

Кинетика и катализ, 2022, T. 63, № 2, стр. 234-246
Влияние добавок солей и фосфинов на состав активных комплексов палладия в реакции Мицороки–Хека с ангидридами ароматических кислот
Е. В. Ларина a, А. А. Курохтина a, Н. А. Лагода a, А. Ф. Шмидт a, *
a Иркутский государственный университет, химический факультет
Иркутск, Россия
* E-mail: aschmidt@chem.isu.ru
Поступила в редакцию 27.07.2021
После доработки 02.09.2021
Принята к публикации 27.09.2021
- EDN: XUMIGV
- DOI: 10.31857/S0453881122020058
Аннотация
В работе представлены результаты исследований закономерностей изменения состава комплексов палладия, ответственных за активацию субстратов в реакции Мицороки–Хека между ангидридами ароматических кислот и алкенами. Закономерности дифференциальной селективности реакции в условиях конкуренции пары ангидридов или алкенов указывают на каталитическую активность комплексов палладия, состав которых меняется в зависимости от соотношения концентраций третичного фосфина, галогенной соли щелочного металла, используемой в качестве промотирующей добавки, и загружаемого в систему палладиевого предшественника катализатора. Показано, что при достижении определенных пороговых значений отношения концентраций галогенид-ионов и третичного фосфина определяющий вклад в образование продуктов реакции, несмотря на присутствие в системе фосфина, вносят исключительно бесфосфиновые комплексы палладия, содержащие в своей координационной сфере анионы галогенной соли. При этом при низких концентрациях соли или ее отсутствии в состав активных комплексов могут входить молекулы третичного фосфина (в фосфинсодержащих каталитических системах), растворителя, а также анионы кислотного остатка ароматической кислоты, формирующиеся в результате конверсии исходного ангидрида. В “неконкурентных” условиях реакции Мицороки–Хека установлено, что природа катиона и аниона добавляемой в систему соли оказывает ключевое влияние на дифференциальную селективность формирования классического “хековского” продукта арилирования алкена и карбонилсодержащего продукта (халкона), образующегося в результате реализации параллельного маршрута реакции. Продемонстрировано повышение дифференциальной селективности и выхода карбонилсодержащего продукта реакции при использовании добавок бромидных солей, указывающее на принципиальную возможность управления составом активных палладиевых комплексов, ответственных за селективность образования различных типов продуктов реакции Мицороки–Хека с ароматическими ангидридами.
ВВЕДЕНИЕ
Катализируемая соединениями палладия реакция арилирования алкенов различными арилирующими реагентами, в первую очередь, арилгалогенидами, известная как реакция Мицороки–Хека [1], в течение последних десятилетий стабильно является одной из наиболее интенсивно изучаемых реакций в смежных областях катализа и тонкого органического синтеза. Причина высокого исследовательского интереса к этому процессу – его широчайшие синтетические возможности в совокупности с высокой селективностью по целевым продуктам. В результате реакция Мицороки–Хека реализована в качестве одного из этапов многочисленных малотоннажных производств различных продуктов современной химической промышленности, в первую очередь, фармацевтических, агрохимических и полимерных материалов с заданными свойствами [2, 3]. При этом растущие экологические требования, стимулирующие переход к “зеленому” химическому производству, делают перспективным и востребованным для любого химического процесса разработку каталитических и субстратных систем, применение которых минимизирует объем утилизируемых и/или перерабатываемых побочных продуктов. Использование в качестве субстратов реакции Мицороки–Хека ангидридов ароматических кислот, впервые предложенное в 1998 г. [4], является предпочтительной альтернативой традиционно применяемым арилгалогенидам. Это обусловлено отсутствием в составе каталитической системы стехиометрического количества основания для нейтрализации галогеноводородной кислоты, формирующейся в качестве побочного продукта в реакции с галогенсодержащими реагентами, а также тем, что образующаяся в ходе реакции ароматическая карбоновая кислота может быть вновь превращена в ангидрид с последующим вовлечением в реакцию арилирования (схема 1 ). Кроме того, использование ангидридов карбоновых кислот позволяет получать наряду с классическими “хековскими” продуктами (дизамещенные алкены типа 1) карбонилсодержащие непредельные соединения типа 2 (схема 1 ) [5, 6], синтез которых с помощью арилгалогенидов в литературе описан только в условиях избыточного давления монооксида углерода в реакционной системе [7, 8].
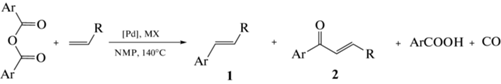
Схема 1 . Реакция Мицороки–Хека с использованием ангидридов ароматических кислот в качестве арилирующих реагентов.
Уже в первых работах, демонстрирующих возможность вовлечения ароматических ангидридов в реакцию Мицороки–Хека, было показано, что добавки неорганических галогенных солей приводят к повышению активности простейших “безлигандных” каталитических систем, т.е. систем, не содержащих добавок фосфиновых, аминовых, карбеновых или иных сильных органических лигандов [4, 5]. При этом достоверные данные о роли добавляемых солей в таких системах в литературе долгое время отсутствовали. Ранее на примере реакции Мицороки–Хека с ароматическими ангидридами в условиях реального катализа при использовании фосфинсодержащих каталитических систем было продемонстрировано образование в ходе процесса комплексов палладия, содержащих в своей координационной сфере одновременно галогенид-ионы и молекулы трифенилфосфина [9]. Однако кинетические данные, в частности, закономерности изменения каталитической активности в ходе реакции, свидетельствовали о том, что такие комплексы палладия не являются активными и находятся за пределами основного каталитического цикла образования продукта реакции – дизамещенного алкена. Позже с помощью исследований закономерностей дифференциальной селективности было показано [10], что в условиях конкуренции пары ароматических ангидридов или пары алкенов в реакции Мицороки–Хека состав активных комплексов палладия меняется при варьировании катиона и аниона галогенных солей, добавляемых в бесфосфиновую каталитическую систему. Такой результат объяснялся анионной природой галогенсодержащих каталитически активных соединений Pd(0) и Pd(II), образующих тесные ионные пары с присутствующими в системе катионами. При этом добавление в реакционную систему триарилфосфинов в концентрациях, не превышающих типичных для катализа реакции Мицороки–Хека 2 эквивалентов в расчете на загружаемый в систему палладий [11], не сказывалось на величине дифференциальной селективности, позволяя сделать вывод о том, что истинные каталитически активные соединения и в этом случае являются анионными комплексами палладия, не содержащими в своем составе фосфиновых лигандов [12]. Однако необходимо учитывать, что сформулированные выводы об определяющей роли в катализе анионных бесфосфиновых комплексов Pd(0) и Pd(II) в [10, 12] основаны на результатах, полученных в условиях присутствия избыточных количеств галогенных солей по отношению к фосфину.
