

Кинетика и катализ, 2022, T. 63, № 2, стр. 160-177
Экспериментальное и расчетно-теоретическое исследование пиролиза и окисления метана в отраженных ударных волнах с учетом сажеобразования
П. А. Власов a, *, А. Р. Ахуньянов a, В. Н. Смирнов a
a ФИЦ Химической физики им. Н.Н. Семенова РАН
119991 Москва, ул. Косыгина, 4, Россия
* E-mail: shocktube@yandex.ru
Поступила в редакцию 21.06.2021
После доработки 02.11.2021
Принята к публикации 10.11.2021
- EDN: XWDSZR
- DOI: 10.31857/S0453881122020149
Аннотация
Выполнено экспериментальное и расчетно-теоретическое изучение процессов пиролиза и окисления метана и образования микрогетерогенных дисперсных частиц сажи в отраженных ударных волнах. В расчетно-теоретическом исследовании использовалась предложенная нами ранее единая кинетическая модель сажеобразования. Эта модель позволяет количественно описывать процессы расходования исходных молекул метана и образования вторичных продуктов при пиролизе и окислении различных смесей с аргоном и кислородом. Проведено сравнение результатов экспериментов, полученных на ударных трубах различной конструкции, оснащенных различными методами регистрации исходных компонентов и вторичных продуктов, с результатами наших кинетических расчетов. В целом кинетическая модель продемонстрировала хорошее согласие расчетов и экспериментов разных авторов по измерению расходования молекул метана и образованию основных и вторичных продуктов пиролиза и окисления метана на различных ударных трубах.
ВВЕДЕНИЕ
В последнее время процесс изменения климата на Земле стал настолько очевидным, что привлек пристальное внимание ученых, занимающихся физико-химическими и технологическими проблемами горения углеводородного топлива. Простое сжигание метана дает в качестве продуктов воду Н2О и углекислый газ СО2. При этом остро встает вопрос о том, как его можно утилизировать. Образование большого количества углекислого газа, который является ярким представителем парниковых газов, ведущих к изменению климата, крайне нежелательно. В настоящее время существуют различные подходы к утилизации СО2, выделяющегося в процессе горения углеводородного топлива. Одним из них является использование СО2 в качестве окислителя метана в процессе его горения. Пиролиз метана позволяет полностью избежать появления СО2. В этом случае в качестве полезного продукта образуются экологически чистый водород и твердый углерод (частицы сажи), который легче утилизировать по сравнению с другими способами утилизации СО2. Однако процесс пиролиза сильно эндотермичен и требует больших затрат энергии. В этой связи обсуждается использование солнечной энергии для осуществления пиролиза метана [1–5], а также применение низкотемпературной плазмы для его проведения в бескислородной среде [6].
Весьма перспективный способ избежать образования СО2 – это конверсия метана в синтез-газ, который и сам представляет собой экологически более чистое топливо, чем различные углеводороды, и также является исходным компонентом для производства различных жидких углеводородов, в частности, метанола. Технология получения метанола из синтез-газа чаще всего реализуется в виде каталитического процесса. Появление в ходе этого процесса твердой микрогетерогенной фазы углерода (частиц сажи) ведет к отравлению катализатора и поэтому крайне нежелательно. В связи с этим необходимо определить условия, при которых образуется минимальное количество твердых частиц сажи, но при этом сохраняется требуемое соотношение Н2/CO в производимом синтез-газе.
Наряду с возрастающей важностью получения новых экспериментальных результатов, с развитием современных детальных кинетических механизмов пиролиза и окисления углеводородов возрастает роль численных экспериментов, которые позволяют извлечь необходимую информацию в тех областях параметров и условиях протекания процесса, где постановка надежного эксперимента затруднительна или просто невозможна.
Современные детальные кинетические механизмы пиролиза и окисления различных углеводородов в газовой фазе разработаны как для самых легких, таких, например, как метан, так и для весьма тяжелых углеводородов. Эти механизмы включают сотни компонентов, в том числе и различные изомеры сложных углеводородов, которые участвуют в десятках тысяч элементарных реакций. При определенных условиях при окислении различных углеводородов образуются частицы новой фазы – сажи. В этом случае возникает проблема адекватного описания процесса образования зародышей и частиц микрогетерогенной дисперсной фазы, в частности, частиц сажи.
В наших предыдущих работах была предложена единая кинетическая модель сажеобразования, позволяющая количественно описать основные параметры процесса формирования частиц сажи при пиролизе и окислении всех простейших углеводородов, начиная с метана и заканчивая простейшими ароматическими соединениями [7].
Основной мотивацией предложить свою кинетическую модель было желание включить процесс образования частиц сажи, который не учитывается в большинстве опубликованных кинетических механизмов. Даже если авторы включали в кинетическую модель появление полиароматических соединений, до образования зародышей и частиц сажи эти механизмы доведены не были. В результате наших усилий удалось разработать единую кинетическую модель для процесса сажеобразования из различных простейших углеводородов – как алифатических, так и ароматических [7]. Модель описывает пиролиз и окисление всех простейших углеводородов, включая метан, с последующим формированием зародышей и частиц сажи. Для этого необходимо было разработать комбинированный механизм, включающий как реакции в газовой фазе, так и гетерогенные реакции, ведущие к возникновению зародышей и последующему превращению их в частицы сажи.
В настоящей работе основное внимание сосредоточено на процессе пиролиза метана при относительно высоких температурах. Основная цель – это демонстрация возможностей единой кинетической модели образования частиц сажи для количественного описания процесса пиролиза и окисления метана при относительно высоких температурах, реализующихся за фронтом отраженной ударной волны, и формирования микрогетерогенной дисперсной фазы углерода (частиц сажи). Проведено прямое сравнение результатов детального кинетического моделирования основных продуктов пиролиза и окисления метана и концентрации и выхода частиц сажи с данными экспериментальных измерений в ударных трубах различной конструкции.
ОПИСАНИЕ ЭКСПЕРИМЕНТАЛЬНЫХ УСТАНОВОК
Наши эксперименты проводили на установке “ударная труба”, изготовленной из нержавеющей стали (внутренний диаметр – 75 мм, длина секции низкого давления – 3.2 м, длина секции высокого давления – 1.5 м). Подробное описание и схема экспериментальной установки приведены в [7]. Параметры газа за проходящей и отраженной ударными волнами вычисляли, исходя из начального давления, состава исследуемой смеси и скорости падающей ударной волны в рамках теории идеального течения в ударной трубе [8]. Скорость падающей ударной волны измеряли на двух базах с помощью трех последовательно установленных пьезоэлектрических датчиков давления, изготовленных из цирконата титаната свинца. Ударная волна генерировалась спонтанным разрывом одной или нескольких алюминиевых диафрагм толщиной 0.05–0.2 мм в зависимости от требуемых условий за падающей или отраженной ударной волной. Толкающим газом служили смеси гелия с воздухом различного состава (вплоть до 50% воздуха): чем ниже была требуемая температура, тем больше воздуха вводили в толкающую смесь.
Для одновременного измерения временных профилей выхода сажи и ее эффективной температуры за отраженной ударной волной использовали двухлучевой абсорбционно-эмиссионный метод. Подробное описание этого метода со всеми необходимыми расчетными формулами приведено в [9, 10]. Выход сажи находили как отношение концентрации атомов углерода, входящих в состав ансамбля частиц сажи, к полной концентрации атомов углерода в исходной системе. При определении выхода сажи предполагали, что поглощение оптического излучения частицами сажи описывается в рамках теории Ми с использованием макроскопического комплексного показателя преломления, который не зависит от размера частицы сажи. Поскольку размер частиц сажи намного меньше длины волны зондирующего света [11], ослабление луча зондирующего света с хорошей точностью может быть описано в рэлеевском приближении. Измерения температуры частиц сажи в настоящей работе проводили с применением двух пространственно разделенных каналов для регистрации излучения и поглощения ансамбля частиц сажи, в отличие от традиционной схемы пирометрических измерений [9, 10]. Когда характерная скорость тепловой релаксации частиц намного выше характерной скорости их нагрева за счет экзотермических реакций роста и охлаждения за счет излучения, а также когда интенсивность излучения хемолюминесцентных реакций намного меньше, чем интенсивность теплового излучения частиц, эффективная температура близка к реальной температуре газа и частиц сажи. В наших экспериментах эти условия выполняются достаточно хорошо.
Недавно пиролиз метана вновь привлек пристальное внимание исследователей. Появились новые экспериментальные данные [12], которые мы будем использовать для тестирования нашей кинетической модели. Важно понимать, что эти экспериментальные данные были получены разными экспериментальными методами, которые были реализованы на ударных трубах различной конструкции.
В работе [12] измерения продуктов пиролиза метана проводили на трех ударных трубах: ударной трубе высокого давления, одноимпульсной ударной трубе, а измерения поглощения лазерного излучения на заданной длине волны осуществляли на третьей ударной трубе постоянного диаметра. Подробные описания всех трех ударных труб можно найти в работах [13–15].
