

Кинетика и катализ, 2022, T. 63, № 2, стр. 147-159
Синтез метилацетата карбонилированием диметилового эфира на цеолитах
М. А. Кипнис a, *, Э. А. Волнина a
a ФГБУН Институт нефтехимического синтеза им. А.В. Топчиева РАН
119991 Москва, Ленинский просп., 29, Россия
* E-mail: kipnis@ips.ac.ru
Поступила в редакцию 30.08.2021
После доработки 30.09.2021
Принята к публикации 21.10.2021
- EDN: TQHXZW
- DOI: 10.31857/S0453881122020034
Аннотация
В обзоре проанализированы особенности синтеза метилацетата карбонилированием диметилового эфира на цеолитных катализаторах. Рассмотрены структурные характеристики цеолитов, в частности, морденита и феррьерита, влияющие на карбонилирование. Мостиковые гидроксильные группы (Al–OH–Si) цеолитов функционируют как кислотные центры Бренстеда и взаимодействуют с диметиловым эфиром. Для реакции карбонилирования характерен индукционный период, в течение которого молекулы диметилового эфира, взаимодействуя с Бренстедовскими кислотными центрами, образуют метоксигруппы. Внедрение СО в метоксигруппу приводит к формированию ацетильного интермедиата. Метилацетат появляется в результате взаимодействия молекулы диметилового эфира с ацетильным интермедиатом. Реакции способствует эффект конфайнмента, присущий небольшим порам цеолита, в частности, восьмичленным карманам морденита. Синтез метилацетата идет при умеренных температурах (около 200°С) в смеси СО/диметиловый эфир с высокой селективностью и существенной конверсией диметилового эфира. Наряду с целевой реакцией синтеза метилацетата наблюдаются побочные реакции образования углеводородов, снижающие стабильность работы катализатора. Эти реакции в случае морденита связывают с центрами, присутствующими в двенадцатичленных каналах. Стабильность работы катализаторов можно повысить при использовании специальных приемов для нейтрализации дезактивирующих центров цеолита. Эффект может быть достигнут при адсорбции пиридина или при ионном обмене соответствующих протонов структуры морденита на органические катионы или катионы металлов. Введение ионов цинка и, дополнительно, меди существенно ингибирует активность центров морденита, на которых протекают побочные реакции. Новым направлением исследований является применение многофункциональных катализаторов, позволяющих вести синтез метилацетата непосредственно из синтез-газа.
ВВЕДЕНИЕ
Производство метилового эфира уксусной кислоты осуществляется разными способами: этерификацией уксусной кислоты, взаимодействием метанола и уксусного ангидрида, реакцией перкарбоновой кислоты с ацетоном, пиролизом древесины. Метилацетат (МА) может быть гидрирован далее в этанол – важное химическое вещество и присадку для топлива [1].
Подробно изучены жидкофазные реакции карбонилирования метанола или диметилового эфира (ДМЭ) оксидом углерода с получением уксусной кислоты и МА, протекающие в присутствии растворимых карбонильных комплексов родия (процесс Monsanto, более 50% промышленного производства уксусной кислоты) или иридия (процесс BP CativaTM) [2, 3]. Недостатками технологии с использованием гомогенных катализаторов на основе карбонильных комплексов являются коррозионная активность и высокая стоимость катализаторов.
Селективное карбонилирование ДМЭ в МА на цеолитных катализаторах привлекло большое внимание после публикации работ Иглесиа (E. Iglesia) с сотр. [4, 5]. Применение цеолитов для синтеза МА перспективно ввиду простоты отделения продуктов, возможности регенерации катализатора, проведения процесса при атмосферном давлении и умеренных температурах.
Форма или топология внутренней пористой структуры цеолита могут значительно влиять на селективность при химических превращениях, катализируемых цеолитом. Физические размеры и конфигурацию пор в цеолитах возможно изменить с использованием приемов ионного обмена, деалюминирования, варьирования соотношения атомов Si/Al или любой иной модификации, которая воздействует на размер пор или геометрию каркаса. На селективность также оказывает влияние эффект конфайнмента (нахождение молекул вещества в порах, сопоставимых по размеру с размером самих молекул) [6, 7]. Согласно расчетам энергия адсорбции, обусловленная фактором удержания субстрата в порах цеолита, существенно возрастает [7].
Анализ литературы по каталитическим реакциям ДМЭ с различными реагентами (алкены, ароматические соединения, СО и т.д.), проведенный в [8], позволил сделать следующие выводы. В образовании продуктов в реакциях на основе ДМЭ участвуют интермедиаты в виде поверхностных метильных групп, формирующихся при взаимодействии ДМЭ с Бренстедовскими кислотными центрами (БКЦ). Стабильность и активность этих интермедиатов определяются температурой и кислотностью катализатора. Распределение и содержание БКЦ в структуре цеолита являются основными параметрами, влияющими на эффективность карбонилирования ДМЭ в МА.
Количество типов цеолитов, используемых в карбонилировании, невелико. В [4, 5] показано, что ряд цеолитов в Н-форме и, прежде всего, морденит (Н-MOR) и феррьерит (H-FER), демонстрируют в карбонилировании ДМЭ в МА при относительно низких температурах (423–513 К) заметную активность и высокую селективность.
Основной недостаток применения цеолитов H-MOR и Н-FER в карбонилировании ДМЭ – постепенная дезактивация катализатора. Соответственно, актуальны исследования по модификации цеолитов с целью повышения их стабильности.
В [9] приведены данные по карбонилированию ДМЭ на гомогенных катализаторах, гетеропо-ликислотах, цеолитах. Тематика карбонилирования ДМЭ на цеолитах представляет самостоятельный интерес, что требует ее отдельного рассмотрения.
Цель обзора – анализ факторов, влияющих на протекание реакции карбонилирования ДМЭ в присутствии цеолитов. Поскольку наиболее эффективным и исследованным цеолитом в карбонилировании ДМЭ является морденит, большая часть материала связана именно с ним.
НЕКОТОРЫЕ ОСОБЕННОСТИ КАРБОНИЛИРОВАНИЯ ДМЭ НА ЦЕОЛИТАХ
Ключевой стадией карбонилирования ДМЭ в МА на цеолитах является разрыв С–О-связи в ДМЭ. По аналогии с карбонилированием спиртов (реакция Коха) [10], для карбонилирования метанола или ДМЭ применяют твердые кислоты: цеолиты и гетерополикислоты.
Фуджимото (K. Fujimoto) с сотр. впервые сообщили о карбонилировании метанола в уксусную кислоту на цеолитах и высказали предположение об участии в реакции поверхностных метильных групп [11]. В настоящее время общепризнано [8], что ДМЭ первоначально реагирует с БКЦ каркаса цеолита. При этом появляется поверхностный метил и одновременно высвобождается метанол. Затем CO внедряется в связь C–O между метильной группой и кислородом каркаса цеолита, формируя ацетильную группу. Наконец, ацетильная группа взаимодействует с другой молекулой ДМЭ с образованием МА и одновременной регенерацией адсорбированной метильной группы. Стадия возникновения ацетильных групп является лимитирующей. В случае присутствия воды при взаимодействии с ней ацетильной группы синтезируется уксусная кислота.
Цеолиты, проявляющие активность в карбонилировании ДМЭ, представляют собой структуры, содержащие поры соответствующего размера и конфигурации. При температурах реакции 150–300°C цеолит H-MOR является наиболее активной и селективной системой, тогда как цеолит H-FER при высокой селективности характеризуется низкой активностью. Хотя другие типы цеолитов, такие как OFF, CHA, ECR, MAZ и имеют поры, ограниченные восьмичленными кольцами, их активность и селективность в карбонилировании ДМЭ неудовлетворительны. Конверсия ДМЭ, выход продуктов, селективность, срок службы катализаторов напрямую связаны с характером, количеством и распределением кислотных центров в структуре цеолита, типом и химическим составом цеолита.