В настоящей работе описаны результаты экспериментов по установлению состава каталитически активных соединений реакции Мицороки–Хека с ангидридами ароматических кислот для фосфинсодержащих и бесфосфиновых каталитических систем в условиях близких мольных соотношений добавок фосфинов, галогенных солей и палладиевого предшественника катализатора. Необходимо отметить, что использование в качестве субстратов ангидридов ароматических кислот в сравнении с реакцией с применением арилгалогенидов несколько упрощает задачу определения роли присутствующих в системе соединений, способных к координации к палладию, как потенциальных лигандов. Это обусловлено отсутствием в реакции Мицороки–Хека с ароматическими ангидридами основания – необходимого компонента реакции с арилгалогенидами, анионы которого способны входить в координационную сферу активных комплексов палладия, что было неоднократно подтверждено экспериментально [13, 14]. С учетом этого использование в качестве модели для изучения состава активных комплексов палладия более простой реакционной системы с ароматическими ангидридами является предпочтительным. В качестве основного экспериментально измеряемого параметра нами была выбрана величина дифференциальной селективности реакции. Из-за протекания в каталитических системах реакции Мицороки–Хека множества сопряженных последовательно-параллельных процессов внутри и за пределами каталитического цикла с участием различных сосуществующих форм катализатора [15], концентрация форм катализатора нестационарная. В связи с вышесказанным применение методов установления природы истинного катализатора, основанных на измерении каталитической активности, которая непосредственно зависит от практически всегда неизвестной концентрации активного катализатора, не всегда способно давать однозначные результаты. Для получения надежных данных нами ранее был предложен и апробирован на ряде реакций кросс-сочетания, в том числе реакции Мицороки–Хека, комплекс подходов на основании анализа закономерностей дифференциальной селективности (ДС), величина которой не зависит от концентрации активного катализатора и определяется исключительно его природой [16, 17]. При этом использование для оценки ДС пары близких по химическим свойствам конкурирующих субстратов является предпочтительным, поскольку позволяет уверенно предполагать одинаковые механизмы образования продуктов в конкурирующих реакциях, что является необходимым условием независимости величины ДС от количества катализатора. Для оценки величины ДС нами было предложено применение так называемых фазовых траекторий конкурирующих реакций, представляющих собой зависимости выходов продуктов конкурирующих реакций друг от друга [18]. Тангенс угла наклона касательной к любой точке фазовой траектории равен отношению скоростей накопления продуктов/групп продуктов конкурирующих реакций, однозначно характеризующему величину ДС. Таким образом, построение фазовых траекторий при варьировании условий проведения реакции позволяет проводить качественный и количественный анализ закономерностей ДС. Важным преимуществом оценки ДС по фазовым траекториям является то, что для его применения достаточно интегральных экспериментальных данных о зависимостях концентраций продуктов реакции от времени, которые могут быть получены с использованием стандартного оснащения химической лаборатории без дорогостоящего специального оборудования, необходимого для определения дифференциальных кинетических зависимостей.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Пробы реакционной смеси анализировали на газожидкостном хроматографе Кристалл 5000 (“Хроматэк”, Россия, ДИП, колонка HP-5 15 м) и хромато-масс-спектрометре GCMS QP-2010 Ultra (“Shimadzu”, Япония) с ионизацией электронным ударом (энергия ионизации − 70 эВ, колонка GsBP-5MS размером 0.25 мкм × 0.25 мм × 30 м, газ-носитель − гелий) с программированным нагревом от 100 до 250°С. Полученные масс-спектры сравнивали с библиотечными (библиотеки сравнения Wiley, NIST, NIST05). Количественный состав проб вычисляли методом внутреннего стандарта (внутренний стандарт − нафталин) с применением калибровки по аутентичным образцам. Математическую обработку кинетических данных и построение полиномиальных аппроксимирующих зависимостей (фазовых траекторий) осуществляли с помощью средств программы “Microsoft Excel 2007” [19].
Каталитические эксперименты
Реакцию Мицороки−Хека в условиях конкуренции пары ангидридов ароматических кислот с использованием фосфинсодержащей каталитической системы проводили, смешивая при комнатной температуре в 5 мл NMP стирол (5 ммоль, 1 М), хлорид лития (0.08−0.64 ммоля, 1−8 экв в расчете на загружаемый в систему палладий, если добавляли) и нафталин (1 ммоль, 0.2 М) в качестве внутреннего стандарта для хроматографии. Полученный раствор продували аргоном и помещали в предварительно отвакуумированный и заполненный аргоном круглодонный стеклянный реактор, снабженный резиновой мембраной и магнитной мешалкой, содержащий ангидриды бензойной и 4-метоксибензойной кислот (по 2.5 ммоля каждого, 0.5 М), третичный фосфин (0.08−0.32 ммоля, 1−4 экв в расчете на загружаемый в систему палладий) и PdCl2 (0.08 ммоля, 16 мМ) в качестве предшественника катализатора. Реакцию начинали, помещая реактор в предварительно нагретую до 140°С масляную баню при перемешивании (480 об/мин). Пробы реакционной смеси для хроматографического анализа периодически отбирали из реактора с помощью шприца с металлической иглой. Каждый эксперимент проводили 3 раза для проверки воспроизводимости.