Ударная труба высокого давления имела постоянный внутренний диаметр 90 мм и длину секций высокого и низкого давлений 6.4 и 6.1 м соответственно, между которыми помещалась алюминиевая диафрагма. Столь внушительные размеры ударной трубы (секций высокого и низкого давления) позволяли существенно увеличить время наблюдения вплоть до 15 мс за счет изменения молекулярного веса толкающего газа (смеси гелия и аргона) [14].
Одноимпульсная ударная труба представляла собой ударную трубу из нержавеющей стали с внутренним диаметром 80 мм, длиной секций высокого и низкого давлений 2.5 и 6.3 м соответственно. Она была оснащена приемным резервуаром объемом 0.35 м3, заполненным аргоном при таком же давлении (50–150 мбар), как и начальное давление исследуемой смеси в секции низкого давления [13].
Третья ударная труба являлась обычной ударной трубой из нержавеющей стали с внутренним диаметром 80 мм и длиной секции высокого и низкого давлений 3.5 и 5.7 м соответственно. Она была оснащена оптическими кварцевыми вводами и выводами.
На всех трех ударных трубах скорость падающей ударной волны измеряли на трех интервалах с помощью четырех пьезоэлектрических датчиков давления Kistler 603B (“Kistler Group”, США) или PCB 112A03 или 112A05 (“PCB Piezotronics, Inc.”, США). Температуру T5 и давление Р5 за фронтом отраженной ударной волны рассчитывали на основе измеренной скорости падающей ударной волны с использованием одномерной модели идеального течения в ударной трубе с погрешностью определения температуры порядка 15 K. Рабочие смеси готовили манометрическим способом в 50-литровом сосуде из нержавеющей стали и выдерживали не менее 12 ч перед проведением эксперимента. Все смеси содержали 10 мол. % метана в Ar. Для проведения хроматографических и масс-спектрометрических измерений в исследуемые смеси добавляли 2% Ne (в качестве внутреннего стандарта), 1 мол. % CO (для измерений температуры) и 20 мол. % He (для ускорения колебательной релаксации).
В экспериментах на ударной трубе высокого давления молекулярная масса толкающего газа (смесь гелия и аргона) подбиралась таким образом, чтобы полезное время наблюдения до момента охлаждения изучаемой смеси после отражения ударной волны от торца ударной трубы составляло примерно 12 мс. Пробы газа отбирали с помощью быстро открывающегося клапана, встроенного в торец ударной трубы (Pulse Valve Series 9, “Parker Hannifin”, США). Затем пробы исследуемой смеси анализировали хроматографически (GC System 7890 A, MSD 5975C с колонкой J&W HP-PLOTQ, “Agilent Technologies”, США).
В одноимпульсной ударной трубе времена наблюдения были существенно короче, чем в ударной трубе высокого давления, и изменялись от 2 до 4 мс. Все концентрации продуктов пиролиза метана определяли также хроматографически.
Эндотермичность процесса пиролиза исследуемых смесей (10 мол. % CH4 в Ar) приводит к заметному изменению со временем температуры реагирующей смеси по сравнению с начальной температурой T50 сразу за фронтом отраженной ударной волны. Поэтому информация об изменении температуры со временем необходима для сравнения результатов измерений с результатами детального кинетического моделирования. Кинетические расчеты проводили в приближение постоянной плотности. При этом температура за фронтом отраженной волны изменялась в соответствие с законами сохранения энергии. Детали экспериментального метода измерения температуры подробно описаны в [12].
Для тестирования нашей кинетической модели мы также использовали данные из работы [16] по пиролизу и окислению метана, полученные в экспериментах на ударных трубах. В работе [16] использовались две ударные трубы с внутренним диаметром 4.1 см. Первая ударная труба могла работать как в обычном режиме для спектроскопических исследований временных профилей продуктов пиролиза метана, так и в режиме одноимпульсной ударной трубы [17–19]. Профили концентраций метана CH4 и образующихся продуктов его пиролиза и окисления, CO2 и H2O, определяли путем измерения ИК-излучения на разных длинах волн: CH4 (3.48 мкм), СО2 (4.24 мкм), H2O (2.56 мкм). Согласно методике [19], относительная интенсивность излучения на длине волны 3.48 мкм была определена также и для других компонентов: C2H2 (ацетилен), C2H4 (этилен), C2H6 (этан), aC3H4 (аллен), pC3H4 (пропин), CH2CO (кетен), CH2O (формальдегид) и CO2 (двуокись углерода). Вклад в интенсивность ИК-излучения на длине волны 4.24 мкм дает не только CO2, но также CH2CO и CO. Относительная интенсивность излучения этих соединений в зависимости от температуры была измерена в [20].
Смеси прореагировавших газов отбирали в предварительно откачанный сосуд (50 см3) через клапан рядом с торцом ударной трубы и анализировали хроматографически с помощью трех последовательно соединенных газовых хроматографов.
Вторая стандартная ударная труба была оборудована средствами измерения лазерного поглощения и ИК-излучения [21]. Для регистрации ИК-излучения ударная труба была оснащена окнами из CaF2. Концентрацию метана определяли методом поглощения лазерного излучения на длине волны 3.39 мкм. Исследовали смеси метана с аргоном (5% CH4 в Ar), а также смеси метана с кислородом в аргоне.
ДЕТАЛЬНАЯ КИНЕТИЧЕСКАЯ МОДЕЛЬ ПИРОЛИЗА МЕТАНА И ОБРАЗОВАНИЯ ЧАСТИЦ САЖИ
Для детального кинетического моделирования результатов экспериментов по пиролизу и окислению метана необходимо построить кинетическую модель, количественно описывающую расходование метана, изменение температуры в процессе пиролиза и окисления метана, и временные зависимости концентрации образующихся промежуточных и конечных продуктов пиролиза и окисления. Поскольку при пиролизе и окислении богатых смесей метана могут возникать зародыши и частицы конденсированной дисперсной фазы (частицы сажи), то кинетическая модель газофазных реакций должна включать процессы образования полиароматических и полииновых соединений, из которых согласно нашей модели формируются зародыши частиц сажи. Для проведения прямого сравнения результатов расчетов и экспериментов по измерению температуры и выхода частиц сажи наша кинетическая модель, помимо механизма реакций в газовой фазе, должна включать также механизм гетерогенных реакций образования и трансформации зародышей и частиц сажи.
В процессе создания механизма газофазных реакций мы использовали подходы, аналогичные приведенным в работе [22] для построения нового кинетического механизма, описывающего высокотемпературные режимы горения различных углеводородных топлив от метана до изооктана с учетом образования молекул бензола (как предвестника зародышей частиц сажи). При создании столь больших кинетических схем важна определенная самосогласованность и непротиворечивость кинетических параметров различных компонентов с их термодинамическими данными и другими модельными предположениями. Наша кинетическая модель основана на объединении нескольких кинетических механизмов, каждый из которых тестировался относительно определенных групп компонентов [23–26]. Слияние столь разных кинетических моделей в единый кинетический механизм должно проводиться с большой осторожностью, поскольку кинетические параметры и термодинамические данные для одних и тех же реакций могут отличаться.
Моделирование образования частиц сажи проводили с использованием кинетического механизма реакций, развитого в [10]. Кинетический механизм реакций в газовой фазе основан на модели USC-Mech II [23], которая описывает окисление H2 и CO и высокотемпературный пиролиз и окисление углеводородов C1–C4.
Кинетический механизм, предложенный в работе [23] для низших углеводородов, таких как метан (CH4) и углеводороды C2–C4, был взят за основу нашего единого кинетического механизма [7]. Механизм из [23] был расширен нами за счет добавления реакций для C5- и C7-компонентов [24, 25]. Были также учтены реакции с участием ароматических компонентов (бензола и толуола [24, 27]) и высших алканов (н-гексана и н-гептана [28]). При расширении механизма реакций мы включали в него только те новые реакции и компоненты, которые отсутствовали в нашем механизме. Хотя такой подход не может полностью гарантировать абсолютное соответствие всех кинетических параметров модели, он, тем не менее, безусловно, улучшает предсказательные способности новой кинетической модели. Константы скорости в предложенном нами механизме реакций не варьировали за исключением отдельных случаев, описанных ниже, для того чтобы обеспечить лучшее совпадение с результатами экспериментальных измерений.