СТРУКТУРА МОРДЕНИТА И ФЕРРЬЕРИТА
Карбонилирование ДМЭ в МА на цеолитах – это типичная реакция, протекающая в пространственно ограниченной среде – порах цеолита. Морденит – цеолит, наиболее предпочтительный для карбонилирования ДМЭ. Составу природного морденита отвечает формула Na8Al8Si40O96 ⋅ nH2O [12] с соотношением SiO2/Al2O3 (силикатным модулем) равным 10.
Простейшим структурным элементом цеолита является тетраэдр, вершины которого занимают атомы кислорода. В центре каждого тетраэдра расположен атом кремния или алюминия. Баланс зарядов (отрицательных у кислорода и положительных у кремния и алюминия) обеспечивается присутствием дополнительных атомов, например, натрия или протона (Н-форма цеолита). Тетраэдры, сочленяясь вершинами, создают каркас структуры цеолита, в котором можно выделить такие пространственные образования, как окна, каналы, карманы. Окно, сформированное сочетанием 12, 8 или 6 тетраэдров, в литературе обозначается, как 12-MR, 8-MR или 6-MR (MR – member ring). Сочетание таких окон в пространстве дает соответствующий канал.
Согласно [13], каркас морденита может быть представлен в виде гофрированных листов, параллельных плоскости (010), связанных между собой четырехчленными кольцами тетраэдров. Большие эллипсоидальные 12-членные кольца (12-MR) имеют сечение размером 7 × 6.5 Å, а сильно сжатые восьмичленные кольца (8-MR) – 5.7 × 2.6 Å. Эти кольца образуют каналы, расположенные вдоль оси с (рис. 1а).
Рис. 1.
Структура морденита: а – в виде гофрированных листов, соединенных вдоль оси b четырехчленными кольцами тетраэдров (выделены черным), образующих двенадцатичленные и восьмичленные кольцевые каналы, идущие вдоль оси c; б – в виде “каналов” [13]. (Разрешение на публикацию получено 7 мая 2021, © 2004 Mineralogical society of America).
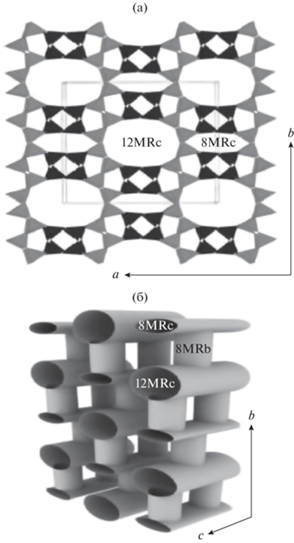
С другой стороны, пространственную структуру морденита удобно изображать в виде системы трубчатых каналов: каналы, идущие в направлении оси с (12-MRc), соединены карманами из восьмичленных колец (8-MRb) с размером в сечении: 3.4 × 4.8 Å (рис. 1б). Феррьерит содержит 10-MR-каналы (5.4 × 4.2 Å) вдоль оси с, пересекающиеся 8-MR-каналами (4.8 × 3.5 Å) вдоль оси b, и 6-MR-каналы параллельно оси с [9].
Малые молекулы в цеолите H-MOR движутся по двумерной системе каналов – большим каналам параллельно оси с и узким каналам перпендикулярно оси b. Поскольку при движении в полостях и каналах молекулы сталкиваются со стенками пор, диффузия в цеолитах протекает в кнудсеновской области [14]. Если речь идет о перемещении молекул, диаметр которых лишь немного меньше сечения каналов, то прочная адсорбция молекулы или димеризация двух молекул полностью перекрывают движение в канале. В узкопористых цеолитах каталитические процессы сильно тормозятся диффузией.
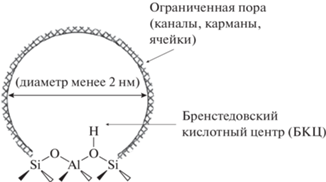
В обзоре [15] рассмотрены природа кислотных центров, распределение атомов Al в структуре различных цеолитов. Отмечено, что на характер протекающих реакций влияет пространственная ограниченность соответствующих центров реакции. Эффект конфайнмента характерен для цеолитов и может быть проиллюстрирован рис. 2.
Рис. 2.
Пространственная ограниченность в структурах цеолита согласно [16]. (Разрешение на публикацию получено 26 апреля 2021, © 2016 Royal Society of Chemistry).
РАСПРЕДЕЛЕНИЕ БКЦ В СТРУКТУРЕ МОРДЕНИТА И ФЕРРЬЕРИТА
В цеолитах могут присутствовать как Бренстедовские, так и Льюисовские кислотные центры. Рассмотрим местоположение БКЦ в элементарной ячейке морденита, в которой имеется ряд структурно неэквивалентных центров, связанных с размещением компенсирующего катиона (иона натрия, протона, метильной группы). Это: T1 в 12-MR канале, T2 и T4 на пересечении каналов 12-MR и 8-MR, и T3 в канале 8-MR [17]. В случае морденита с соотношением SiO2/Al2O3 = 20 на элементарную ячейку приходится 4 атома алюминия с формальным зарядом +3. С учетом того, что каждый атом алюминия связан с четырьмя атомами кислорода, в элементарной ячейке такого цеолита имеется 16 позиций для размещения компенсирующего катиона.
Энергетика связи протонов и метильных групп в структуре морденита рассмотрена в [18]. Расчеты проведены относительно наиболее стабильных позиций протонов и метильных групп. Протоны почти одинаково стабильны на центрах T1-О1 и T1-O4 (табл. 1). В позициях T3 адсорбция предпочтительна на T3-O3, который более стабилен, чем T3-O8 и T3-O9. Аналогичная тенденция наблюдается и для метильных групп: центры T1-O4 и T3-O3 более предпочтительны для адсорбции по сравнению с другими.
Распределение БКЦ между восьмичленными карманами и двенадцатичленными каналами в Н-мордените зависит от соотношения Na/H в структуре цеолита: рост содержания натрия приводит к снижению доли БКЦ в 8-членных карманах (табл. 2).
Таблица 2.
Влияние содержания Na на распределение ОН-групп в цеолитах Н-MOR [16]2
| Цеолит | Производитель | Si/Al | Na/Al | OH8-MR, % | OH12-MR, % |
|---|---|---|---|---|---|
| H100Na0MOR | Tosoh Corporation | 8.9 | 0.001 | 78 | 22 |
| H100Na0MOR | Sud-Chemie | 10.1 | 0.001 | 60 | 40 |
| H100Na0MOR | Zeolyst International | 10.0 | 0.001 | 56 | 44 |
| H83Na17MOR | Zeolyst International | 10.0 | 0.17 | 36 | 64 |
| H73Na27MOR | Zeolyst International | 10.0 | 0.27 | 27 | 73 |
| H59Na41MOR | Zeolyst International | 10.0 | 0.41 | 20 | 80 |
| H45Na55MOR | Zeolyst International | 10.0 | 0.55 | 13 | 87 |
Возможное расположение атомов Al в структуре феррьерита по центрам Т1–Т4 при различных значениях Si/Al приведено в [19]. При соотношениях Si/Al, равных 8.6, 10.8, 20, 27, атомы Al связаны, в основном, с центрами Т2, Т4. При этом согласно [5] в коммерческих образцах цеолитов H-MOR с отношением Si/Al ≈ 10, произведенных разными производителями (“Tosoh Corporation”, “Sud-Chemie”, “Zeolyst International”), около 20% атомов Al находятся во внерешеточном положении.