Реакцию Мицороки−Хека в условиях конкуренции пары ангидридов ароматических кислот с использованием бесфосфиновой каталитической системы осуществляли без добавления третичного фосфина аналогично процедуре, описанной выше, включая процедуры предварительного вакуумирования и заполнения реактора аргоном. Каждый эксперимент выполняли 3 раза для проверки воспроизводимости.
Реакцию Мицороки−Хека в условиях конкуренции пары алкенов в присутствии фосфинсодержащей каталитической системы проводили, смешивая при комнатной температуре в 5 мл NMP стирол и н-бутилакрилат (по 2.5 ммоля каждого, 0.5 М), хлорид лития (0.08−0.64 ммоля, 1−8 экв в расчете на загружаемый в систему палладий, если добавляли) и нафталин (1 ммоль, 0.2 М) в качестве внутреннего стандарта для хроматографии. Полученный раствор продували аргоном и помещали в предварительно отвакуумированный и заполненный аргоном круглодонный стеклянный реактор, снабженный резиновой мембраной и магнитной мешалкой, содержащий ангидрид бензойной кислоты (5 ммоль, 1 М), третичный фосфин (0.08−0.16 ммоля, 1−2 экв в расчете на загружаемый в систему палладий) и PdCl2 (0.08 ммоля, 16 мМ) в качестве предшественника катализатора. Реакцию начинали, помещая реактор в предварительно нагретую до 140°С масляную баню при перемешивании (480 об/мин). Пробы реакционной смеси для хроматографического анализа периодически отбирали из реактора с помощью шприца с металлической иглой. Каждый эксперимент проводили 3 раза для проверки воспроизводимости.
Реакцию Мицороки−Хека в условиях конкуренции пары алкенов с использованием бесфосфиновой каталитической системы осуществляли без добавления третичного фосфина аналогично процедуре, описанной выше, включая процедуры предварительного вакуумирования и заполнения реактора аргоном. Каждый эксперимент выполняли 3 раза для проверки воспроизводимости.
Реакцию Мицороки−Хека в “неконкурентных” условиях в присутствии бесфосфиновой каталитической системы проводили, смешивая при комнатной температуре в 5 мл NMP стирол (5 ммоль, 1 М), галогенную соль (0.32−3.2 ммоля, 4−40 экв в расчете на загружаемый в систему палладий) и нафталин (1 ммоль, 0.2 М) в качестве внутреннего стандарта для хроматографии. Полученный раствор помещали в круглодонный стеклянный реактор, снабженный резиновой мембраной и магнитной мешалкой, содержащий ангидрид бензойной кислоты (5 ммоль, 1 М) и PdCl2 (0.08 ммоля, 16 мМ) в качестве предшественника катализатора. Реакцию начинали, помещая реактор в предварительно нагретую до 140°С масляную баню при перемешивании (480 об/мин). Пробы реакционной смеси для хроматографического анализа периодически отбирали из реактора с помощью шприца с металлической иглой. Каждый эксперимент проводили 3 раза для проверки воспроизводимости.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Для установления влияния добавок третичных фосфинов и солей на состав активных комплексов палладия нами были выполнены исследования закономерностей ДС реакции Мицороки−Хека в условиях конкуренции пары ароматических ангидридов или пары алкенов. Необходимо отметить, что одним из ключевых преимуществ использования ДС как инструмента для установления тонких деталей механизмов сложных химических процессов является то, что ее значение, в отличие от каталитической активности, определяется относительно небольшим набором элементарных стадий. В предельном случае практически полной необратимости стадии, в которой осуществляется конкуренция субстратов, – только самой этой необратимой стадией [17]. Поэтому при выборе типа пары конкурирующих субстратов появляется уникальная возможность анализа именно тех элементарных стадий, в которых участвуют конкурирующие субстраты, в том числе, удается получить информацию о том, какие факторы влияют на состав активных интермедиатов каталитического цикла, взаимодействующих с конкурирующими субстратами. Таким образом, анализ закономерностей ДС по конкурирующим ангидридам приводит к получению данных о составе каталитически активных соединений, участвующих в стадии активации ангидридов. Поскольку активация ароматических ангидридов, согласно общепринятым представлениям [1, 20, 21], осуществляется в стадии их окислительного присоединения к комплексам нольвалентного палладия (схема 2 , стадия А), такими соединениями должны быть комплексы Pd(0). В случае использования пары конкурирующих алкенов зависимости ДС, построенные по концентрациям классических “хековских” продуктов, будут характеризовать природу соединений Pd(II), активирующих алкены, согласно общепринятой схеме механизма каталитического цикла, в стадиях координации и внедрения алкена (схема 2 , стадия Б), либо иной альтернативной последовательности стадий, тем не менее также начинающейся со стадии координации [22]. Здесь важно подчеркнуть, что вывод о неизменности природы активного катализатора, участвующего в стадиях с конкурирующими субстратами, в случае совпадения ДС при варьировании условий проведения процесса (либо, напротив, ее изменения при изменении ДС) не зависит от принимаемой гипотезы этих стадий в рамках той или иной альтернативной модели механизма реакции. Единственным необходимым условием для корректного применения ДС в целях получения информации о природе активного катализатора является выполнение предположения об одинаковых механизмах превращений конкурирующих субстратов, которое, в случае использования пары ароматических ангидридов, отличающихся заместителем в пара-положении бензольного кольца, или пары алкенов, отличающихся заместителем при двойной связи, не вызывает сомнений.
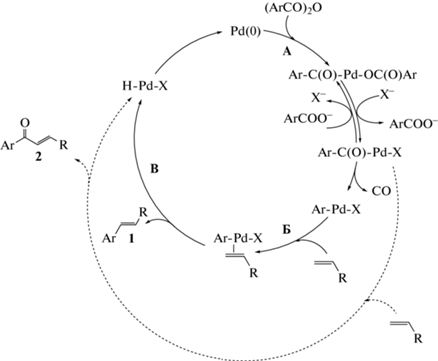
Схема 2 . Общепринятый механизм каталитических циклов образования продуктов в реакции Мицороки–Хека с использованием ангидридов ароматических кислот (лиганды при палладии опущены).