Поскольку наш механизм содержит компоненты и реакции, уже включенные в различные более ограниченные механизмы реакций, конструирование кинетической схемы должно осуществляться очень тщательно и аккуратно. Мы при этом придерживались следующих правил: любые компоненты и реакции, отсутствовавшие в кинетическом механизме, просто добавлялись в него; в случае одинаковых реакций предпочтение отдавалось той, что уже присутствовала в старом механизме. Новым аспектом в предложенном в настоящей работе кинетическом механизме является то, что основной блок реакций, взятый из работы [29], который использовался в нашем старом механизме, был полностью заменен на механизм из работы [28]. Термодинамические данные были также существенно обновлены, и их основу составили данные из этой же работы [28]. Для частиц, отсутствующих в механизме реакций и файле термодинамических данных из [28], термодинамические данные брали из других механизмов [24–26], включенных как составные части в наш полный механизм реакций в газовой фазе. Механизм, взятый из работы [28], предусматривает ряд новых путей реакций, в частности, с участием винилидена C2HH. Одновременно введены целый ряд дополнительных каналов образования и роста молекул полиароматических углеводородов (вплоть до коронена) и реакции с участием углеводородов C3, C5 и C7 [24, 25]. В частности, новый механизм включает:
1) последовательный рост молекул полиароматических углеводородов по механизму HACA (H abstraction/C2H2 addition);
2) реакции комбинации фенильных радикалов с молекулами бензола C6H6;
3) реакции рекомбинации циклопентадиенила;
4) реакции замыкания в кольцевые структуры высших алифатических углеводородов.
Поскольку химия алкилпероксидов важна при низких и средних температурах, в наш механизм добавлены реакции с участием метилпероксидов (CH3O2), этилпероксидов (C2H5O2) и пропилпероксидов (C3H7O2) для корректного описания задержек воспламенения в смесях CH4 + O2 + Ar и промотирующего эффекта добавок пропана в смеси с метаном [8]. Полный модифицированный механизм газофазных реакций включает 3320 прямых и обратных реакций с участием 274 компонентов; константы скорости ряда важных реакций имеют зависимость от давления.
Детальное кинетическое моделирование процессов образования зародышей и частиц сажи проводилось нами с использованием дискретного метода Галеркина. Детали метода изложены в нашей работе [7]. Коротко они заключаются в следующем.
Процессы образования, роста, окисления и коагуляции зародышей и собственно частиц сажи описывали с помощью дискретного метода Галеркина. Он был предложен для решения систем обыкновенных дифференциальных уравнений, получающихся при рассмотрении прямой кинетической задачи кинетики гетерогенной полимеризации. Метод основан на разложении функции распределения по размерам гетерогенных компонентов по ортогональным полиномам, зависящим от дискретной переменной, в частности, от числа мономеров в гетерогенной частице. Погрешность такого разложения может быть строго оценена в рамках формализма дискретного метода Галеркина. На основе весовых функций, которые определяются типом химического процесса, строятся ортогональные полиномы, зависящие от дискретной переменной n. Например, в случае радикальной полимеризации весовая функция представляет собой распределение Шульца–Флори. Этот тип весовых функций связан с ортогональными полиномами Лагерра с дискретной переменной n. Если известна весовая функция, можно вычислить первые статистические моменты функции распределения по размерам и определить величину среднего размера частиц. Изменение во времени функции распределения частиц по размерам, которое зависит от химической кинетики соответствующих компонентов, позволяет получить полную систему обыкновенных дифференциальных уравнений для каждого коэффициента разложения. Этот подход дает возможность сохранить дискретный характер элементарного изменения микрогетерогенной частицы в любой реакции с участием газофазных и гетерогенных частиц и описывать такие изменения в виде обычных элементарных химических реакций для микрогетерогенных частиц любых размеров.
Согласно нашей модели сажеобразования, зародышами частиц сажи являются полиароматические структуры, образующиеся из небольших полиароматических молекул и радикалов (включающих два–три ароматических кольца). Реакции образования зародышей частиц сажи предполагаются необратимыми. Постулируется, что реакции поверхностного роста зародышей частиц сажи происходят на активных центрах, которые возникают за счет отрыва атомов водорода при взаимодействии с атомами водорода из газовой фазы. Таким образом, в модели рассматривается два ансамбля зародышей разных типов: с активными центрами и без них.
Зародыши частиц сажи активируются в реакциях с радикалами H• и OH• и деактивируются в реакциях с H, H2 и H2O. Активированные зародыши частиц сажи растут за счет присоединения из газовой фазы молекул C2H2, C4H2 и C6H2 (концентрация этих частиц относительно высока при пиролизе и окислении богатых смесей алифатических и ароматических углеводородов) и взаимодействия с полиароматическими молекулами и радикалами, а также за счет реакций друг с другом (коагуляции). Зародыши частиц сажи окисляются в реакциях с радикалами O• и OH• и трансформируются в частицы сажи без участия газовой фазы, которые, в свою очередь, растут за счет присоединения молекул алифатических углеводородов C2H2, C4H2, C6H2 и полиароматических углеводородов и их радикалов. Частицы сажи участвуют в реакциях коагуляции. Окисление частиц сажи происходит при вдаимодействии с радикалами O• и OH•.
Основное отличие механизма газофазных реакций, используемого в настоящей работе, от механизма, разработанного нами ранее [30, 31], заключается в том, что новый механизм включает новый блок реакций, описывающий горение этилена и ацетилена, предложенный в работе [28], и новый блок реакций гидратирования полииновых молекул, ведущих к расщеплению молекул высших полиинов и образованию более мелких углеводородов [27, 32]. Высшие полиины (C10H2 и C12H2) были исключены из кинетической схемы реакций. Принимая во внимание результаты экспериментов, представленные в работе [33], блок реакций конденсации полиинов с образованием зародышей частиц сажи из работ [30, 31], не был включен в рассматриваемую модель сажеобразования.
Все основные идеи дискретного метода Галеркина одновременно с возможностью решения большого числа дифференциальных уравнений для элементарных газофазных реакций впервые были реализованы в программе MACRON. Существенно переработанная и расширенная версия этой программы использована в наших кинетических расчетах процесса образования частиц сажи. Программа позволяет непосредственно вычислить выход сажи как отношение количества атомов углерода в ансамбле частиц сажи к полному числу атомов углерода в исходной смеси.
Сравнительный анализ наших экспериментальных данных с результатами кинетических расчетов процесса сажеобразования при пиролизе и окислении всех простейших алифатических и ароматических углеводородов, представленных в [7], позволяют нам говорить о создании единой кинетической модели сажеобразования. Эту модель тестировали с использованием как собственных экспериментальных данных, так и данных других авторов по задержкам воспламенения для различных углеводородов, а также экспериментально измеренным временным зависимостям концентраций различных компонентов. Еще одним новым тестом для нашего кинетического механизма стало количественное описание процесса химической ионизации, наблюдающейся экспериментально при окислении различных углеводородов, включая метан [34, 35].
Основной и практически единственной реакцией, в которой образуются первичные ионы и электроны, в наших исследуемых условиях является химическая (ассоциативная) ионизация CH• + O = CHO+ + e–. Именно протекание этой важной реакции наряду с обратной реакцией диссоциативной рекомбинации и определяют временные зависимости концентрации электронов, измеряемые в эксперименте. Поэтому для достижения совпадения результатов экспериментов и кинетических расчетов для профиля свободных электронов необходимо, чтобы кинетическая модель давала бы адекватный профиль радикалов СН• и атомов кислорода О. Наиболее чувствителен профиль свободных электронов именно к временной зависимости радикалов СН•. Поэтому мы рассматриваем процесс химической ионизации как дополнительный источник информации о кинетике образования и расходования радикалов СН• в реагирующей системе.
С другой стороны, включение реакций с участием заряженных частиц напрямую не влияет на кинетику образования и расходования радикалов, в частности, радикалов СН•. Концентрация заряженных частиц в наших экспериментах и кинетических расчетах довольно низкая, порядка 1010–1012 см–3, и заметного влияния на процесс пиролиза и окисления метана ионизация (химическая и термическая) оказать не может. Вклад процесса термической ионизации возможных легкоионизирующихся примесей, например, NaCl, с образованием ионов Na+ в общий баланс заряженных частиц для низких и средних температур пренебрежимо мал и возрастает лишь при самых высоких температурах [34, 35], которые не достигались в экспериментах, представленных в настоящей работе. Оценки возможных концентраций примесей NaCl в экспериментах на ударной трубе приведены в [35]. Процесс термической ионизации был включен в нашу кинетическую модель для придания ей еще более универсального характера. Тестовые эксперименты продемонстрировали, что термическая ионизация не оказывает влияния на временные зависимости свободных электронов, измеряемые в наших экспериментах в исследуемом интервале температур и давлений. Кинетические расчеты с учетом наличия примесей NaCl с концентрацией, рекомендованной в [35], также свидетельствуют о незначительном влиянии термической ионизации атомов натрия в объеме на концентрацию ионов и электронов, образующихся в процессе химической ионизации.
Выполненные в наших работах [34, 35] эксперименты по химической ионизации при окислении метана, ацетилена, н-гексана и ацетона в отраженных ударных волнах и их детальное кинетическое моделирование с помощью нашей единой кинетической модели [7] показали, что необходимо внести определенную коррекцию в механизм пиролиза и окисления метана [34, 35].
