 452.
Yang F., Bao X., Zhao Y., Wang X., Cheng G., Luo W. // J. Mater. Chem. A. 2019. V. 7. № 18. P. 10936.
Gao L., Wang Y., Li H., Li Q., Ta N., Zhuang L., Fu Q., Bao X. // Chem. Sci. 2017. V. 8. № 8. P. 5728.
Wang T., Wang M., Yang H., Xu M., Zuo C., Feng K., Xie M., Deng J., Zhong J., Zhou W., Cheng T., Li Y. // Energy Environ. Sci. 2019. V. 12. № 12. P. 3522.
Song F., Li W., Yang J., Han G., Liao P., Sun Y. // Nat. Commun. 2018. V. 9. № 1. P. 4531.
Pan Y., Hu G., Lu J., Xiao L., Zhuang L. // J. Energy Chem. 2019. V. 29. P. 111.
Yang Y., Sun X., Han G., Liu X., Zhang X., Sun Y., Zhang M., Cao Z., Sun Y. // Angew. Chemie – Int. Ed. 2019. V. 58. № 31. P. 10644.
Oshchepkov A.G., Bonnefont A., Pronkin S.N., Cherstiouk O.V., Ulhaq-Bouillet C., Papaefthimiou V., Parmon V.N., Savinova E.R. // J. Power Sources. 2018. V. 402. P. 447.
Davydova E.S., Manikandan M., Dekel D.R., Sunde S. // ACS Appl. Energy Mater. 2021. V. 4. № 4. P. 3404.
Bates M.K., Jia Q., Ramaswamy N., Allen R.J., Mukerjee S. // J. Phys. Chem. C. 2015. V. 119. № 10. P. 5467.
Sheng W., Gasteiger H.A., Shao-Horn Y. // J. Electrochem. Soc. 2010. V. 157. № 11. P. 1529.
Zheng J., Zhou S., Gu S., Xu B., Yan Y. // J. Electrochem. Soc. 2016. V. 163. № 6. P. F499.
Simonov P.A., Cherstiouk O.V., Kuznetsov A.N., Zaikovskii V.I., Kardash T.Y., Oshchepkov A.G., Bonnefont A., Savinova E.R. // J. Electroanal. Chem. 2019. V. 852. № 3. P. 113551.
Ni W., Krammer A., Hsu C.S., Chen H.M., Schüler A., Hu X. // Angew. Chemie – Int. Ed. 2019. V. 58. № 22. P. 7445.
Zhuang Z., Giles S.A., Zheng J., Jenness G.R., Caratzoulas S., Vlachos D.G., Yan Y. // Nat. Commun. 2016. V. 7. № 1. P. 10141.
Veziroglu N.T., Sherif S.A., Barbir F. // Environ. Solut. 2005. P. 143.
Niaz S., Manzoor T., Pandith A.H. // Renew. Sustain. Energy Rev. 2015. V. 50. P. 457.
Soloveichik G.L. // Beilstein J. Nanotechnol. 2014. V. 5. № 1. P. 1399.
Adli N.M., Zhang H., Mukherjee S., Wu G. // J. Electrochem. Soc. 2018. V. 165. № 15. P. J3130.
Finkelstein D.A., Imbeault R., Garbarino S., Roué L., Guay D. // J. Phys. Chem. C. 2016. V. 120. № 9. P. 4717.
Zadick A., Dubau L., Demirci U.B., Chatenet M. // J. Electrochem. Soc. 2016. V. 163. № 8. P. F781.
Olu P.Y., Deschamps F., Caldarella G., Chatenet M., Job N. // J. Power Sources. 2015. V. 297. P. 492.
Braesch G., Bonnefont A., Martin V., Savinova E.R., Chatenet M. // Electrochim. Acta. 2018. V. 273. P. 483.
Zadick A., Petit J.F., Martin V., Dubau L., Demirci U.B., Geantet C., Chatenet M. // ACS Catal. 2018. V. 8. № 4. P. 3150.
Li B., Yan Q., Song C., Yan P., Ye K., Cheng K., Zhu K., Yan J., Cao D., Wang G. // Appl. Surf. Sci. 2019. V. 491. P. 659.
Saha S., Ganguly S., Banerjee D., Kargupta K. // Int. J. Hydrogen Energy. 2015. V. 40. № 4. P. 1760.
Guo M., Cheng Y., Yu Y., Hu J. // Appl. Surf. Sci. 2017. V. 416. P. 439.
Santos D.M.F.F., Eugénio S., Cardoso D.S.P.P., Šljukić B., Montemor M.F. // Russ. J. Phys. Chem. A. 2015. V. 89. № 13. P. 2449.
Song C., Wang G., Li B., Miao C., Ma K., Zhu K., Cheng K., Ye K., Yan J., Cao D., Yin J. // Electrochim. Acta. 2019. V. 299. P. 395.
Wu C., Zhu J., Wang H., Wang G., Chen T., Tan Y. // ACS Catal. 2020. V. 10. № 1. P. 721.