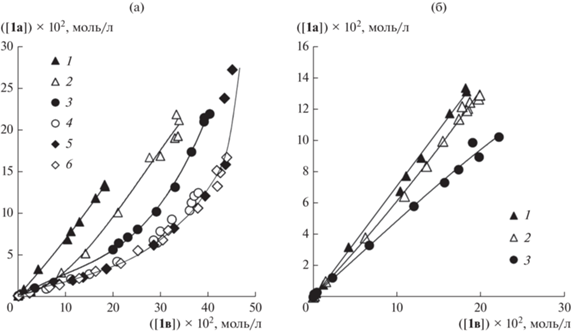
Поскольку при проведении реакции Мицороки–Хека с ангидридами ароматических кислот в качестве предшественника катализатора применялся дихлорид палладия (PdCl2), даже в условиях отсутствия в системе добавок хлоридных солей в реакционной смеси всегда присутствовало, как минимум, два эквивалента хлорид-ионов в расчете на загружаемый в реактор палладий. Также очевидно, что в используемой нами реакционной среде (полярный апротонный растворитель NMP, содержание воды в котором составляет около 0.1%) в реакционной системе всегда имеется некоторое количество анионов кислотных остатков ароматической кислоты, ангидрид которой служит в качестве арилирующего реагента. Такие анионы тоже способны координироваться к палладию, в том числе отвечающему за каталитическую активность. Поэтому для проверки возможного изменения состава комплексов палладия, активных в стадии окислительного присоединения, при изменении концентраций галогенной соли, добавки третичного фосфина, а также загружаемого в систему палладиевого предшественника катализатора нами было проведено сравнение величин ДС реакции Мицороки–Хека в условиях конкуренции бензойного и 4-метоксибензойного ангидридов (схема 3 ) при использовании каталитической системы на основе дихлорида палладия без добавления галогенных солей и при последовательном увеличении количества добавляемой соли (хлорида лития, как наиболее сильно повышающей каталитическую активность в исследуемой системе [5]). При этом для оценки величины ДС фазовые траектории были построены в координатах суммарных концентраций продуктов, образующихся из каждого конкурирующего ангидрида. Как следует из полученных данных (рис. 1а), ДС менялась, монотонно возрастая в направлении преимущественного образования продуктов превращения бензойного ангидрида, при изменении количества хлорид-ионов в небольшом интервале путем добавления соли (3–8.25 экв на загружаемый в систему палладий; здесь и далее количество эквивалентов хлорид-ионов указано с учетом 2 атомов хлора в молекуле предшественника катализатора). При увеличении количества хлорид-ионов с 8.25 экв до 10.0 в расчете на загружаемый в систему палладий изменения фазовой траектории не фиксировались (рис. 1а). Полученные данные позволяют предположить, что при достижении соотношения концентраций Cl–/Pd = = 8.25 комплексы палладия, содержащие в своей координационной сфере исключительно хлорид-ионы, вносят определяющий вклад в катализ. С учетом формально нулевой степени окисления палладия, участвующего в стадии окислительного присоединения к молекуле ароматического ангидрида (схема 2 , стадия А), и заряда аниона галогена, такие комплексы должны иметь анионную природу, что согласуется с обнаруженным ранее влиянием некоординирующегося катиона галогенной соли на ДС реакции Мицороки–Хека по конкурирующим ароматическим ангидридам [10]. При этом отличающиеся фазовые траектории в экспериментах с более низкими концентрациями галогенид-ионов позволяют предположить участие в катализе смешанно-лигандных комплексов палладия, содержащих в своей координационной сфере помимо анионов хлора, образующихся из молекулы предшественника катализатора и добавки хлорида лития, анионы кислотного остатка ароматической кислоты и/или молекулы растворителя, либо одновременное протекание катализа на нескольких типах активных комплексов, содержащих эти лиганды. При увеличении концентрации хлорид-ионов до соотношения Cl–/Pd = 8.25 более слабые лиганды полностью вытесняются из координационной сферы каталитически активных комплексов, приводя к формированию активных галогенидных анионных комплексов Pd(0), состав которых не изменяется при дальнейшем повышении концентрации добавляемой галогенной соли.
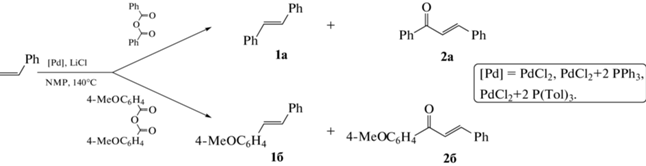
Схема 3 . Реакция Мицороки–Хека в условиях конкуренции пары ароматических ангидридов.
Рис. 1.
Фазовые траектории конкурентной реакции Мицороки–Хека с ангидридами бензойной и 4-метоксибензойной кислот: а – при варьировании количества добавки LiCl до соотношений Cl–/Pd равных: 2 (1), 2.5 (2), 3 (3), 8.25 (4), 10 (5); б – при варьировании количества добавки третичного фосфина в отсутствие добавки LiCl до соотношений PAr3/Pd = 0 (1), PPh3/Pd = 1 (2), PPh3/Pd = 2 (3), PPh3/Pd = 4 (4), P(Tol)3/Pd = 2 (5).