Для детального кинетического моделирования процесса химической ионизации при окислении углеводородов, в частности метана, мы использовали единый кинетический механизм, который ранее применялся для описания процесса сажеобразования [9, 10]. Этот механизм газофазных реакций был расширен за счет включения ряда процессов химической ионизации при окислении метана и ацетилена, представленных в табл. 1. Особое внимание было уделено образованию и расходованию радикалов ${\text{CH}}_{3}^{ \bullet },$ ${\text{CH}}_{2}^{ \bullet }$ и CH•, а также атомарного кислорода O, которые участвуют в реакциях, предшествующих химической ионизации CH• + O = CHO+ + e–. После введения дополнительных реакций, представленных в [34, 35], и в табл. 2, модифицированный механизм газофазных процессов включал 3559 прямых и обратных реакций между 309 различными компонентами. Константы скорости некоторых важных реакций зависели от давления (Приложения 1 и 2 ).
Таблица 1.
Основные реакции ионизации при окислении метана и ацетилена. Константы скорости представлены в виде аррениусовской зависимости k = ATn exp(–E/RT)*
| № | Реакция | A | E | n | Ссылка** |
|---|---|---|---|---|---|
| I | CH + O = CHO+ + e– | 2.530 × 1011 | 7.060 | 0.00 | Schofield, 2008 |
| II | CHO+ + H2O = H3O+ + CO | 9.030 × 1015 | 0.000 | 0.00 | Karasevich, 2009 |
| III | H3O+ + e– = H2O + H | 4.577 × 1022 | 0.000 | –1.60 | Karasevich, 2009 |
| IV | NaCl + M = Na + Cl + M | 5.000 × 1014 | 334.9 | 0.00 | Hartig et al., 1968 |
| V | Na + M = Na+ + e– + M | 1.870 × 1015 | 494.0 | 0.50 | Karasevich, 2009 |
| VI | NaCl + M = Na+ + Cl– + M | 4.940 × 1022 | 561.0 | –2.00 | Schneider, Park, 1975 |
| VII | H3O+ + Na = Na+ + H2O + H | 6.620 × 1015 | 0.000 | 0.00 | Karasevich, 2009 |
* Примечание. A – предэкспоненциальный коэффициент, учитывающий вероятность и число столкновений; T – температура, К; n – безразмерный показатель степени у температуры в выражении для константы скорости k = ATn exp(–E/RT); E – энергия активации, кДж/моль; R – универсальная газовая постоянная, 8.314 Дж моль–1 К–1. В выражении для константы скорости k = ATn exp(–E/RT) используются следующие единицы измерения: моль, см3, с, кДж/моль.
Таблица 2.
Дополнительные реакции термического разложения и окисления метана. Константы скорости представлены в виде аррениусовской зависимости k = ATn exp(–E/RT)*
| № | Реакция | A | E | n | Ссылка** |
|---|---|---|---|---|---|
| I | CH3 + M = CH2 + H + M | 2.000× 1016 | 382.6 | 0.00 | Markus et al., 1992 |
| II | CH3 + M = CH + H2 + M | 1.000 × 1016 | 356.9 | 0.00 | Rohrig et al., 1997 |
| III | CH3 + OH = CH2O + H2 | 3.160 × 1012 | 45.25 | –0.53 | Dean, Westmoreland, 1987 |
| IV | CH3 + OH = CH3O + H | 5.750 × 1012 | 58.31 | –0.23 | Dean, Westmoreland, 1987 |
| V | CH3 + OH = CH2OH + H | 2.630 × 1019 | 33.78 | –1.80 | Dean, Westmoreland, 1987 |
| VI | CH3 + H = CH2 + H2 | 6.030 × 1013 | 63.21 | 0.00 | Baulch et al., 1994 |
| VII | CH2 + M = CH + H + M | 5.620 × 1015 | 375.2 | 0.00 | Bauerle et al., 1995 |
| VIII | CH2 + M = C + H2 + M | 1.580 × 1014 | 267.9 | 0.00 | Markus et al., 1994 |
| IX | CH2 + CH2 = C2H2 + H + H | 2.000 × 1014 | 46.00 | 0.00 | Bauerle et al., 1995 |
| X | CH2 + CH2 = C2H3 + H | 2.000 × 1013 | 0.00 | 0.00 | Frank, Just, 1984 |
| XI | CH2 + O2 = CH2O + O | 4.000 × 1010 | 0.00 | 0.00 | Dombrowsky et al., 1992 |
| XII | CH2 + O = CH + OH | 3.160 × 1014 | 49.90 | 0.00 | Frank, Just, 1984 |
| XIII | CH2 + O = CO + H2 | 1.000 × 1013 | 0.00 | 0.00 | Frank, Just, 1984 |
| XIV | CH2 + O = CO + H + H | 6.310 × 1013 | 0.00 | 0.00 | Baulch et al., 1994 |
| XV | CH2 + H = CH + H2 | 7.940 × 1012 | 0.00 | 0.00 | Frank et al., 1986 |
| XVI | CH + M = C + H + M | 3.160 × 1014 | 301.4 | 0.00 | Markus et al., 1994 |
| XVII | CH + O2 = CO + OH | 5.010 × 1013 | 0.00 | 0.00 | Lichtin et al., 1984 |
| XVIII | CH + O2 = CO2 + H | 2.000 × 1013 | 0.00 | 0.00 | Hidaka et al., 1999 |
| XIX | C2H + O2 = HCCO + O | 6.030 × 1011 | 0.00 | 0.00 | Tsang, Hampson, 1986 |
| XX | C2H + O2 = CO + CO + H | 1.820 × 1013 | 0.00 | 0.00 | Hidaka et al., 1999 |
* Примечание. A – предэкспоненциальный коэффициент, учитывающий вероятность и число столкновений; T – температура, К; n – безразмерный показатель степени у температуры в выражении для константы скорости k = ATn exp(–E/RT); E – энергия активации, кДж/моль; R – универсальная газовая постоянная, 8.31 Дж моль–1 К–1. В выражении для константы скорости k = ATn exp(–E/RT) используются следующие единицы измерения: моль, см3, с, кДж/моль.
Изначально нам не удалось описать наши эксперименты по химической ионизации при окислении метано-кислородных смесей. Кинетическая модель, которую мы разработали, значительно (в разы) переоценивала скорость образования и концентрацию свободных электронов в области максимума по сравнению с нашими экспериментальными результатами. При этом время достижения максимума концентрации свободных электронов кинетическая модель предсказывала правильно. В то же время для смесей ацетилена с кислородом наблюдалось удовлетворительное согласие измеренных и расчетных значений концентрации свободных электронов и времен достижения их максимальных значений.
Окисление метана – это наиболее простой и прямой путь образования радикалов ${\text{CH}}_{3}^{ \bullet },$ ${\text{CH}}_{2}^{ \bullet }$ и CH• из исходного углеводорода CH4. Наше кинетическое моделирование показало высокую чувствительность концентрации свободных электронов к скорости термического разложения метана, в частности, к константе скорости обратной реакции (${\text{CH}}_{3}^{ \bullet }$ + H• (+ M) = CH4 (+ M)). Вариации коэффициента скорости этой реакции вызывают существенные изменения временного профиля концентрации электронов. Учитывая, что в области достижения максимума расчетные концентрации свободных электронов существенно превышали экспериментально измеренные, необходимо было найти способы уменьшения расчетной концентрации радикалов CH•. Включение дополнительных каналов расходования радикалов ${\text{CH}}_{3}^{ \bullet },$ ${\text{CH}}_{2}^{ \bullet }$ и СН•, взятых из опубликованных кинетических механизмов (табл. 2 в настоящей работе), позволяет снизить концентрацию радикалов СН• и, как следствие, концентрацию свободных электронов без существенного изменения положения максимума. При этом наблюдается значительное улучшение (на десятки процентов) совпадения расчетных и экспериментально измеренных временных зависимостей концентраций электронов [34, 35].
Полученную кинетическую модель, помимо описания результатов наших экспериментов по химической ионизации [34, 35], тщательно тестировали с использованием прямого сравнения результатов кинетических расчетов с результатами экспериментальных измерений временных зависимостей концентраций исходных углеводородов в процессе их расходования, различных промежуточных радикалов и продуктов реакции. Для пиролиза ацетилена данные такого сравнения приведены в нашей работе [10]. Также указанную модель применяли для описания собственных экспериментов по образованию частиц сажи при пиролизе и окислении различных углеводородов (метана, этана, этилена, ацетилена, пропана, пропилена, бензола, толуола и ряда других углеводородов). Показано, что она способна количественно описать температурную зависимость выхода сажи для всех указанных выше углеводородов без изменения каких-либо параметров единой кинетической модели [7].
Как уже отмечалось выше, реакция ${\text{CH}}_{3}^{ \bullet }$ + H• (+ M) = CH4 (+ M) играет определяющую роль в механизме пиролиза и окисления метана. Поэтому задание адекватной константы скорости этой реакции в детальном кинетическом механизме приобретает особое значение. В этой связи нами был проведен анализ температурных зависимостей для констант скорости этого процесса, полученных разными авторами и включенных в различные детальные кинетические механизмы. На рис. 1 приведены имеющиеся в литературе [36, 37, 40] температурные зависимости константы скорости бимолекулярной реакции термической диссоциации молекул метана CH4 + M (M = Ar). На рис. 1 также показаны экспериментальные результаты прямого измерения этой константы [38, 39].