Grinberg V.A., Mayorova N.A., Korlyukov A.A., Pasynskii A.A. // Russ. J. Electrochem. 2010. V. 46. № 11. P. 1289.
Tsivadze A.Y., Tarasevich M.R., Titova V.N., Yavich A.A., Petrova N.V. // Dokl. Phys. Chem. 2007. V. 414. № 1. P. 107.
Liu B.H., Li Z.P., Suda S. // J. Power Sources. 2008. V. 175. № 1. P. 226.
Ma J., Sahai Y., Buchheit R.G. // J. Power Sources. 2010. V. 195. № 15. P. 4709.
Šljukić B., Martins M., Kayhan E., Balčiūnaitė A., Şener T., Sequeira C.A.C., Santos D.M.F. // J. Electroanal. Chem. 2017. V. 797. P. 23.
Mahmoodi R., Hosseini M.G., Rasouli H. // Appl. Catal. B: Environ. 2019. V. 251. P. 37.
Cao D., Gao Y., Wang G., Miao R., Liu Y. // Int. J. Hydrogen Energy. 2010. V. 35. № 2. P. 807.
Ma X., Ye K., Wang G., Duan M., Cheng K., Wang G., Cao D. // Appl. Surf. Sci. 2017. V. 414. P. 353.
Tamašauskaitė-Tamašiūnaitė L., Balčiūnaitė A., Šimkūnaitė D., Selskis A. // J. Power Sources. 2012. V. 202. P. 85.
Duan D., Liang J., Liu H., You X., Wei H., Wei G., Liu S. // Int. J. Hydrogen Energy. 2015. V. 40. № 1. P. 488.
Martins M., Šljukić B., Sequeira C.A.C., Soylu G.S.P., Yurtcan A.B., Bozkurt G., Sener T., Santos D.M.F. // Appl. Surf. Sci. 2018. V. 428. P. 31.
Šljukić B., Milikić J., Santos D.M.F., Sequeira C.A.C. // Electrochim. Acta. 2013. V. 107. P. 577.
Gyenge E., Atwan M., Northwood D. // J. Electrochem. Soc. 2006. V. 153. № 1. P. 150.
Tamašauskaite-Tamašiunaite L., Čekaviciute R., Šimkunaite D., Selskis A. // Int. Conf. Power Energy Syst. Eur. 2011. P. 467.
Tamasauskaite-Tamasiunaite L., Balčiunaite A., Zabielaite A., Vaičiuniene J., Selskis A., Pakštas V., Norkus E. // J. Electroanal. Chem. 2013. V. 707. P. 31.
Martins M., Milikić J., Šljukić B., Soylu G.S.P., Yurtcan A.B., Bozkurt G., Santos D.M.F. // Micropor. Mesopor. Mater. 2019. V. 273. P. 286.
Hasan M., Newcomb S.B., Razeeb K.M. // ECS Trans. 2013. V. 45. № 20. P. 111.
Liu B.H., Suda S. // J. Alloys Compd. 2008. V. 454. № 1–2. P. 280.
Paschoalino W.J., Ticianelli E.A. // Int. J. Hydrogen Energy. 2013. V. 38. № 18. P. 7344.
Santos D.M.F., Šljukić B., Amaral L., Macciò D., Saccone A., Sequeira C.A.C. // J. Electrochem. Soc. 2014. V. 161. № 5. P. F594.
Santos D.M.F., Šljukić B., Amaral L., Milikić J., Sequeira C.A.C., Macciò D., Saccone A. // Electrochim. Acta. 2016. V. 190. P. 1050.
Hosseini M.G., Abdolmaleki M., Nasirpouri F. // Electrochim. Acta. 2013. V. 114. P. 215.
Zhang D., Cheng K., Shi N., Guo F., Wang G., Cao D. // Electrochem. Commun. 2013. V. 35. P. 128.
Romanova V.O., Romanov A.V., Churikov A.V., Gamayunova I.M., Churikov M.A. // Electrochem. Energ. 2014. V. 14. № 2. P. 57.
Oshchepkov A.G., Braesch G., Rostamikia G., Bonnefont A., Janik M.J., Chatenet M., Savinova E.R. // Electrochim. Acta. 2021. V. 389. P. 138721.
Lima F.H.B., Pasqualeti A.M., Molina Concha M.B., Chatenet M., Ticianelli E.A. // Electrochim. Acta. 2012. V. 84. P. 202.
Concha B.M., Chatenet M., Maillard F., Ticianelli E.A., Lima F.H.B., de Lima R.B. // Phys. Chem. Chem. Phys. 2010. V. 12. № 37. P. 11507.
Pasqualeti A.M., Olu P.Y., Chatenet M., Lima F.H.B. // ACS Catal. 2015. V. 5. № 5. P. 2778.
Braesch G., Oshchepkov A.G., Bonnefont A., Asonkeng F., Maurer T., Maranzana G., Savinova E.R., Chatenet M. // ChemElectroChem. 2020. V. 7. № 7. P. 1789.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