Для оценки кислотности цеолитов помимо расчетных применяют разнообразные экспериментальные методы: термокалориметрию, термопрограммируемую десорбцию молекул-зондов, твердотельный ЯМР 1H, 13C и 17O, инфракрасную и рамановскую колебательные спектроскопии [20, 21]. Характеристики молекул-зондов, используемых в инфракрасной спектроскопии (кинетический диаметр, полосы поглощения) приведены в [21]. Отметим, что согласно [20] обычные методы, основанные на адсорбции молекулы-зонда, не позволяют оценить истинную силу кислотных центров цеолита из-за взаимодействия молекул-зондов с кислородным каркасом цеолита.
БКЦ, существующие в структуре цеолита, проявляются характерными полосами валентных колебаний ОН-групп в ИК-спектрах. Сочетание адсорбции ряда молекул (аммиак, легкие углеводороды, пиридин) с приемами обработки ИК-спектров позволяет провести количественную оценку содержания БКЦ в разных структурных положениях. Молекула аммиака, благодаря своим малым размерам (кинетический диаметр 0.26 нм), с легкостью проникает в узкие каналы. Пиридин имеет кинетический диаметр около 0.5 нм и с его помощью можно исследовать кислотные центры в 12-MR-каналах, тогда как кислотные центры в 8‑MR-каналах для пиридина недоступны.
При исследовании ИК-спектров морденитов для описания области ОН-колебаний (~3600 см–1) необходим учет вклада ряда полос, отвечающих разным структурным положениям протонов. Так, согласно [22] паре полос (3590 и 3582 см–1) соответствуют БКЦ, локализованные в 8-MR-каналах. Пара полос 3610 и 3600 см–1 связана с центрами, находящимися на пересечении 8-MR- и 12-MR-каналов. Полосы 3625 и 3617 см–1 характеризуют БКЦ в 12-MR-каналах. Авторы [22] использовали морденит (Si/Al = 7.3, “Zeolyst International”) в натриевой форме, из которого путем замены ионов натрия на ион аммония с последующей прокалкой был получен Н-MOR. С помощью ионного обмена из Н-MOR были синтезированы 4 образца с разной степенью обмена на Na (<0.01, 18, 37 и 52%). Ионы натрия при ионном обмене прежде всего занимают положения в 8-MR-каналах, что согласуется с выводами, сделанными при рассмотрении данных табл. 2.
Иглесиа (E. Iglesia) с соавт. [23] изучали ИК-спектры цеолитов H-MOR и H-FER (“Zeolyst International”) при адсорбции пропана, н-гексана, пиридина и 2,6-диметилпиридина. Использование деконволюции ИК-спектров в области поглощений, соответствующих колебаниям ОН-групп, позволило оценить концентрации протонов в 8-MR- и 12-МR-каналах H-MOR и в 8-MR- и 10-МR-каналах H-FER. Обработка спектров показала, что 0.55 (±0.05) от всего количества центров H+ в H-MOR (Si/Al = 10) находится в пределах 8-МR-каналов. Для H-FER (Si/Al = 33.5) соответствующее значение равно 0.11 (±0.04).
В [24] методом ИК-Фурье-спектроскопии с помощью слабого основания CD3CN как молекулы-зонда были исследованы кислотные свойства морденита. Для молекул ацетонитрила доступны все кислотные центры в мордените: в ИК-спектрах при его адсорбции пропадают кислотные полосы ОН-групп. Их исчезновение сопровождается появлением трех полос, которые относятся к колебаниям ацетонитрила, связанного с кислотными центрами в каналах 8-MR, 12-MR и концевыми группами SiOH. Исходя из большого сдвига частоты ν (C≡N) в случае канала 8-MR по сравнению с каналом 12-MR, авторы делают вывод о том, что кислотные центры в канале 8-MR сильнее.
МОДИФИКАЦИЯ РАСПРЕДЕЛЕНИЯ БКЦ В СТРУКТУРЕ ЦЕОЛИТОВ: ВЛИЯНИЕ НА АКТИВНОСТЬ И СТАБИЛЬНОСТЬ
Управляемый синтез является основным подходом к повышению концентрации протонов в цеолитах даже с низким содержанием алюминия. По данным спектров 1H MAS ЯМР (ядерный магнитный резонанс с вращением под магическим углом на ядрах 1H) и термопрограммированной десорбции аммиака [25] количество БКЦ в образцах цеолита Н-MOR, синтезированных с применением тетраэтиламмоний гидроксида в качестве структурообразующего агента выше, чем в коммерческом мордените (отношение Si/Al составляет от 8.2 до 11.8 в синтезированных образцах и 8.2 – в коммерческом мордените).
Роль положения атомов алюминия в различных местах решетки морденита рассмотрена в [26] на примере синтезированных гидротермально морденитов с соотношением Si/Al от 6.5 до 10.2. Повышение отношения Si/Al приводит к преимущественному размещению атомов Al в позициях Т3, Т4. При этом центры Т3 были активны в карбонилировании ДМЭ, тогда как центры Т4 – в реакциях, ведущих к коксообразованию. Атомы Al в позициях Т1, Т2 в реакциях не участвуют.
На основании проведенных квантово-химических расчетов в [17] предположено, что только БКЦ, находящиеся в позиции Т3-О33, являются селективными в карбонилировании ДМЭ, и эта селективность обусловлена ориентацией метоксигруппы вдоль оси 8-MR-канала. С такими выводами согласуется корреляция скорости синтеза МА с количеством кислотных центров в боковых карманах 8-MR (рис. 3).
Рис. 3.
Корреляция скорости синтеза МА при карбонилировании ДМЭ (483 К) с количеством Н+-центров в боковых карманах 8-MR морденита [16]. (Разрешение на публикацию получено 28 апреля 2021, © 2016 Royal Society of Chemistry).

Цеолит MOR (Si/Al = 10.1) с частицами миллиметрового размера получен с применением тетраэтиламмоний гидроксида в [27]. Сопоставление каталитических характеристик синтезированного морденита с коммерческим образцом (Si/Al = 6.5) (использована Н-форма) показало его высокую эффективность. Цеолит характеризуется высокой концентрацией БКЦ в боковых карманах 8-MR (0.76 ммоль/г), что в 3 раза выше, чем в каналах 12-MR. В коммерческом цеолите концентрация БКЦ как в боковых карманах 8-MR, так и в 12-MR-каналах равна 0.39 ммоль/г. Эксперименты по карбонилированию ДМЭ проводили в проточном реакторе при 200°C, предварительно обрабатывая образец смесью 1.3% пиридина в азоте (пиридин применяли для блокировки БКЦ в 12-MR-каналах). Использовали смесь, об. %: 5 ДМЭ, 35 CO, 60 H2. Объемная скорость (GHSV) – 1500 мл ${\text{г}}_{{{\text{кат}}}}^{{ - 1}}$ ч–1, давление – 2.0 MПa. Селективность оказалась близкой к 100% как на коммерческом, так и на синтезированном цеолитах. Конверсия ДМЭ после индукционного периода на синтезированном цеолите была около 90%, тогда как на коммерческом – около 20%. Рассчитанная скорость синтеза МА относительно центров в 8‑MR кармане составила для синтезированного цеолита 3.6 моль ${\text{моль}}_{{{\text{центров}}}}^{{ - 1}}$ ч–1, а для коммерческого – 1.75 моль ${\text{моль}}_{{{\text{центров}}}}^{{ - 1}}$ ч–1. Таким образом, блокирование пиридином БКЦ в 12-MR-каналах способствовало повышению стабильности работы катализатора, что согласуется с данными [28].