Рис. 1.
Сравнение температурных зависимостей константы скорости бимолекулярной реакции термической диссоциации молекул метана CH4 + M (M = Ar) при давлении Р = 1.7 бар. Представленные зависимости построены для констант скорости, используемых в кинетических механизмах различных авторов. На рис. 1 также приведены экспериментальные результаты прямого измерения этой константы, полученные в работах [38, 39].
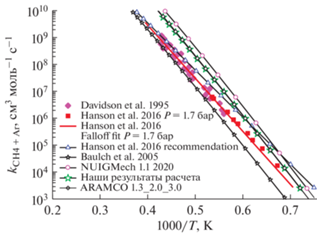
Представленные на рис. 1 и 2 температурные зависимости соответственно константы скорости бимолекулярной реакции термической диссоциации молекул метана CH4 + M (M = Ar) и константы скорости мономолекулярного процесса термической диссоциации молекул метана были получены нами с использованием программы OpenChem Workbench [41].
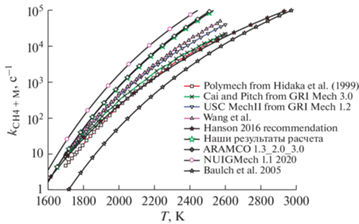
Рис. 2.
Сравнение температурных зависимостей константы скорости мономолекулярного процесса термической диссоциации молекул метана для давления Р = 1.5 бар. Представленные зависимости построены для констант скорости, используемых в кинетических механизмах различных авторов.
Как видно из рис. 1, наименьшие значения для константы скорости бимолекулярной реакции термической диссоциации молекул метана CH4 + M (M = Ar) приводятся в работе [40], а максимально высокие – в механизме NUIGMech 1.1 2020 [37], который является последней версией широко известного механизма ARAMCO. Как известно, существует три различных версии данного механизма (ARAMCO 1.3, 2.0, 3.0). Поскольку в основе нашего кинетического механизма лежит механизм USC-Mech II [23], на рис. 2 приведены результаты для константы скорости мономолекулярного процесса термической диссоциации молекул метана для этого механизма. Также на рис. 1 и 2 представлены результаты для механизма ARAMCO 1.3. Параметры для рассматриваемых констант скорости для механизмов ARAMCO 1.3, 2.0, 3.0 идентичны. Кроме того, на рис. 1 показано, как влияет температура на константу скорости бимолекулярной реакции термической диссоциации молекул метана, используемую в предложенном нами кинетическом механизме с учетом небольших изменений, сделанных для улучшения описания наших экспериментальных результатов по химической ионизации для смесей метана с кислородом. Эта зависимость практически совпадает с той, что была получена для вышеуказанной константы из механизма ARAMCO 1.3. На рис. 1 также представлена температурная зависимость данной константы скорости, найденная из экспериментальных измерений концентрации радикалов ${\text{CH}}_{3}^{ \bullet }$ в ударных трубах [38, 39] при давлении 1.7 бар.
Экспериментальные зависимости из этих работ весьма близки к теоретическим, полученным для констант скорости из механизма ARAMCO и нашего механизма. В работе [38] авторы определили свои параметры для описания скорости бимолекулярной реакции термической диссоциации молекул метана CH4 + M (M = Ar), которые практически идеально ложатся на их экспериментальные точки, а также предложили температурную зависимость для вышеуказанной константы в более широком диапазоне температур. Если в работе [38] рекомендуемая температурная зависимость данной константы скорости идеально совпадает с результатами эксперимента при высоких температурах и несколько расходится при низких, то для нашего механизма наблюдается противоположная картина, то есть наилучшее совпадение с результатами эксперимента отмечается именно при низких температурах, которые и будут, главным образом, нас интересовать в дальнейшем.
При рассмотрении данной реакции как бимолекулярного процесса CH4 + M (M = Ar) константа скорости зависит от давления (рис. 1). Как показывает теория РРКМ, в случае прямой пропорциональности константы скорости процесса термического распада молекулы величине давления рассматриваемая константа близка к пределу низких давлений. При полном отсутствии зависимости от давления константа скорости находится в пределе высоких давлений. Если все же наблюдается определенная зависимость константы скорости от давления, мы находимся в переходной области по давлению.
На рис. 2 представлены результаты рассмотрения данной реакции как мономолекулярного процесса CH4 + M (M = Ar). Результаты, приведенные на рис. 2, для температурных зависимостей константы скорости мономолекулярного процесса термической диссоциации молекул метана для давления Р = 1.5 бар во многом аналогичны результатам на рис. 1. Минимальные значения для данной температурной зависимости получаются для параметров, взятых из работы [40], максимальные значения – для параметров из работы [3].
Все остальные проанализированные механизмы дают довольно близкие результаты. Проведенный анализ позволяет заключить, что параметры для описания зависимости от давления процесса CH3 + H (+M) = CH4 (+M), используемые в нашем кинетическом механизме, дают температурную зависимость для константы скорости обратного процесса, которая весьма близка к температурным зависимостям, получающимся для параметров из ряда других детальных кинетических механизмов.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Рассмотрим результаты сравнения экспериментов из работы [12] с результатами наших кинетических расчетов.
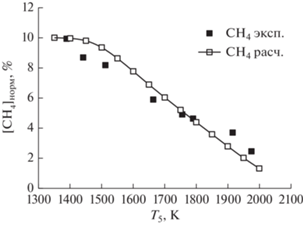
В работе [12] состав продуктов пиролиза метана в интервале температур 1285–2400 К и давлении 1.5 и 30 бар анализировали объединенным методом газовой хроматографии–масс-спектрометрии. При давлениях 1.5 бара наблюдалась хорошая воспроизводимость экспериментальных результатов, и углеродный баланс соблюдался с точностью до 100 ± 5% для всех температур. Как показали эксперименты в работе [12] и наши кинетические расчеты, основными продуктами пиролиза метана (в порядке уменьшения их относительной концентрации) являются ацетилен (C2H2), этилен (C2H4), аллен (aC3H4), пропин (pC3H4), диа-цетилен (C4H2), винилацетилен (C4H4) и бензол (A1).
Как можно видеть из рис. 3, превращение метана CH4 начинается при ∼1600 K. С повышением температуры концентрации ацетилен C2H2 и этилена C4H2 увеличиваются, достигая значений ∼3 и ∼0.2% при 2400 K соответственно. Для остальных продуктов (рис. 4) наибольшие концентрации наблюдаются при температурах 2000–2200 К. Исключение составляет этилен C2H4, максимальное содержание которого зарегистрировано при ∼1800 К. При температуре 2400 К расходуется примерно 80% исходного метана. Результаты расчетов, представленные на рис. 3 и 4, показывают, что наша кинетическая модель хорошо предсказывает расходование исходных молекул CH4, Также хорошо воспроизводится образование молекул ацетилена C2H2 во всем исследованном температурном диапазоне. Наблюдается некоторое расхождение экспериментальных и расчетных концентраций молекул этилена C2H4 в интервале температур 1800–2200 К. Следующими по значимости продуктами пиролиза метана являются бензол (A1) и диацетилен (C4H2).
Рис. 3.
Сравнение экспериментальных температурных зависимостей концентраций исходного углеводорода (метана) и основных продуктов его пиролиза (этилена и ацетилена) с результатами наших кинетических расчетов по единой кинетической модели для смеси 10% CH4 в Ar при давлении за фронтом отраженной ударной волны 1.5 бар.
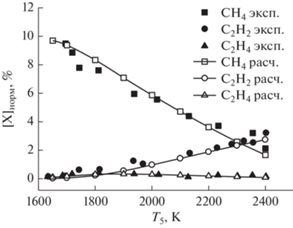
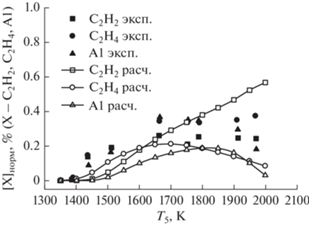
Рис. 4.
Сравнение экспериментальных температурных зависимостей концентраций вторичных продуктов пиролиза метана (диацетилена C4H2, винилацетилена C4H4, бензола (A1), этилена C2H4 и аллена aC3H4) с результатами наших расчетов по единой кинетической модели для смеси 10% CH4 в Ar при давлении за фронтом отраженной ударной волны 1.5 бар. Заштрихованные символы – результаты экспериментов; незаштрихованные символы и линии – результаты наших кинетических расчетов.
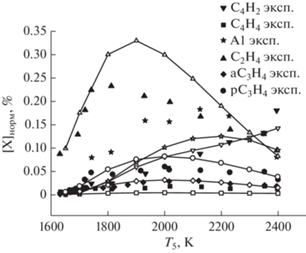
На рис. 4 приведены результаты сравнения экспериментальных температурных зависимостей концентраций вторичных продуктов пиролиза метана (диацетилена C4H2, винилацетилена C4H4, бензола (А1), этилена C2H4, аллена aC3H4) с результатами наших расчетов по единой кинетической модели для смеси 10% CH4 в Ar при давлении за фронтом отраженной ударной волны 1.5 бар. Видно, что наша кинетическая модель удовлетворительно описывает изменение во времени этих компонентов, которые очень важны для образования зародышей частиц сажи.