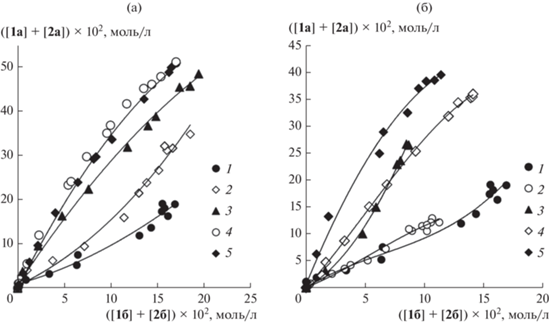
Ранее было показано, что в присутствии 8.25 экв хлорид-ионов по отношению к палладию добавки 2 экв третичных фосфинов, несмотря на уменьшение активности формирующейся каталитической системы, не приводили к изменению ДС, а, следовательно, и природы каталитически активных комплексов, указывая на отсутствие фосфинового лиганда в их координационной сфере [12]. Тем не менее, представленные на рис. 1а данные об изменении состава активных комплексов палладия при значительном снижении концентрации галогенной соли позволяют предположить, что в такой ситуации фосфиновые лиганды при их добавлении в реакционную систему смогут войти в координационную сферу каталитически активных соединений Pd(0), вытесняя слабокоординирующиеся анионы кислотного остатка ароматической кислоты и/или молекулы растворителя. Действительно, введение трифенилфосфина в каталитическую систему, не содержащую добавок галогенной соли, приводило к тому, что вид фазовых траекторий менялся (рис. 1б). При этом характер их изменения при повышении концентрации трифенилфосфина (постепенное увеличение ДС по продуктам превращения бензойного ангидрида) также позволяет предположить участие в селективностьопределяющей стадии смешанно-лигандных комплексов палладия, содержащих в своей координационной сфере молекулы третичного фосфина, растворителя, а также хлорид-анионы, образующиеся в данном случае из молекулы предшественника катализатора, и анионы кислотного остатка ароматической кислоты, возникающие при конверсии исходного ароматического ангидрида. Кроме того, аналогичные закономерности могут наблюдаться и при одновременном протекании катализа на нескольких типах активных комплексов палладия, включающих различные присутствующие в системе лиганды. Замена PPh3 на P(Tol)3 также приводила к изменению вида фазовой траектории (рис. 1б), подтверждая вывод об участии комплексов, содержащих наряду с имеющимися в системе анионами хлора и ароматической кислоты триарилфосфиновые лиганды, в активации ароматических ангидридов в реакции Мицороки–Хека в условиях отсутствия в системе добавок галогенных солей.
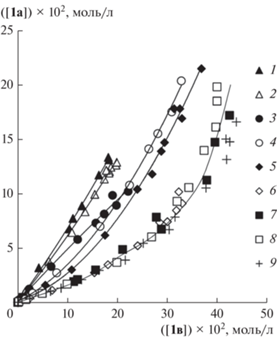
Совместный анализ полученных ранее [10, 12], а также представленных на рис. 1 результатов позволяет предположить конкурентное образование фосфиновых и галогенидных каталитически активных комплексов Pd(0), участвующих в соответствии с принятым механизмом в стадии окислительного присоединения ангидрида, вклад которых в катализ определяется соотношением количеств применяемых в качестве добавок галогенных солей и триарилфосфина. В серии дополнительных экспериментов, направленных на установление состава формирующихся активных комплексов в условиях использования сопоставимых мольных количеств галогенид-ионов, триарилфосфина и палладиевого предшественника катализатора, была установлена чувствительность ДС к варьированию соотношений Cl–/PPh3/Pd (рис. 2). При увеличении отношения между количествами как галогенной соли, так и фосфина к загружаемому в систему палладию вид фазовой траектории постепенно менялся от эксперимента, проводимого без добавления соли и фосфина, достигая фазовой траектории, описывающей ДС в опыте в присутствии 8.25 экв хлорид-ионов по отношению к палладию в бесфосфиновой каталитической системе. При этом в эксперименте с использованием 2 экв PPh3 по отношению к PdCl2 наличия 4 экв хлорид-ионов по отношению к палладию оказывалось достаточным для полного совпадения фазовой траектории с траекторией в опыте без добавок фосфина (рис. 2). Это позволяет уверенно утверждать протекание катализа на бесфосфиновых комплексах палладия, в том числе и в присутствии добавок фосфина, поскольку одинаковая дифференциальная селективность комплексов с фосфинами и без них – крайне маловероятное событие. Таким образом, при достижении соотношения Cl–/Pd = 4/1 в условиях реакции Мицороки–Хека при использовании как бесфосфиновых, так и фосфинсодержащих (как минимум, при соотношениях PPh3/Pd ≤ 4) каталитических систем определяющий вклад в активацию ароматических ангидридов вносят анионные комплексы палладия, в координационной сфере которых отсутствуют фосфины и присутствуют только хлорид-ионы. Следует подчеркнуть, что полученные результаты ни в коей мере не исключают существование в каталитической системе фосфиновых комплексов палладия. Более того, нет сомнений, что доля фосфинсодержащих комплексов в силу их большей стабильности значительно превышает долю комплексов, не включающих фосфиновые лиганды. Однако, учитывая, что введение в каталитическую систему фосфинов при соотношениях PPh3/Pd ≤ 4 не сказывается на ДС, а следовательно, и на природе каталитически активных соединений, можно обоснованно предположить, что истинно каталитически активные соединения не содержат фосфиновых лигандов, хоть и находятся в относительно низкой в сравнении с фосфиновыми комплексами концентрации. Их формирование обусловлено протеканием самопроизвольных быстрых процессов диссоциации–координации различных лигандов за пределами основного каталитического цикла.
Рис. 2.
Фазовые траектории конкурентной реакции Мицороки–Хека с ангидридами бензойной и 4-метоксибензойной кислот при различных соотношениях добавляемых LiCl и PPh3: Сl–/PPh3/Pd = 2/0/1 (1), Сl–/PPh3/Pd = 2/1/1 (2), Сl–/PPh3/Pd = 2/2/1 (3), Сl–/ PPh3/Pd = 2/4/1 (4), Сl–/PPh3/Pd = 2.5/0/1 (5), Сl–/ PPh3/Pd = 2.5/2/1 (6), Сl–/PPh3/Pd = 3/0/1 (7), Сl–/ PPh3/Pd = 3/2/1 (8), Сl–/PPh3/Pd = 8.25/0/1 (9), Сl–/ PPh3/Pd = 8.25/2/1 (10), Сl–/PPh3/Pd = 10/0/1 (11), Сl–/PPh3/Pd = 4/4/1 (12).