Влияние типа органического темплата на распределение кислотных центров в каналах морденита рассмотрено в [29]. Цеолиты синтезировали гидротермальным способом. В качестве структурообразующего агента использовали гомопиперазин, гексаметиленимин, 4-метилпиперидин, циклогексиламин. Для сравнения был приготовлен цеолит без добавления органического структурообразующего агента. После прокалки при 550°С, обмена с NH4NO3 и повторной прокалки при 500°С цеолиты были переведены в Н-форму. Реакцию карбонилирования ДМЭ проводили при 200°С, давлении 1.5 МПа, в смеси ДМЭ/СО (1/49), объемной скорости 6000 ч–1. Временные зависимости выхода МА аналогичны для всех образцов: рост в течение первых двух часов и затем быстрый спад. Отмечена линейная корреляция максимальных значений выхода МА с количеством БКЦ в 8-MR-каналах.
Модификация морденита различными алкилимидазолиевыми ионами предложена в [30]. Введение 1,3-диметилимидазолия позволило селективно подавить кислотные центры в 12-MR-каналах, на которых идет образование углеводородов, и существенно повысить стабильность и активность цеолита.
Роль различных центров морденита в карбонилировании ДМЭ рассмотрена в [24]. Исходный коммерческий морденит с Si/Al = 17 (содержание Na – 1.9 мас. %) был переведен сначала в Н-форму, затем проведен обмен протонов на ионы натрия. Далее ионы натрия селективно обменяли на ионы тетраметиламмония (ТМА+). Ионы ТМА+ в силу пространственных ограничений замещали ионы натрия только в 12-MR-каналах. После прокалки при 550°С и разложения ТМА+ были получены образцы, содержащие как ионы Na, так и протоны в соответствующих центрах морденита. 68% кислотных центров морденита были локализованы в каналах 12-MR, 32% – в каналах 8-MR. Ион ТМА+ сохраняет стабильность при нагреве, по крайней мере, до 300°С. Цеолиты, синтезированные с замещением протонов на ионы ТМА+ в каналах 12-MR, демонстрировали бóльшую стабильность в карбонилировании ДМЭ, чем исходный Н-MOR, за счет подавления реакций образования углеводородов.
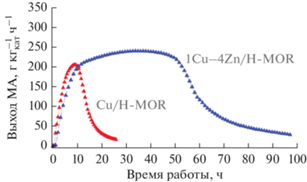
В [31] предложено повышать стабильность работы морденита, вводя одновременно с ионами меди ионы цинка. Использован цеолит Na-MOR (Si/Al = 6.5, “Zeolyst International”), в котором ионным обменом Na+ заменен на ${\text{NH}}_{4}^{ + },$ а затем ион аммония частично обменен на ионы меди и цинка. Окончательно образцы прокаливали при 823 К в течение 3 ч. Катализатор перед каталитическим тестированием восстанавливали в смеси 10% H2/Ar при 598 К в течение 2 ч. Условия тестирования: навеска – 0.3 г, смесь, об. %: 50.0 CO/2.4 ДМЭ/2.9 H2/44.7 He, давление – 2 МПа, температура – 483 К, расход – 15 Нмл/мин (стандартные условия). Данные по карбонилированию ДМЭ в присутствии Cu–Zn-морденитов приведены в табл. 3.
Таблица 3.
Данные по карбонилированию ДМЭ на Cu–Zn морденитах [31]
| Катализатор | Cu, мас. % | Zn, мас. % |
Выход МА, кгМА${\text{кг}}_{{{\text{кат}}}}^{{ - 1}}$ |
Максимальная активность, гМА${\text{кг}}_{{{\text{кат}}}}^{{ - 1}}$ч–1 |
Время жизни, ч* |
|---|---|---|---|---|---|
| H-MOR | – | – | 3.57 | 213 | 25 |
| Cu/H-MOR | 3.21 | – | 2.51 | 206 | 20 |
| 2Cu–1Zn/H-MOR | 2.04 | 0.92 | 5.53 | 238 | 37 |
| 1Cu–1Zn/H-MOR | 1.61 | 1.57 | 7.12 | 246 | 41 |
| 1Cu–4Zn/H-MOR | 0.57 | 2.47 | 14.24 | 240 | 86 |
| Zn/H-MOR | – | 3.05 | 6.0 | 217 | 45 |
Показатели работы H-MOR и Cu/H-MOR различались незначительно. Однако введение Cu и Zn в соотношении 1/4 способствовало повышению активности и, что важно, времени стабильной работы катализатора (рис. 4). Отметим, что для всех образцов на участках стабильной работы селективность образования МА была около 100%. Уменьшение активности сопровождалось снижением селективности образования МА и ростом селективности образования метанола. Дезактивированный катализатор был регенерирован в токе Н2 с постепенным подъемом температуры до 823 К. Если на исходном образце максимальная конверсия ДМЭ составляла 72%, то после восстановительной регенерации – 55%. Согласно [32] причина увеличения срока службы катализатора связана с тем, что Zn2+ ингибирует активность центров T4 морденита, на которых протекают побочные реакции.
Рис. 4.
Выход МА при карбонилировании ДМЭ на медьсодержащих морденитах [31]. (Разрешение на публикацию получено 26 апреля 2021, © 2016 American Chemical Society).
В [33] рассматривали влияние типа структурообразующего органического катиона на распределение кислотных центров в каналах 8-MR феррьерита, содержание внерешеточных атомов алюминия (AlВР) и активность в синтезе МА при 473 К (табл. 4).
Таблица 4.
Характеристики ряда образцов на основе феррьерита [33]
| Цеолит* | Скорость синтеза МА, моль(г-атом Al)–1 ч–1 | Si/Al | Доля AlВР, % | Доля кислотных центров в 8-MR-каналах, % | Содержание БКЦ, ммоль Al8-MR/гкат |
|---|---|---|---|---|---|
| FER-OH | 0.9 | 10 | 3.8 | 53 ± 6 | 0.85 |
| FER+Pyr | 1.3 | 13.2 | 4.0 | 89 ± 2 | 1.08 |
| FER + Pyr + TMA | 0.8 | 17 | 2.1 | 84 ± 3 | 0.8 |
| FER + HMI + TMA | менее 0.1 | 17.2 | 20.0 | 27 ± 5 | 0.21 |
Влияние условий получения феррьерита на стабильность карбонилирования ДМЭ изучено в [34]. Синтезы осуществляли гидротермальным способом с применением в качестве затравки коммерческого феррьерита (Si/Al = 10.4). Каталитические тесты проводили в проточном реакторе при 220°С, 1 МПа, объемной скорости 2000 л ${\text{кг}}_{{{\text{кат}}}}^{{ - 1}}$ ч–1, в смеси ДМЭ/СО/N2 = 5/45/50. Найдено, что при использовании в синтезе 15 вес. % затравки конверсия ДМЭ стабильна в течение 100 ч. В то же время при синтезе с меньшим (5 вес. %) или бóльшим (20 вес. %) количеством затравки, а также на коммерческом феррьерите стабильность работы образцов не достигается, а их активность ниже.
Временные зависимости конверсии ДМЭ характеризуются заметным начальным периодом роста, что связано с протекающими с участием БКЦ реакциями образования метильных групп [4, 5]:
(I)
${\text{С}}{{{\text{Н}}}_{{\text{3}}}}{\text{ОС}}{{{\text{Н}}}_{{\text{3}}}} + {{{\text{Н}}}^{ + }}* = {\text{СН}}_{3}^{*} + \,\,{\text{С}}{{{\text{Н}}}_{{\text{3}}}}{\text{ОН}},~$(II)
${\text{С}}{{{\text{Н}}}_{{\text{3}}}}{\text{ОН}} + {{{\text{Н}}}^{ + }}* = {\text{СН}}_{3}^{*} + {{{\text{Н}}}_{{\text{2}}}}{\text{О}}{\text{.}}$“Звездочка” означает, что данный ион связан со структурой цеолита.