При повышенном давлении Р5 = 30 бар эксперименты из работы [12] и наши кинетические расчеты показывают (рис. 5), что молекулы CH4 начинают разлагаться при более низкой температуре (∼1400 K), и примерно 80% метана расходуется при температуре около 2000 К. В результате пиролиза метана образуется не только водород, но и микрогетерогенные частицы сажи. В экспериментах работы [12] углеродный баланс, который определялся на основе анализа продуктов, образующихся в газовой фазе, начинал уменьшаться от 100% при температуре 1650 К до 50% при температуре 2000 К. Поскольку специальных методов измерения частиц сажи в работе [12] не применялось, то изменение углеродного баланса рассматривалось как косвенное подтверждение формирования частиц сажи.
Рис. 5.
Сравнение экспериментальных температурных зависимостей концентраций исходного углеводорода (метана) с результатами наших расчетов по единой кинетической модели для смеси 10% CH4 в Ar при давлении за фронтом отраженной ударной волны 30 бар.
Как видно из рис. 6, при повышенном давлении основными стабильными продуктами пиролиза метана были ацетилен C2H2, этилен C2H4 и бензол (A1), относительные концентрации которых превышали 0.2%. На этом рисунке представлены результаты сравнения экспериментальных температурных зависимостей концентраций вторичных продуктов пиролиза метана (ацетилена C2H2, этилена C2H4, бензола (А1)) с результатами наших расчетов по единой кинетической модели для смеси 10% CH4 в Ar при давлении за фронтом отраженной ударной волны 30 бар. Наша кинетическая модель дает несколько более высокие значения концентрации ацетилена и несколько меньшие значения концентрации этилена при температуре выше 1800 К. С другой стороны, модель правильно предсказывает поведение молекул бензола во всем диапазоне температур, несколько занижая абсолютные значения концентрации. Бензол (A1) начинает расходоваться при температуре ∼1650 K (рис. 6), что согласуется, по мнению авторов работы [12], с наблюдаемым в их экспериментах образованием сажи.
Рис. 6.
Сравнение экспериментальных температурных зависимостей концентраций вторичных продуктов пиролиза метана (ацетилена C2H2, этилена C2H4, бензола (А1)) с результатами наших расчетов по единой кинетической модели для смеси 10% CH4 в Ar при давлении за фронтом отраженной ударной волны 30 бар.
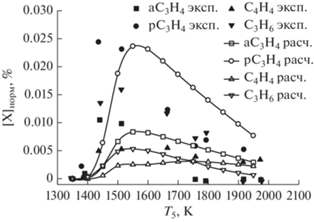
При повышенном давлении вторичными продуктами пиролиза метана (<0.03%) являются аллен (aC3H4), пропин (pC3H4), винилацетилен (C4H4) и пропен (C3H6), концентрации которых показаны на рис. 7. Концентрации этих продуктов достигают максимума уже при низких температурах, а их расход начинается при 1500 К. Рис. 7 демонстрирует удовлетворительное согласие экспериментальных температурных зависимостей концентраций вторичных продуктов пиролиза метана (аллена aC3H4, пропина pC3H4, винилацетилена C4H4, и пропена C3H6) и наших расчетных температурных зависимостей.
Рис. 7.
Сравнение экспериментальных температурных зависимостей концентраций вторичных продуктов пиролиза метана (аллена aC3H4, пропина pC3H4, винилацетилена C4H4 и пропилена C3H6) с результатами наших расчетов по единой кинетической модели для смеси 10% CH4 в Ar при давлении за фронтом отраженной ударной волны 30 бар.
В работе [12] были проведены измерения температуры в зависимости от времени при пиролизе смеси 10% CH4 + 1% CO + 20% He + 69% Ar за отраженными ударными волнами при температурах 1895–2573 K и давлении ∼2.2 бар. Общая погрешность экспериментальных измерений температуры в работе [12] оценивалась в 2%.
На рис. 8 показаны результаты сравнения экспериментально измеренных и рассчитанных зависимостей от времени температур за фронтом отраженной ударной волны при пиролизе смеси 10% CH4 + 1% CO + 20% He в Ar при различных начальных температурах и давлениях: T5 = 1895 K и Р5 = 2.46 бар, T5 = 2147 K и Р5 = 2.31 бар, T5 = = 2383 K и Р5 = 2.01 бар.
Рис. 8.
Сравнение экспериментально измеренной и рассчитанной температуры за фронтом отраженной ударной волны при пиролизе смеси 10% CH4 + 1% CO + 20% He в Ar при различных начальных температурах и давлениях: T5 = 1895 K и Р5 = 2.46 бар (а); T5 = 2147 K и Р5 = 2.31 бар (б); T5 = 2383 K и Р5 = 2.01 бар (в).
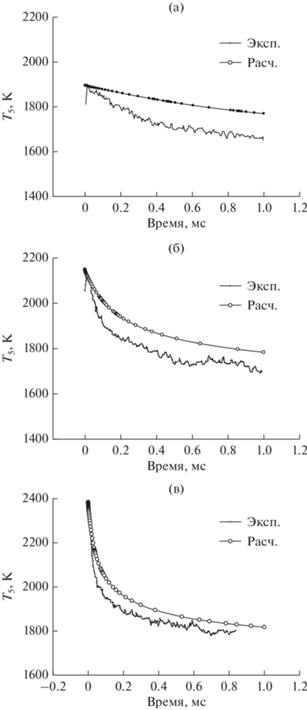
Из рис. 8 видно, что эндотермичность процесса пиролиза метана значительно снижает температуру за время наблюдения. Этот эффект усиливается с повышением температуры за фронтом отраженной ударной волны T5. При T5 = 1895 K наблюдается максимальное расхождение между результатами экспериментальных измерений и наших кинетических расчетов температуры T5, которое уменьшается с ростом начальной температуры T50 непосредственно за фронтом отраженной ударной волны.
Результаты наших кинетических расчетов также сравнивали с результатами экспериментов по пиролизу и окислению метана в отраженных ударных волнах в одноимпульсной ударной трубе, проведенных другими авторами в работе [16].
На рис. 9 представлены результаты сравнения экспериментальных температурных зависимостей концентраций исходного углеводорода (метана) и продукта пиролиза (ацетилена) с результатами наших расчетов по единой кинетической модели для смеси 10% CH4 в Ar при давлении за фронтом отраженной ударной волны 1.5 бар. Все профили были нормированы на исходную концентрацию метана в исследуемой смеси.
Рис. 9.
Сравнение экспериментальных температурных зависимостей концентраций исходного углеводорода (метана) и продукта пиролиза (ацетилена) с результатами наших расчетов по единой кинетической модели для смеси 10% CH4 в Ar при давлении за фронтом отраженной ударной волны 1.5 бар. Все профили были нормированы на исходную концентрацию метана в исследуемой смеси.
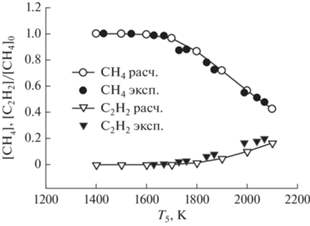
Как видно из рис. 9 наблюдается очень хорошее совпадение экспериментальных измерений, проведенных в работе [16], и наших кинетических расчетов для расходования исходных молекул метана и образования основного продукта пиролиза метана – молекул ацетилена.
На рис. 10 приведены результаты сравнения экспериментальных температурных зависимостей концентраций вторичных продуктов пиролиза метана (этилена C2H4, аллена aC3H4, пропина pC3H4, диацетилена C4H2 и этана C2H6) с результатами наших расчетов по единой кинетической модели для смеси 5% CH4 + 95% Ar для интервала давлений за фронтом отраженной ударной волны от 2.0 до 3.8 бар в одноимпульсной ударной трубе. В этих экспериментах каждой температуре соответствовало свое характерное время реакции: 1400 K (2120 мкс), 1500 K (1950 мкс), 1600 K (1770 мкс), 1700 K (1600 мкс), 1800 K (1420 мкс), 1900 K (1240 мкс), 2000 K (1070 мкс) и 2100 K (890 мкс). Все профили были нормированы на исходную концентрацию метана в исследуемой смеси.
Рис. 10.
Сравнение экспериментальных температурных зависимостей концентраций вторичных продуктов пиролиза метана (этилена C2H4, аллена aC3H4, пропина pC3H4, диацетилена C4H2 и этана C2H6) с результатами наших расчетов по единой кинетической модели для смеси 5% CH4 в 95% Ar для интервала давлении за фронтом отраженной ударной волны от 2.0 до 3.8 бар в одноимпульсной ударной трубе [16].