Для установления закономерностей изменения состава активных комплексов Pd(II), активирующих алкены в реакции Мицороки–Хека (схема 2 , стадия Б), нами были проведены аналогичные эксперименты по исследованию закономерностей ДС по конкурирующим алкенам (схема 4 , карбонилсодержащие продукты не представлены на схеме реакции, поскольку в реакциях с участием н-бутилакрилата их выходы были незначимыми, не превышая 4% от теоретически возможного). В бесфосфиновой каталитической системе ДС, как и в случае конкуренции ароматических ангидридов, оказывалась чувствительной к введению добавки LiCl и ее концентрации (рис. 3а). При этом вид фазовых траекторий при постепенном увеличении количества хлорид-ионов до 5 экв по отношению к PdCl2 также менялся постепенно, и дальнейшее повышение концентрации хлорид-ионов не приводило к изменению фазовой траектории. Соответственно, можно уверенно предполагать, что при достижении соотношения Cl–/Pd = 5/1 активация конкурирующих алкенов осуществляется комплексами палладия, содержащими в своей координационной сфере исключительно хлорид-анионы. Следует отметить, что, в отличие от комплексов Pd(0), активирующих ароматические ангидриды в каталитическом цикле реакции Мицороки–Хека, комплексы Pd(II) с координированными анионами хлора формально могут не быть анионными. Тем не менее, учитывая данные о зависимости дифференциальной селективности реакции Мицороки–Хека по конкурирующим алкенам от природы катиона присутствующей в системе галогенной соли [10], полученные при более высоких концентрациях последней, можно предположить анионную природу комплексов и при более низких концентрациях соли с учетом неизменности фазовой траектории при дальнейшем увеличении ее концентрации (рис. 3а).
Схема 4 . Реакция Мицороки–Хека в условиях конкуренции пары алкенов.
Рис. 3.
Фазовые траектории конкурентной реакции Мицороки–Хека со стиролом и н-бутилакрилатом: а – при варьировании количества добавки LiCl до соотношений Cl–/Pd равных: 2 (1), 3 (2), 4 (3), 5 (4), 8.25 (5), 10 (6); б – при варьировании количества добавки PPh3 в отсутствие добавки LiCl до соотношений PPh3/Pd равных 0 (1), 1 (2), 2 (3).
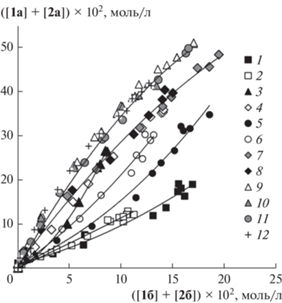
Поскольку проведенные в условиях конкуренции ароматических ангидридов эксперименты показали, что в отсутствие в каталитической системе добавок галогенных солей третичный фосфин может входить в состав каталитически активных комплексов палладия, возможное содержание фосфина в активных комплексах Pd(II) в реакционных системах без добавления галогенид-ионов также требовало проверки. Действительно, фазовые траектории по конкурирующим алкенам менялись при введении 1 и 2 экв трифенилфосфина в расчете на загружаемый в систему палладий (рис. 3б), указывая на участие в активации конкурирующих алкенов комплексов палладия, в составе которых находятся молекулы трифенилфосфина. При этом, с учетом постепенного изменения наклона фазовой траектории при изменении концентрации трифенилфосфина, можно предположить участие в катализе в этом случае смешанных комплексов, содержащих как трифенилфосфиновые лиганды, так и анионы хлора и/или кислотного остатка бензойной кислоты, а также молекулы растворителя, либо одновременное протекание катализа на нескольких типах активных комплексов, включающих эти лиганды. Для проверки выдвигаемой гипотезы ДС по конкурирующим алкенам оценивали в серии экспериментов с варьированием концентрации добавляемой галогенной соли в каталитическую систему в присутствии 2 экв PPh3. Увеличение количества LiCl вновь приводило к постепенному изменению фазовой траектории, которая при достижении соотношения Cl–/Pd = 5/1 переставала меняться, совпадая с фазовой траекторией бесфосфиновой системы (рис. 4). Поскольку одинаковая дифференциальная селективность фосфинсодержащих и бесфосфиновых активных комплексов палладия является крайне маловероятной, такой результат, на наш взгляд, позволяет сделать вывод о том, что безотносительно наличия в системе добавок третичного фосфина пятикратного избытка хлорид-ионов по отношению к палладию оказывается достаточным для того, чтобы за каталитическую активность отвечали исключительно анионные хлоридные комплексы одинаковой природы.
Рис. 4.
Фазовые траектории конкурентной реакции Мицороки–Хека со стиролом и н-бутилакрилатом при различных соотношениях добавляемых LiCl и PPh3: Сl–/PPh3/Pd = 2/0/1 (1), Сl–/PPh3/Pd = 2/1/1 (2), Сl–/PPh3/Pd = 2/2/1 (3), Сl–/PPh3/Pd = 3/2/1 (4), Сl–/PPh3/Pd = 4/2/1 (5), Сl–/PPh3/Pd = 5/2/1 (6), Сl–/ PPh3/Pd = 8.25/2/1 (7), Сl–/PPh3/Pd = 10/2/1 (8), Сl–/ PPh3/Pd = 10/0/1 (9).
Таким образом, результаты исследования закономерностей ДС в условиях конкуренции пары ароматических ангидридов или пары алкенов в присутствии добавок галогенных солей позволили сделать вывод о формировании и определяющем вкладе в катализ бесфосфиновых анионных комплексов палладия, содержащих в своей координационной сфере только анионы галогена, даже при наличии в реакционной системе третичного фосфина в количестве 2 экв по отношению к загружаемому в систему палладию. Полученные в настоящей работе данные об отсутствии фосфинового лиганда в составе активных комплексов палладия, вносящих определяющий вклад в конверсию субстратов, в совокупности с имеющимися в литературе свидетельствами о катализе реакции Мицороки–Хека бесфосфиновыми соединениями палладия [10, 23, 24] указывают на принципиальную возможность создания эффективных бесфосфиновых систем для осуществления этой реакции. Очевидно, фосфиновые лиганды принимают участие только в процессах, протекающих за пределами основного каталитического цикла образования целевых продуктов реакции, подтверждением чему являются значительные колебания каталитической активности, происходящие при введении в реакцию добавок фосфинов, свидетельствующие об изменениях количества активного катализатора, а также детектирование фосфинсодержащих комплексов палладия посредством ЯМР-мониторинга реакции в реальных каталитических условиях [9]. Соответственно, с точки зрения управления процессами за пределами основного каталитического цикла (например, повышения стабильности комплексов палладия в растворенной фазе) применение фосфинсодержащих каталитических систем может быть оправдано, поскольку интенсивное развитие процессов дезактивации катализатора в случае бесфосфиновых систем, приводящее к падению каталитической активности в ходе реакции, зачастую является основной причиной неполных конверсий субстратов [25]. Однако, учитывая, что присутствие в каталитической системе фосфинов не сказывается на природе каталитически активных соединений, можно обоснованно предположить постоянное протекание за пределами основного каталитического цикла обменных процессов между комплексами палладия с различными лигандами и преимущественное участие в катализе не фосфиновых, а хлоридных комплексов палладия, не содержащих фосфиновых лигандов, что тем не менее позволяет достигать высоких степеней конверсии исходных веществ и выходов продуктов.