Скорость синтеза МА в расчете на один кислотный центр невысока. Так, в [4] для цеолита H-MOR (Si/Al = 10) при температуре 438 К и давлении СО 500 кПа стационарная скорость синтеза МА составляет 0.5 моль (гатом Al)–1 ч–1. Согласно [5], для цеолита H-MOR (Si/Al = 9.8) при температуре 438 К и давлении СО 930 кПа этот показатель равен 0.82 моль (гатом Al)–1 ч–1, что соответствует примерно 0.1 гМА${\text{г}}_{{{\text{цеолита}}}}^{{ - 1}}$ ч–1. Близкие результаты получены для активных образцов феррьерита. Приведенные в [9] данные об активности разных цеолитов при температурах <210°С не превышают значений 0.8 гМА${\text{г}}_{{{\text{цеолита}}}}^{{ - 1}}$ ч–1.
ОСОБЕННОСТИ АДСОРБЦИИ ДМЭ И СО НА ЦЕОЛИТАХ
Для определения каталитических характеристик цеолитных катализаторов в карбонилировании ДМЭ необходимо знать, могут ли диффундировать реагенты к активным центрам, а образовавшиеся продукты – из пористой структуры цеолита вовне. Адсорбция СО, ДМЭ и диффузия МА на ряде цеолитов, содержащих по крайней мере один восьмичленный кольцевой канал, рассмотрена с использованием расчетных методов в [35]. Для молекул CO и ДМЭ характерна более высокая плотность вероятности нахождения в канале цеолита, чем в газе. Примечательно, что свободная энергия адсорбированного СО в канале цеолита выше, чем у ДМЭ. Следовательно, высокое соотношение CO/ДМЭ необходимо для улучшения проникновения CO в поры цеолита. Восьмичленные боковые карманы являются критически важными центрами реакции не только из-за низкого барьера активации, создаваемого ограниченной средой, но также из-за агрегации молекул CO вокруг активных центров, тогда как двенадцатичленные каналы обеспечивают транспорт реагентов и продуктов.
В [35] сопоставлены свойства ряда цеолитов в отношении агрегации СО, диффузии МА и реакционной способности. Активационный барьер в боковых 8-MR-карманах морденита ниже, чем внутри 12-MR-каналов. Это, а также высокая агрегация CO в 8-MR-карманах в сочетании с существенной диффузией МА в 12-MR-каналах приводят к сверхвысокой реакционной способности MOR при карбонилировании. Схожие результаты можно наблюдать и для феррьерита в случае 10-MR-каналов. В цеолитах GON и ATS молекулы СО агрегируются в 12-MR-каналах с небольшой плотностью. В цеолите IRN плотность адсорбции молекул СО в 8-MR-канале достаточно заметна, но отмечается медленная диффузия МА. Таким образом, авторы [35] приходят к выводу, что морденит является наилучшим кандидатом для карбонилирования ДМЭ. Цеолиты FER и IRN менее пригодны.
МЕХАНИЗМ РЕАКЦИИ СИНТЕЗА МЕТИЛАЦЕТАТА
На основании DFT-расчетов в [18] представлен маршрут реакции, по которому ацетил образуется из метильной группы и СО на центре T3-O3 в такой геометрической конфигурации, которая позволяет ему реагировать с молекулой ДМЭ только внутри бокового кармана. Этот маршрут требует значительно меньшей энергии активации (1.0 эВ) по сравнению с ранее проведенными расчетами (2.2 эВ [36]). Он реализуется практически полностью внутри бокового кармана 8-MR, где расположены кислотные центры T3-O3. Реакция между метанолом и ацетилом протекает с нулевым энергетическим барьером в основных каналах и с очень низким – 0.02 эВ – в боковых карманах 8-MR. Это указывает на то, что если в системе присутствует метанол, он быстрее, чем ДМЭ, реагирует с ацетилом. При карбонилировании ДМЭ на центре Т4 может идти реакция синтеза триметилоксоний катиона. Данный катион такой же нестабильный, как и ацетил, поэтому он быстро взаимодействует с другими частицами с образованием углеводородов. В [18] показано, что МА и уксусная кислота, по сравнению с ДМЭ и метанолом, адсорбируются сильнее на БКЦ в основных каналах, участвующих в реакциях, связанных с дезактивацией. Это, возможно, объясняет, почему добавление в реакционную смесь МА и уксусной кислоты ингибирует появление углеводородов, в частности, метана, при карбонилировании ДМЭ на мордените [37]. При этом МА адсорбируется на БКЦ в боковых карманах 8-MR так же хорошо, как ДМЭ и метанол.
Исходя из экспериментальных исследований кинетики и теоретических расчетов авторы [18] пришли к выводу, что МА ингибирует реакцию, возможно, из-за стерических препятствий для атаки СО поверхностной метильной группы в боковых карманах. Для скорости образования МА (rMA) получено уравнение:
Скорость образования МА прямо пропорциональна давлению СО (PСО) и зависит от давлений МА (PMA) и ДМЭ (PДМЭ). Ингибирование реакции образующимся МА учитывается членами знаменателя. k1, K2, K3 – константы скоростей элементарных стадий: образования ацетила, блокировки метоксигруппы МА, образования МА по реакции ДМЭ с ацетилом. Более сложные выражения, принимающие во внимание элементарные стадии реакции с возможным участием воды и метанола, рассмотрены в [5].
Скоростьконтролирующей стадией реакции обычно принято считать стадию образования ацетила: внедрение СО в связь метильной группы с кислородом. В [38] рассмотрены различные варианты выражений для скорости реакции, учитывающие, что скоростьконтролирующей стадией может быть диссоциация ДМЭ или образование МА.
МЕТАЛЛСОДЕРЖАЩИЕ ЦЕОЛИТЫ
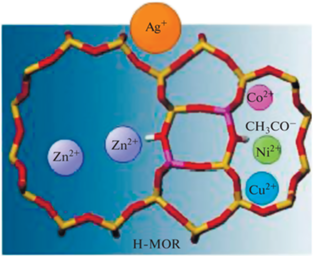
Для изменения кислотности и распределения кислотных центров в цеолитах, применяемых для карбонилирования ДМЭ, используется частичный ионный обмен протонов на ионы меди, кобальта, никеля, цинка или серебра [39]. Введение ионов металлов позволяет адсорбировать большее количество СО. Так как радиусы ионов Cu2+, Co2+, Ni2+, Zn2+ или Ag+ заметно разнятся, эти ионы координируются в разных каналах H-MOR (рис. 5), что приводит к различиям в каталитических характеристиках. При одинаковых зарядах меньшие по размерам обменные катионы обладают большей способностью принимать электроны. Соответственно, по кислотной силе их можно расположить в следующем порядке Zn2+ > Cu2+ > > Ni2+ > Co2+ (некоторые кислотные центры Бренстеда в результате ионного обмена протонов на ионы металлов превращаются в кислотные центры Льюиса [40]). Механизм карбонилирования ДМЭ в восьми- и двенадцатичленных кольцевых каналах различный. Промежуточный ацетил при карбонилировании ДМЭ в основном образуется в восьмичленном кольцевом канале. Так как конверсия ДМЭ при карбонилировании на Zn/H-MOR и H-MOR практически одинакова, это означает, что ион Zn2+, имеющий большой радиус, может обмениваться с протонами только в двенадцатичленном кольце. Радиусы ионов Cu2+, Ni2+, Co2+ меньше, и они могут легко обмениваться с протонами и в восьмичленном кольце. Кислотные центры в случае Ag/H-MOR имеют однородное распределение по силе, что приводит к уменьшению конверсии ДМЭ. Физико-химические и каталитические характеристики металлсодержащих образцов на основе морденита и возможные положения различных ионов металлов, координированных в каналах H-MOR, рассмотрены в [39] на примере коммерческого H-MOR (SiO2/Al2O3 = 18, “Tosoh Corporation”), в который ионным обменом из нитратов солей вводили Cu, Ni, Co, Zn или Ag. После процедуры обмена образцы прокаливали на воздухе при 500°С в течение 5 ч. Содержание металлов в них по данным элементного анализа составило 1.3–1.74 мас. %. По данным просвечивающей электронной микроскопии в прокаленных образцах присутствовали мелкодисперсные (2–3 нм) частицы оксидов металлов. Введение металлов примерно вдвое снизило содержание внерешеточного алюминия. Каталитическая активность в карбонилировании ДМЭ оценивалась при 210°С, давлении 1.8 МПа, соотношении СО/ДМЭ = 19 и объемных скоростях 3960 и 5160 мл г–1 ч–1. Присутствие меди привело к заметному росту активности катализатора. Временная зависимость конверсии ДМЭ качественно не изменилась: наблюдался рост активности на начальном участке (индукционный период), краткий стационарный участок и падение активности после 5 ч работы. Однако при наличии меди заметно выросла конверсия ДМЭ. Так, при объемной скорости поступающего газа 3960 мл г–1 ч–1 она составила 93.7 и 53.6% для Cu/H-MOR и H-MOR соответственно. Содержание углеродистых отложений в Cu/H-MOR было существенно ниже по сравнению с H-MOR (данные термопрограммированного окисления катализаторов после карбонилирования).