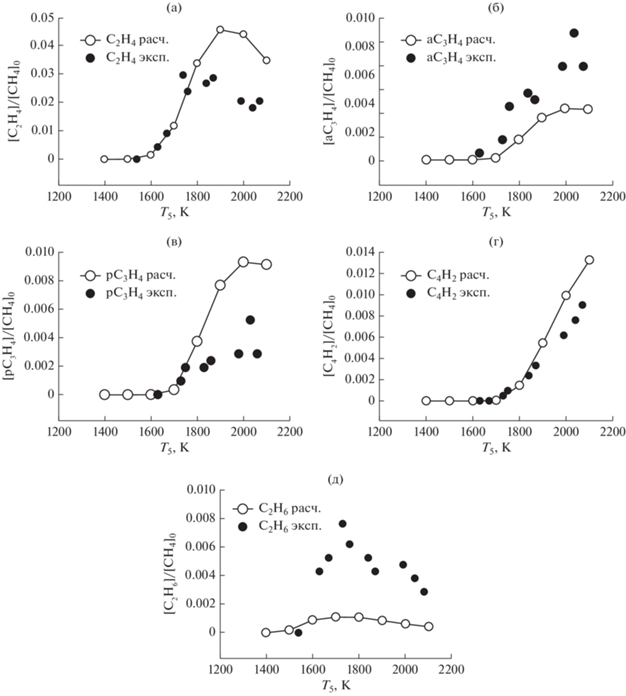
Рисунок 10 демонстрирует хорошее совпадение расчетных и экспериментально измеренных концентраций этилена C2H4, аллена aC3H4 и диацетилена C4H2 до температур порядка 1800 К. Несколько худшее совпадение наблюдается для молекул пропина pC3H4 и удовлетворительное – для молекул этана C2H6.
В работе [16] были также проведены эксперименты с богатыми смесями метана с кислородом (φ = 4 и 10). В этом случае отмечается протекание так называемого окислительного пиролиза метана, сопровождающегося образованием частиц сажи.
На рис. 11 представлены результаты сравнения экспериментальных температурных зависимостей концентраций метана и продуктов его окисления (C2H2, CO) с результатами наших расчетов по единой кинетической модели для богатой смеси 5% CH4 + 1% O2 + 94% Ar (φ = 10) для интервала давлений за фронтом отраженной ударной волны от 2.2 до 3.9 бар в одноимпульсной ударной трубе [16]. В этих экспериментах каждой температуре соответствовало свое характерное время реакции: 1500 K (1800 мкс), 1600 K (1650 мкс), 1700 K (1500 мкс), 1800 K (1350 мкс), 1900 K (1190 мкс), 2000 K (1040 мкс), 2100 K (890 мкс). Все профили были нормированы на исходную концентрацию метана в исследуемой смеси.
Рис. 11.
Сравнение экспериментальных температурных зависимостей концентраций метана и продуктов его окисления (ацетилена C2H2, окиси углерода CO) с результатами наших расчетов по единой кинетической модели для богатой смеси 5% CH4 + 1% O2 + 94% Ar (φ = 10) для интервала давлении за фронтом отраженной ударной волны от 2.2 до 3.9 бар в одноимпульсной ударной трубе [16].
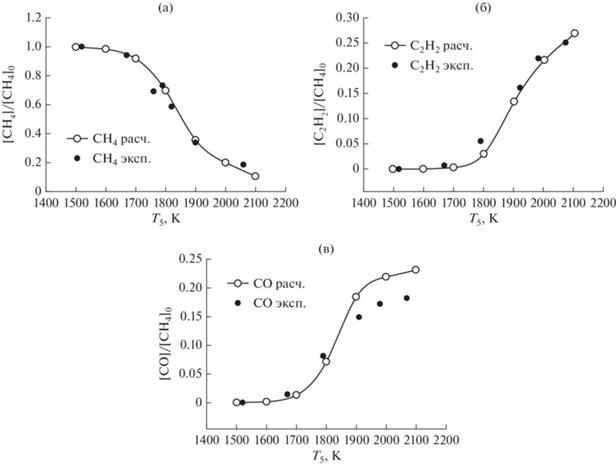
Наблюдается очень хорошее совпадение экспериментально измеренных и расчетных профилей концентраций метана (CH4), ацетилена (C2H2) и CO. Для столь богатой смеси образования молекул CO2 не отмечалось (рис. 11).
На рис. 12 приведены результаты сравнения экспериментальных температурных зависимостей концентраций метана и продуктов его окисления (C2H2, CO) с результатами наших расчетов по единой кинетической модели для богатой смеси 5% CH4 + 2.5% O2 + 92.5% Ar (φ = 4) для интервала давления за фронтом отраженной ударной волны от 2.3 до 3.6 бар в одноимпульсной ударной трубе [16]. В этих экспериментах каждой температуре соответствовало свое характерное время реакции: 1500 K (2010 мкс), 1600 K (1790 мкс), 1700 K (1560 мкс), 1800 K (1340 мкс), 1900 K (1120 мкс), 2000 K (900 мкс). Все профили были нормированы на исходную концентрацию метана в исследуемой смеси.
Рис. 12.
Сравнение экспериментальных температурных зависимостей концентраций метана и продуктов его окисления (ацетилена C2H2, окиси углерода CO) с результатами наших расчетов по единой кинетической модели для богатой смеси 5% CH4 + 2.5% O2 + + 92.5% Ar (φ = 4) для интервала давлении за фронтом отраженной ударной волны от 2.3 до 3.6 бар в одноимпульсной ударной трубе [16].
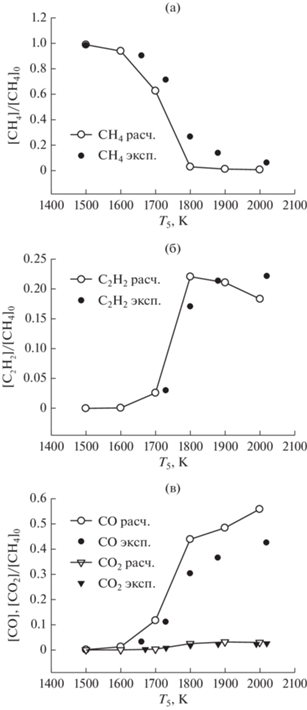
С увеличением концентрации кислорода в смеси (φ = 4) эксперименты и расчеты показывают отличное согласие для молекул метана (CH4), ацетилена (C2H2) и CO, при этом начинается образование молекул CO2 (рис. 12).
Изучение закономерностей процесса сажеобразования при пиролизе и окислении метана важно не только с теоретической, но и с практической точки зрения. Как известно, метан является основным компонентом природного газа, широко применяемого в промышленности и в быту. При горении метана выделяется наименьшее количество вредных веществ, загрязняющих окружающую среду как в газовой фазе, так и в виде гетерогенных частиц сажи, что связано с максимальным среди всех углеводородов отношением Н/С. Тем не менее, количественные измерения показывают, что даже в пламенах метана обнаруживаются весьма высокие концентрации полиароматических углеводородов, которые при определенных условиях образуют частицы сажи. Кроме того, неполное окисление метана в газовой фазе в настоящее время рассматривается как один из перспективных способов получения синтез-газа с заданным отношением H2/CO. Некаталитическое неполное окисление метана в газовой фазе происходит только при повышенных температурах, обеспечивающих максимальный выход полезного продукта (H2 и CO). В этих условиях нежелательный процесс образования частиц сажи весьма вероятен.
Процесс сажеобразования при пиролизе метана имеет весьма длинный период индукции (более 500 мкс), что связано с необходимостью прохождения целого ряда стадий роста углеводородных фрагментов от исходной молекулы метана до полиароматических фрагментов и зародышей частиц сажи. Наши эксперименты и расчеты показывают, что падение температуры за фронтом отраженной ударной волны для смесей метана с аргоном значительно выше, чем для смесей метана с аргоном с добавками кислорода: 200–300 и 50–100 K соответственно. Эта разница в изменении температуры объясняет, почему периоды индукции становятся короче, а максимум температурной зависимости выхода сажи смещается в сторону низких температур в присутствии добавок кислорода.
На рис. 13 представлены результаты сравнения экспериментально измеренных в работе [7] и рассчитанных по единой кинетической модели температурных зависимостей выхода частиц сажи при пиролизе (5% CH4 + 95% Ar и 10% CH4 + 90% Ar) и окислении метана (5% CH4 + 1.1% O2 + 93.9% Ar) в отраженных ударных волнах для интервала давлений P5 = 4.5–6.7 бар и времени реакции τреак = = 1.5 мс. Темные символы – наши экспериментальные измерения, светлые символы – наш детальный кинетический расчет, линии – нелинейная аппроксимация расчетных точек.
Рис. 13.
Сравнение экспериментально измеренных в [7] и рассчитанных по единой кинетической модели температурных зависимостей выхода частиц сажи при пиролизе (5% CH4 + 95% Ar и 10% CH4 + 90% Ar) (а) и окислении метана (5% CH4 + 1.1% O2 + 93.9% Ar) (б) в отраженных ударных волнах для интервала давлений P5 = 4.5–6.7 бар и времени реакции τреак = 1.5 мс. Темные символы – наши экспериментальные измерения; светлые символы – результаты нашего детального кинетического расчета; линии – нелинейная аппроксимация расчетных точек.