Применение бесфосфиновой каталитической системы на основе хлорида палладия приводило к получению не только “классического” продукта реакции Мицороки–Хека, то есть дизамещенного алкена типа 1, но и продукта, имеющего в своем составе карбонильную группу, то есть халкона типа 2 (схема 1 ). Халкон и его производные относятся к классу флавоноидов и обладают широким спектром биологической активности [26], что делает очевидным актуальность разработки их методов синтеза. Представленные в литературе способы получения производных такого типа обычно предполагают присутствие в системе избыточного давления СО [7, 8], что, безусловно, является нежелательным, поскольку предполагает усложнение процедуры проведения синтеза в сравнении с методиками, не предполагающими применения газообразных реагентов и высокого давления.
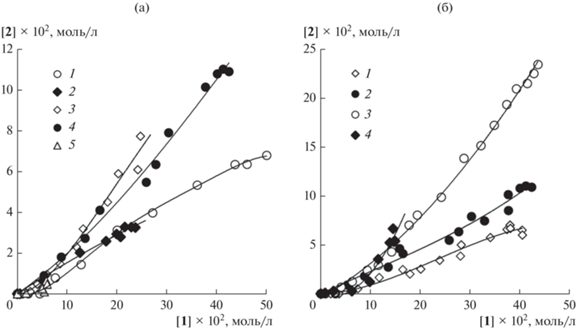
Для определения чувствительности селективности образования карбонилсодержащих продуктов реакции Мицороки–Хека к природе добавки галогенной соли нами были изучены закономерности ДС, которую оценивали по фазовым траекториям “неконкурентных” реакций между ангидридом бензойной кислоты и стиролом, построенным в координатах количеств халкона 2 и классического “хековского” продукта 1 (схема 1 ). Использование разных галогенных солей щелочных металлов и тетрабутиламмония давало отличающиеся друг от друга фазовые траектории, подтверждая вхождение галогенид-ионов в состав активных комплексов палладия, определяющих селективность (рис. 5а). При этом чувствительность к природе добавки проявляла как ДС реакции по халкону, так и конверсия исходных веществ в карбонилсодержащий и классический “хековский” продукты (конверсия может быть оценена по протяженности фазовой траектории). Применение LiCl и NBu4Br в качестве добавки (в количестве 6.25 экв в расчете на загружаемый в систему палладий) приводило к близким значениям суммарных выходов продуктов 1 и 2, однако использование бромистого тетрабутиламмония сопровождалось значительным ростом селективности по карбонилсодержащему продукту (рис. 5а). Близкая ДС наблюдалась и при добавлении бромида лития, позволяя предположить преимущественное образование карбонилсодержащего халкона с участием бромидных комплексов палладия. В случае справедливости такого предположения возрастание концентрации добавляемых в систему бромид-ионов способно повысить селективность и выход халкона. Действительно, увеличение количества NBu4Br до 20 экв в расчете на палладий приводило к значительному росту селективности по продукту 2, обусловленному повышением его выхода (рис. 5б). Однако при дальнейшем увеличении количества NBu4Br до 40 экв отмечалось резкое падение суммарного выхода продуктов 1 и 2 (рис. 5б), что, скорее всего, связано с переходом большей части палладия в каталитически неактивную форму [PdBr4]2–, появление которой наблюдается при использовании в бесфосфиновых каталитических системах реакции Мицороки–Хека добавок бромидов тетраалкиламмония [27, 28], в том числе, при применении ангидридов ароматических кислот в качестве арилирующих реагентов [29]. Таким образом, полученные данные указывают на участие бромид-содержащих комплексов палладия в образовании карбонилсодержащего продукта в условиях реакции Мицороки–Хека и позволяют предположить направления для рациональной “настройки” каталитических систем для синтеза определенных продуктов реакции с высокой селективностью.
Рис. 5.
Фазовые траектории “неконкурентной” реакции Мицороки–Хека, построенные с использованием концентраций стильбена 1a и халкона 2a (схема 3 ): а – при варьировании природы добавки галогенидной соли в количестве 6.25 экв в расчете на Pd: LiCl (1), NaCl (2), LiBr (3), NBu4Br (4), NaI (5); б – при варьировании количества добавки NBu4Br: 4 (1), 6.25 (2), 20 (3), 40 экв (4) в расчете на Pd.
ЗАКЛЮЧЕНИЕ
Представленные в работе данные о закономерностях дифференциальной селективности реакции Мицороки–Хека по конкурирующим ароматическим ангидридам, конкурирующим алкенам, а также карбонилсодержащему и “хековскому” продуктам в неконкурентных условиях однозначно указывают на вхождение в состав активных комплексов палладия, участвующих в селективностьопределяющих стадиях, галогенид-ионов, присутствующих в молекуле предшественника катализатора либо генерирующихся из добавляемой для повышения каталитической активности соли. При использовании добавок фосфинов молекула третичного фосфина входит в координационную сферу активных комплексов палладия только в условиях низких концентраций добавляемой галогенной соли или ее отсутствия. На примере хлорида лития показано, что определяющее влияние на состав истинного катализатора оказывают соотношения концентраций галогенид-иона, третичного фосфина и палладия. Таким образом, молекулы фосфина отсутствуют в интермедиатах каталитического цикла реакции Мицороки–Хека, вносящих определяющий вклад в конверсию субстратов, при обычно применяемых на практике избытках фосфина к палладию в 1–2 эквивалента. При этом добавки фосфинов влияют на процессы превращения катализатора за пределами основного каталитического цикла реакции Мицороки–Хека, в частности, стабилизируя неустойчивые молекулярные комплексы Pd(II).