Рис. 5.
Различные катионы металлов и ацилий-катион при координировании в структуре H-MOR [39]. (Разрешение на публикацию получено 17 августа 2021, © 2015 American Chemical Society).
В [38] использовался коммерческий цеолит NH4-MOR (Si/Al = 8.2, “Yangzhou Zhonghe Petroleum Chemicals Institute Co”), в который ионным обменом вводили медь, после чего прокаливали при 773 K. Образец перед каталитическими испытаниями восстанавливали при 473 К в смеси 10% Н2/N2. По данным просвечивающей электронной микроскопии в восстановленном катализаторе присутствуют частицы меди размером от 0.2 до 1.2 нм. Тестирование проводили при давлении 1.5 МПа, подаче смеси с соотношением CO/ДМЭ = = 47, объемной скорости 4500 ч–1 (расход 150 мл/мин), температуре 473 К. Установлено, что выход МА коррелирует линейно с ростом содержания меди в цеолите. Введение меди в количестве 0.2 ммоль/г увеличивает выход МА примерно в 1.5 раза. Авторы полагают, что в случае Cu/H-MOR ДМЭ диссоциирует на паре Cu–БКЦ, образуя метильную группу на меди и метоксигруппу на БКЦ (последнюю авторы называют “адсорбированным метанолом”). СО, внедряясь в связь Cu–CH3, дает ацетил, реагирующий с “адсорбированным метанолом”, что приводит к синтезу МА и восстановлению БКЦ.
Появление монокарбонилов на атомах меди, присутствующих в мордените, согласуется с данными ИК-Фурье-спектроскопии и 13С MAS ЯМР в работе [41]. Медьсодержащий морденит был приготовлен ионным обменом Н-морденита, полученного прокалкой коммерческого цеолита (SiО2/Al2О3 = 20; CBV20A, “Zeolyst International”). Затем образец прокаливали при 500°С в течение 2 ч. Содержание меди составило 2.5 мас. %. Перед исследованиями катализаторы выдерживали в присутствии СО при 350°С.
Одновременное введение меди и цинка в морденит [31] в количестве 0.57 и 2.47 вес. %, соответственно, увеличивает, как уже отмечалось выше, стабильность его работы. Согласно [42] медь промотирует занятие ионами цинка центров Т4 в каналах 12-MR, что препятствует протеканию реакций образования углеводородов.
Обзор работ, касающихся медьсодержащих цеолитов, проведен в [32]. Отмечено, что наблюдается разнообразие состояний меди, зависящих от количества вводимой меди, способа ввода и характера последующей обработки цеолита.
ПРОБЛЕМЫ ДЕЗАКТИВАЦИИ ЦЕОЛИТОВ
Цеолиты с 8-MR-каналами, в частности H-MOR, показывают высокие начальные активность и селективность в отношении МА (>99%) благодаря эффекту конфайнмента боковых карманов 8-MR, а также введению легирующих добавок. Тем не менее, все они, как отмечено выше, демонстрируют быстрое снижение активности, связанное с отложением углеродистых соединений.
Авторы [36] предположили, что углеводороды появляются при взаимодействии метоксигрупп с ДМЭ с формированием промежуточных триметилоксоний катионов, дальнейшие реакции которых на активных центрах в основных каналах 12-MR морденита приводят к синтезу углеводородов. Вывод о том, что в 12-MR-каналах H-MOR (Si/Al = 10.3) практически не идет образование МА (кинетически затруднено), но протекают реакции синтеза углеводородов, подтверждается результатами in situ 13C CP/MAS ЯМР-спектроскопии (ЯМР с кросс-поляризационным вращением под магическим углом на ядрах 13C) [43].
Серия образцов, приготовленных деалюминированием азотной кислотой Na-морденита (Si/Al = = 6.5, CBV-10A, “Zeolyst International”) с последующим переводом их в Н-форму, изучена в [44]. Удаление алюминия происходило в основном в центрах Т3 и Т4, локализованных в четырехчленных кольцах 8-MR- и 12-MR-каналов. Отмечается, что обработка азотной кислотой приводила вначале к росту активности из-за снижения содержания внерешеточного алюминия. При глубоком деалюминировании активность снижалась, но существенно возрастала стабильность. Однако после удаления половины атомов алюминия катализатор для карбонилирования становился непригоден. Авторы приходят к выводу, что именно центры Т3 ответственны за активность в карбонилировании ДМЭ, тогда как Т1 и Т2 – за дезактивацию.
Природа углеродистых отложений охарактеризована в [45]. Углеродистые частицы в основном представляли собой алкилированные ароматические соединения. Количество БКЦ в основных каналах 12-MR снижалось во время реакции без значительного изменения их числа в восьмичленных каналах. Поскольку последние считаются активными центрами реакции карбонилирования ДМЭ, дезактивация может быть связана с блокированием основных каналов за счет осаждения кокса. Спектры 13C ЯМР твердотельных катализаторов после проведения каталитических испытаний содержали полосы, относящиеся к алкокси- и карбоксильным группам. Кроме того, были обнаружены метильные, метиленовые, а также третичные и четвертичные алифатические группы, что соответствует структуре алкилзамещенных ароматических соединений. Углеродистые образования, дезактивирующие катализатор, удаляются при термопрограммированном окислении до 1000 К. Наличие меди в катализаторе способствует снижению температуры их окисления.
В [28] сопоставлено поведение в карбонилировании ДМЭ кислотных центров в каналах 12-MR и боковых карманах 8-MR морденита (Si/Al = = 7.5). Сравнивали три образца: 1) Н-MOR, полученный ионным обменом натрия на ион аммония; 2) образец, в котором ионный обмен проведен частично (HMOR-MC); 3) Н-MOR, в котором с помощью адсорбции пиридина нейтрализована кислотность в 12-MR-каналах (HMOR-SP). Неполный обмен натрия на протоны (содержание Na – 2.5 против 4.7 мас. % в исходном Na-MOR) позволил, как полагают авторы, синтезировать морденит с кислотными центрами только в 12-MR-каналах. Поведение образцов при каталитическом тестировании существенно различалось. Скорость синтеза МА при использовании катализатора HMOR-SP, в котором активные кислотные центры находятся только в боковых карманах 8-MR, выше, а работа стабильнее, чем в случае катализатора HMOR-MC, в котором активные центры присутствуют только в 12-MR-каналах. Активность исходного цеолита Н-MOR, пройдя начальный максимум, быстро падает во времени из-за протекания побочных реакций.