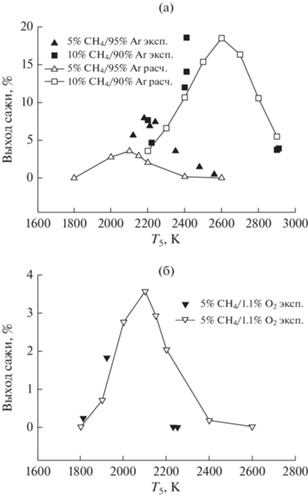
Как видно из рис. 13, единая кинетическая модель правильно описывает колоколообразную форму температурной зависимости выхода сажи, правильно воспроизводит концентрационную зависимость выхода сажи, удовлетворительно описывает положение максимума выхода сажи и абсолютные значения выхода сажи. Рисунок 13 демонстрирует, что при пиролизе метана с ростом концентрации метана в смеси максимум выхода сажи смещается в сторону высоких температур за счет все большего падения температуры. Добавки кислорода в смесь метана с аргоном существенно уменьшают падение температуры, и заметного сдвига выхода сажи в сторону высоких температур не наблюдается.
ЗАКЛЮЧЕНИЕ
В работе была предложена единая кинетическая модель для описания процесса пиролиза и окисления метана и образования конденсированной дисперсной фазы (частиц сажи).
Данная кинетическая модель была протестирована путем прямого сравнения результатов кинетических расчетов с результатами экспериментальных измерений в отраженных ударных волнах процессов пиролиза и окисления метана, образования первичных и вторичных продуктов, а также частиц сажи. В целом кинетическая модель продемонстрировала хорошее согласие результатов расчетов с результатами экспериментов разных авторов по измерению расходования молекул метана и образованию основных и вторичных продуктов пиролиза и окисления метана на различных ударных трубах.
Модель показала хорошее согласие результатов расчетов и собственных экспериментов по измерению процесса образования частиц сажи.
Список литературы
Abanades S., Kimura H., Otsuka H. // Fuel Process. Technol. 2014. V. 122. P. 153.
Rodat S., Abanades S. // Front. Energy Res. 2020. V. 8. P. 206.
Caliot C., Flamant G., Patrianakos G., Kostoglou M., Konstandopoulos A.G. // AIChE J. 2012. V. 58. P. 2545.
Rodat S., Abanades S., Flamant G. // Int. J. Chem. React. Eng. 2010. V. 8. Article A125 P. 1.
Flamant G. // J. Solar Energy Eng. 2011. V. 133. P. 0310011.
Dors M., Nowakowska H., Jasin’ski M., Mizeraczyk J. // Plasma Chem. Plasma Process. 2014. V. 34. P. 313.
Агафонов Г.Л., Билера И.В., Власов П.А., Жильцова И.В., Колбановский Ю.А., Смирнов В.Н., Тереза А.М. // Кинетика и катализ. 2016. Т. 57. № 5. С. 571.
Ступоченко Е.В., Лосев С.А., Осипов А.И. Релаксационные процессы в ударных волнах. М.: Наука, 1965. 328 с.
Агафонов Г.Л., Власов П.А., Смирнов В.Н. // Кинетика и катализ. 2011. Т. 52. № 3. С. 368.
Агафонов Г.Л., Билера И.В., Власов П.А., Колбановский Ю.А., Смирнов В.Н., Тереза А.М. // Кинетика и катализ. 2015. Т. 56. № 1. С. 15.
Haynes B.S., Wagner H.G. // Prog. Energy Combust. Sci. 1981. V. 7. № 4. P. 229.
Nativel D., Shu B., Herzler J., Fikri M., Schulz C. // Proceed. Combust. Inst. 2019. V. 37. P. 197.
Sela P., Shu B., Aghsaee M., Herzler J., Welz O., Fikri M., Schulz C. // Rev. Sci. Instrum. 2016. V. 87. P. 105103.
Cancino L.R., Fikri M., Oliveira A.A.M., Schulz C. // Energy Fuels. 2010. V. 24. P. 2830.
Fikri M., Makeich A., Rollmann G., Schulz C., Entel P. // J. Phys. Chem. A. 2008. V. 112. P. 6330.
Hidaka Y., Sato K., Henmi Y., Tanaka H., Inami K. // Combust. Flame. 1999. V. 118. P. 340.
Hidaka Y., Hattori K., Okuno T., Inami K., Abe T., Koike T. // Combust. Flame. 1996. V. 107. P. 401.
Hidaka Y., Shiba S., Takuma H., Suga M. // Int. J. Chem. Kinet. 1985. V. 17. P. 441.
Hidaka Y., Nakamura T., Miyauchi A., Shiraishi T., Kawano H. // Int. J. Chem. Kinet. 1989. V. 21. P. 643.
Hidaka Y., Kimura K., Hattori K., Okuno T. // Combust. Flame 1996. V. 106. P. 155.
Hidaka Y., Taniguchi T., Tanaka H., Kamesawa T., Inami K., Kawano H. // Combust. Flame. 1993. V. 92. P. 365.
Blanquart G., Pepiot-Desjardins P., Pitsch H. // Combust. Flame. 2009. V. 156. P. 588.
Wang H., You X., Joshi A.V., Davis S.G., Laskin A., Egolfopoulos F., Law C.K. USC Mech Version II, High Temperature Combustion Reaction Model of H2/CO/C1–C4 Compounds, available at <http://ignis.usc.edu/USC-Mech_II.htm, May 2007>
Richter H., Granata S., Green W.H., Howard J.B. // Proc. Combust. Inst. 2005. V. 30. P. 1397.
Skjøth-Rasmussen M.S., Glarborg P., Østberg M., Johannessen J.T., Livbjerg H., Jensen A.D., Christensen T.S. // Combust. Flame. 2004. V. 136. P. 91.
Correa C., Niemann H., Schramm B., Warnatz J. // Proc. Combust. Inst. 2000. V. 28. P. 1607.
Frenklach M., Warnatz J. // Combust. Sci. Technol. 1987. V. 51. P. 265.
Wang H., Dames E., Sirjean B., Sheen D.A., Tangko R., Violi A. 2010. A high-temperature chemical kinetic model of n-alkane (up to n-dodecane), cyclohexane, and methyl-, ethyl-, n-propyl and n-butyl-cyclohexane oxidation at high temperatures. JetSurF version 2.0. http://melchior.usc.edu/JetSurF/JetSurF2.0
Hansen N., Klippenstein S.J., Westmoreland P.R., Kasper T., Kohse-Höinghaus K., Wang J., Cool T.A. // Phys. Chem. Chem. Phys. 2008. V. 10. P. 366.
Vlasov P.A., Warnatz J. // Proc. Combust. Inst. 2002. V. 29. P. 2335.
Naydenova I., Nullmeier M., Warnatz J., Vlasov P.A. // Combust. Sci. Technol. 2004. V. 176. P. 1667.
Frenklach M., Clary D.W., Yuan T., Gardiner W.C., Jr., Stein S.E. // Combust. Sci. Technol. 1986. V. 50. P. 79.
Böhm H., Jander H. // Phys. Chem. Chem. Phys. 1999. V. 1. P. 3775.
Agafonov G.L., Mikhailov D.I., Smirnov V.N., Tereza A.M., Vlasov P.A., Zhiltsova I.V. // Combust. Sci. Technol. 2016. V. 188. P. 1815.
Vlasov P.A., Zhiltsova I.V., Smirnov V.N., Tereza A.M., Agafonov G.L., Mikhailov D.I. // Combust. Sci. Technol. 2018. V. 190. № 1. P. 57.
Metcalfe W.K., Burke S.M., Ahmed S.S., Curran H.J. // Intl. J. Chem. Kinet. 2013. V. 45. P. 638.
Baigmohammadi M., Patel V., Nagaraja S., Ramalingam A., Martinez S., Panigrahy S., Mohamed A., Somers K.P., Burke U., Heufer K.A., Pekalski A., Curran H.J. // Energy Fuels. 2020. V. 34. P. 8808.
Davidson D.F., Hanson R.K., Bowman C.T. // Int. J. Chem. Kinet. 1995. V. 27. P. 305.
Wang S., Davidson D.F., Hanson R.K. // J. Phys. Chem. A. 2016. V. 120. P. 5427.
Baulch D.L., Bowman C.T., Cobos C.J., Cox R.A., Just Th., Kerr J.A., Pilling M.J., Stocker D., Troe J., Tsang W. // J. Phys. Chem. 2005. V. 34. P. 757.
Simmie J.M. // Prog. Energy Combust. Sci. 2003. V. 29. P. 599.
Дополнительные материалы
- скачать ESM_1.txt
- Приложение 1.
- скачать ESM_2.txt
- Приложение 2.
Инструменты
Кинетика и катализ