Также показано, что природа катиона и аниона соли оказывает ключевое действие на дифференциальную селективность и, соответственно, соотношение выходов классического “хековского” продукта арилирования алкена и карбонилсодержащего продукта, образующегося в результате реализации параллельного маршрута реакции в условиях применения бесфосфиновых каталитических систем. Полученные результаты демонстрируют принципиальную возможность повышения селективности таких систем по карбонилсодержащим продуктам реакции Мицороки–Хека.
Список литературы
The Mizoroki–Heck Reaction / Ed. Oestreich M. Munster: John Wiley & Sons Ltd. 2009. 587 p.
Christoffel F., Ward T.R. // Catal. Lett. 2018. V. 148. P. 489.
Ju B., Kong W. // Asian J. Org. Chem. 2020. V. 9. P. 1154.
Stephan M.S., Teunissen A.J.J.M., Verzijl G.K.M., de Vries J.G. // Angew. Chem. Int. Ed. 1998. V. 37. P. 662.
Шмидт А.Ф., Смирнов В.В. // Кинетика и катализ. 2000. Т. 41. С. 820. (Shmidt A.F., Smirnov V.V. // Kinet. Catal. 2000. V. 41. P. 743.)
Kurokhtina A.A., Larina E.V., Yarosh E.V., Schmidt A.F. // J. Mol. Catal. A: Chem. 2016. V. 425. P. 43.
Wu X.-F., Jiao H., Neumann H., Beller M. // ChemCatChem. 2011. V. 3. P. 726.
Wu X.-F., Neumann H., Beller M. // Angew. Chem. Int. Ed. 2010. V. 49. P. 5284.
Шмидт А.Ф., Смирнов В.В. // Кинетика и катализ. 2002. Т. 43. С. 215. (Shmidt A.F., Smirnov V.V. // Kinet. Catal. 2002. V. 43. P. 195.)
Schmidt A.F., Kurokhtina A.A., Larina E.V., Yarosh E.V., Lagoda N.A. // Organometallics. 2017. V. 36. P. 3382.
Gildner P.G., Colacot T.J. // Organometallics. 2015. V. 34. P. 5497.
Лагода Н.А., Ларина Е.В., Ярош Е.В., Курохтина А.А., Шмидт А.Ф. // Изв. Акад. наук. Сер. хим. 2019. Т. 68. С. 817. (Lagoda N.A., Larina E.V., Yarosh E.V., Kurokhtina A.A., Schmidt A.F. // Russ. Chem. Bull. 2019. V. 68. P. 817.)
Amatore C., Carré E., Jutand A., M’Barki M.A., Meyer G. // Organometallics. 1995. V. 14. P. 5605.
Amatore C., Jutand A. // Acc. Chem. Res. 2000. V. 33. P. 314.
Schmidt A.F., Al Halaiqa A., Smirnov V.V. // Synlett. 2006. V. 18. P. 2861.
Schmidt A.F., Kurokhtina A.A., Larina E.V. // Cat. Sci. Technol. 2014. V. 4. P. 3439.
Шмидт А.Ф., Курохтина А.А., Ларина Е.В. // Кинетика и катализ. 2019. Т. 60. С. 555. (Schmidt A.F., Kurokhtina A.A., Larina E.V. // Kinet. Catal. 2019. V. 60. P. 551.)
Шмидт А.Ф., Курохтина А.А., Ларина Е.В. // Кинетика и катализ. 2012. Т. 53. С. 86. (Schmidt A.F., Kurokhtina A.A., Larina E.V. // Kinet. Catal. 2012. V. 53. P. 84.)
Excel for Scientists and Engineers: Numerical Methods / E.J. Billo. John Wiley & Sons. 2007. 480 p.
Gooßen L.J., Rodríguez N., Gooßen K. // Angew. Chem. Int. Ed. 2008. V. 47. P. 3100.
Jutand A., Négri S. // Organometallics. 2003. V. 22. P. 4229.
Schmidt A.F., Kurokhtina A.A., Larina E.V., Vidyaeva E.V., Lagoda N.A., Schmidt E.Y. // ChemCatChem. 2020. V.12. P. 5523.
Schroeter F., Strassner T. // Inorg. Chem. 2018. V. 57. P. 5159.
Carrow B.P., Hartwig J.F. // J. Am. Chem. Soc. 2010. V. 132. P. 79.
Курохтина А.А., Ярош Е.В., Ларина Е.В., Лагода Н.А., Шмидт А.Ф. // Кинетика и катализ. 2018. Т. 59. С. 551. (Kurokhtina A.A., Yarosh E.V., Larina E.V., Lagoda N.A., Schmidt A.F. // Kinet. Catal. 2018. V. 59. P. 564.)
Zhuang C., Zhang W., Sheng C., Zhang W., Xing C., Miao Z. // Chem. Rev. 2017. V. 117. P. 7762.
Reimann S., Stotzel J., Frahm R., Kleist W., Grunwaldt J.-D., Baiker A. // J. Am. Chem. Soc. 2011. V. 133. P. 3921.
Шмидт А.Ф., Аль-Халайка А., Смирнов В.В., Курохтина А.А. // Кинетика и Катализ. 2008. Т. 49. С. 669. (Schmidt A.F., Al-Halaiqa A., Smirnov V.V., Kurokhtina A.A. // Kinet. Catal. 2008. V. 49. P. 638.)
Ларина Е.В., Курохтина А.А., Ярош Е.В., Лагода Н.А., Шмидт А.Ф. // Вестник БурГУ. Химия. Физика. 2016. Вып. 4. С. 26. (Larina E.V., Kurokhtina A.A., Yarosh E.V., Lagoda N.A., Schmidt A.F. // BSU bulletin. Chemistry. Physics. 2016. Is. 4. P. 26.)
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