Изучение дезактивированных в карбонилировании ДМЭ катализаторов H-MOR и Cu/H-MOR проведено в [45]. Синтез образцов H-MOR и Cu/H-MOR (2 мас. % Cu) выполнен по методике [38]. Активность катализаторов в карбонилировании ДМЭ исследовали при 473 К, давлении 1.5 МПа, ДМЭ/CO = 1/49, GHSV = 3000 ч–1. Хотя начальная активность медьсодержащего катализатора была заметно выше, после нескольких часов работы наблюдалась постепенная дезактивация обоих катализаторов. Углеродистые отложения представляли собой в основном алкилированные ароматические соединения. Большая часть углеродистых отложений была локализована в 12-MR-каналах, что приводило к их блокировке. Модификация H-MOR медью способствовала росту его активности как в карбонилировании, так и в реакциях, связанных с дезактивацией.
Согласно [46] частичная замена алюминия на цинк (0.31 мас. % в расчете на оксид) при синтезе морденита несколько замедляет дезактивацию катализатора.
Таким образом, можно сделать вывод, что углеродистые отложения, образующиеся при карбонилировании ДМЭ на H-MOR и Cu-MOR, состоят в основном из алкилированных ароматических соединений, и на их формирование влияет пространственная ограниченность топологии MOR. Большая часть углеродистых отложений расположена в каналах 12-MR, что приводит к блокировке каналов 8-MR и к дезактивации катализатора.
ЦЕОЛИТЫ EU-12, SSZ-13 В КАРБОНИЛИРОВАНИИ ДМЭ
Авторами [47] в 1986 г. с использованием холина в качестве агента, регулирующего органическую структуру, в присутствии ионов Na+ и Rb+ был синтезирован цеолит с мелкими порами, названный EU-12. Установлено, что структура EU-12 имеет двумерную систему каналов 8-MR. Из двух отдельных 8-MR-каналов вдоль оси c меньший соединяется с синусоидальным 8-MR-каналом вдоль оси а, в то время как другой, больший – с синусоидальным каналом, разделяя 8-MR-каналы в плоскости ас [48]. Цеолит Н-EU-12 показал приемлемые характеристики в карбонилировании ДМЭ в МА. Так, в [49] изучили стабильность работы цеолита в ходе длительного 50-часового пробега при 220°С и давлении 1.5 МПа. Использовали газ состава, об. %: 3.03 Ar, 4.13 ДМЭ, 92.84 CO, расход – 20 мл мин–1, навеска – 0.5 г. Конверсия ДМЭ снизилась с 15.7 до 10% в течение 30 ч работы, но далее оставалась постоянной. Стабильность работы в тех же условиях цеолитов H-MOR и H-ZSM-35 ниже. Проведение реакции при повышенных температурах (230 и 240°С) приводит к быстрой дезактивации H-EU-12, более низкой селективности по МА и большему количеству побочных продуктов. Катализатор может быть регенерирован окислительной обработкой с восстановлением активности.
Поведение цеолита со структурой шабазита SSZ-13 (Si/Al = 10) в карбонилировании ДМЭ близко к таковому морденита (CBV 21A, Si/Al = = 10) [50]. Образцы тестировали при 0.1 МПа, 165°С, подавая смесь, об. %: 2 ДМЭ, 3 He, 5 Ar, 95 CO с расходом 75 Нсм3/мин. Предполагается, что активные центры SSZ-13 расположены в 8-MR-окнах.
ПРЯМОЕ ПРЕВРАЩЕНИЕ СИНТЕЗ-ГАЗА В МА
О прямом превращении синтез-газа в МА в присутствии двухслойного катализатора – оксиды Cu–Zn–Al/H-ZSM-5 и морденит (H-MOR) – сообщено в [51]. Авторы исследовали превращение синтез-газа (Н2/СО = 1, давление 3 МПа) в МА, комбинируя в одном реакторе катализаторы с разными функциями. В случае комбинации Cu–Zn–Al/H-ZSM-5 и H-MOR суммарная селективность превращения в МА и уксусную кислоту достигала при температуре 473 К примерно 95% при конверсии CO 4.5%. При удалении кислотных центров в 12-MR-каналах H-MOR даже спустя 100 ч работы дезактивация катализатора была незначительная. При использовании комбинации H-MOR и шпинели ZnAl2O4 селективность превращения в МА и уксусную кислоту при 603–643 К составляла 85% при конверсии СО 11%; ДМЭ являлся ключевым промежуточным продуктом. На оксидном катализаторе Cu–Zn–Al с высокой селективностью производился метанол. Для катализатора Cu–Zn–Al/H-ZSM-5, полученного смешением оксидного катализатора Cu–Zn–Al и H-ZSM-5, наблюдалась 93% селективность по ДМЭ. Двухслойная комбинация оксидного катализатора Cu–Zn–Al и H-MOR катализировала синтез ДМЭ, превращающегося после индукционного периода в МА и уксусную кислоту. Авторы полагают, что появление уксусной кислоты связано с наличием воды, образующейся при синтезе ДМЭ. H-ZSM-5, используемый для дегидратации метанола в ДМЭ, может быть заменен другими типами цеолитов, такими как H-SAPO-34 и H-β, без значительного изменения суммарной селективности по МА и уксусной кислоте.
С другой стороны, замена H-MOR в тройной комбинации Cu–Zn–Al/H-ZSM-5/H-MOR на другие типы цеолитов в синтезе МА и уксусной кислоты значительно снижала конверсию ДМЭ и селективность реакции. МА и уксусная кислота образовывались из синтез-газа при относительно высоких температурах (603–643 К) без значительной дезактивации катализатора при применении каталитических систем на основе оксидов металлов и H-MOR. Было обнаружено, что оксидные катализаторы типа Cu–Zn–Al не подходят для высокотемпературной конверсии (643 К) синтез-газа в оксигенаты: наблюдалось образование метана и С2-углеводородов.
ЗАКЛЮЧЕНИЕ
В настоящее время продолжается интенсивное изучение цеолитов с целью применения их в качестве катализаторов для различных каталитических реакций. Этому способствует создание новых методик синтеза цеолитов, развитие физико-химических методов исследований, усовершенствование квантово-химических методов расчета. Карбонилирование ДМЭ с получением МА на цеолитах типа морденита и, отчасти, феррьерита на сегодняшний день достаточно изучено. Синтез МА протекает в смеси СО/ДМЭ на БКЦ при умеренных температурах (около 200°С) с высокой селективностью и существенной конверсией ДМЭ. Как правило, используются высокие отношения СО/ДМЭ при относительно низких объемных скоростях. При повышении температуры наблюдается быстрая дезактивация, связанная с протеканием побочных реакций, приводящих к отложению в структуре цеолита ароматических соединений.
Структуры ряда цеолитов, так же как и структура морденита, содержат 8-MR-окна, в которых находятся центры, проявляющие активность в карбонилировании ДМЭ. Однако их активность существенно ниже, и выяснение причин этого остается предметом исследований.
Основной проблемой карбонилирования ДМЭ в МА является низкая стабильность используемых катализаторов: активность падает после нескольких часов работы. Хотя дезактивированный катализатор может быть регенерирован окислительной или восстановительной обработкой, для реализации процесса в промышленном масштабе необходимо добиться увеличения длительности пробега. Перспективный прием, повышающий стабильность катализатора, – блокировка в структуре цеолита кислотных центров, на которых идут побочные реакции, которая может быть достигнута при адсорбции пиридина или при ионном обмене соответствующих протонов структуры морденита на органические катионы. Введение ионов Zn2+ также ингибирует активность центров морденита, на которых протекают побочные реакции.
Новым направлением исследований является разработка высокоселективных многофункциональных катализаторов, позволяющих вести синтез МА непосредственно из синтез-газа.
Список литературы
Goldemberg J. // Science. 2007. V. 315. № 5813. P. 808.
Van Leeuwen P.W.N.M. / In: Homogeneous Catalysis. Dordrecht: Springer, 2004. P. 109.
Jones J.H. // Platinum Metals Rev. 2000. V. 44. P. 94.
Cheung P., Bhan A., Sunley G.J., Iglesia E. // Angew. Chem. Int. Ed. 2006. V. 45. P. 1617.
Cheung P., Bhan A., Sunley G.J., Law D.J., Iglesia E. // J. Catal. 2007. V. 245. P. 110.
Derouane E.G. // J. Catal. 1986. V. 100. P. 541.
Derouane E.G., André J.M., Lucas A.A. // J. Catal. 1988. V. 110. P. 58.
Волнина Э.А., Кипнис М.А., Хаджиев С.Н. // Нефтехимия. 2017. Т. 57. № 3. С. 243. (Volnina E.A., Kipnis M.A., Khadzhiev S.N. // Pet. Chem. 2017. V. 57. P. 353.)
Zhan E., Xiong Z., Shen W. // J. Energy Chem. 2019. V. 36. P. 51.
Koch H., Haaf W. // Justus Liebigs Ann. Chem. 1958. V. 618. P. 251.
Fujimoto K., Shikada T., Omata K., Tominaga H. // Chem. Lett. 1984. V. 13. P. 2047.
Alberti A. // Zeolites. 1997. V. 19. P. 411.
Simoncic P., Armbruster T. // Am. Mineral. 2004. V. 89. P. 421.
Terasaki O., Yamazaki K., Thomas J.M., Ohsuna T., Watanabe D., Sanders J.V., Barry J.C. // J. Solid State Chem. 1988. V. 77. P. 72.
Thuy T.L., Chawla A., Rimer J.D. // J. Catal. 2020. V. 391. P. 56.
Gounder R., Iglesia E. // Chem. Commun. 2013. V. 49. P. 3491.
Boronat M., Martınez-Sanchez C., Law D., Corma A. // J. Am. Chem. Soc. 2008. V. 130. P. 16316.
Rasmussen D.B., Christensen J.M., Temel B., Studt F., Moses P.G., Rossmeisl J., Riisager A., Jensen A.D. // Catal. Sci. Technol. 2017. V. 7. P. 1141.
Dědeček J., Sobalik Z., Wichterlová B. // Catal. Rev. Sci. Eng. 2012. V. 54. P. 135.
Boronat M., Corma A. // ACS Catal. 2019. V. 9. 1539.
Palčić A., Valtchev V. // Appl. Catal. A: Gen. 2020. V. 606. 117795.
Cherkasov N., Vazhnova T., Lukyanov D.B. // Vib. Spectrosc. 2016. V. 83. P. 170.
Bhan A., Allian A.D., Sunley G.J., Law D.J., Iglesia E. // J. Am. Chem. Soc. 2007. V. 129. P. 4919.
Liu S., Liu H., Ma X., Liu Y., Zhu W., Liu Z. // Catal. Sci. Technol. 2020. V. 10. P. 4663.
Wang M., Huang S., Lu J., Cheng Z., Li Y., Wang S., Ma X. // Chin. J. Catal. 2016. V. 37. P. 1530.
Huang X., Ma M., Li M., Shen W. // Catal. Sci. Technol. 2020. V. 10. P. 7280.
Li L., Wang Q., Liu H., Sun T., Fan D., Yang M., Tian P., Liu Z. // ACS Appl. Mater. Interfaces. 2018. V. 10. P. 32239.
Zhou H., Zhu W.L., Shi L., Liu H.C., Liu S.P., Ni Y.M., Liu Y., He Y.L., Xu S.L., Li L.N., Liu Z.M. // J. Mol. Catal. A. 2016. V. 417. P. 1.
Li Y., Yu M., Cai K., Wang M., Lv J., Howe R.F., Huang S., Ma X. // Phys. Chem. Chem. Phys. 2020. V. 22. P. 11374.
Liu S., Fang X., Liu Y., Liu H., Ma X., Zhu W., Liu Z. // Catal. Commun. 2020. V. 147. ID 106161.
Reule A.A.C., Semagina N. // ACS Catal. 2016. V. 6. P. 4972.
Reule A.A.C., Prasad V., Semagina N. // Micropor. Mesopor. Mater. 2018. V. 263. P. 220.
Román-Leshkov Y., Moliner M., Davis M.E. // J. Phys. Chem. C 2011. V. 115. P. 1096.
Jung H.S., Ham H., Bae J.W. // Catal. Today. 2020. V. 339. P. 79.
Liu Z., Yi X., Wang G., Tang X., Li G., Huang L., Zheng A. // J. Catal. 2019. V. 369. P. 335.
Boronat M., Martinez C., Corma A. // Phys. Chem. Chem. Phys. 2011. V. 13. P. 2603.
Hazel N.J., Key L.A., Roberts M.S., Sunley J.G. // Pat. USA 8624054. 2014.
Li Y., Huang S., Cheng Z., Wang S., Ge Q., Ma X. // J. Catal. 2018. V. 365. P. 440.
Wang S., Guo W., Zhu L., Wang H., Qiu K., Cen K. // J. Phys. Chem. C. 2015. V. 119. P. 524.
Lezcano-Gonzalez I., Deka U., Arstad B., Van Yperen-De Deyne A., Hemelsoet K., Waroquier M., Van Speybroeck V., Weckhuysen B.M., Beale A.M. // Phys. Chem. Chem. Phys. 2014. V. 16. P. 1639.
Blasco T., Boronat M., Concepcion P., Corma A., Law D., Vidal-Moya J.A. // Angew. Chem. Int. Ed. 2007. V. 46. P. 3938.
Reule A.A.C., Shen J., Semagina N. // ChemPhysChem. 2018. V. 19. P. 1500.
Li B., Xu J., Han B., Wang X., Qi G., Zhang Z., Wang C., Deng F. // J. Phys. Chem. C. 2013. V.117. P. 5840.
Reule A.A.C., Sawada J.A., Semagina N. // J. Catal. 2017. V. 349. P. 98.
Cheng Z., Huang S., Li Y., Cai K., Yao D., Lv J., Wang S., Ma X. // Appl. Catal. A: Gen. 2019. V. 576. P. 1.
Zhang Z., Zhao N., Ma K., Cheng Q., Zhang J., Zheng L., Tian Y., Li X. // Chin. Chem. Lett. 2019. V. 30. 513.
Araya A., Lowe B.M. // Pat. USA 4581211. 1986.
Bae J., Cho J., Lee J.H., Seo S.M., Hong S.B. // Angew. Chem. Int. Ed. 2016. V. 55. P. 7369.
Feng X., Yao J., Li H., Fang Y., Yoneyama Y., Yang G., Tsubaki N. // Chem. Commun. 2019. V. 55. P. 1048.
Lusardi M., Chen T.T., Kale M., Kang J.H., Neurock M., Davis M.E. // ACS Catal. 2020. V. 10. P. 842.
Zhou W., Kang J., Cheng K., He S., Shi J., Zhou C., Zhang Q., Chen J., Peng L., Chen M., Wang Y. // Angew. Chem. Int. Ed. 2018. V. 57. P. 12012.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