

Кинетика и катализ, 2022, T. 63, № 3, стр. 384-393
Роль цеолитов в тепло- и массообмене в гранулированных многофункциональных катализаторах синтеза Фишера–Тропша на основе кобальта
Л. В. Синева a, *, Е. О. Наливайко a, К. О. Грязнов a, В. З. Мордкович a
a ГНЦ ФГБНУ Технологический институт сверхтвердых и новых углеродных материалов (ТИСНУМ)
108840 Москва, Троицк, ул. Центральная, 7а, Россия
* E-mail: sinevalv@tisnum.ru
Поступила в редакцию 24.11.2021
После доработки 20.12.2021
Принята к публикации 20.01.2022
- EDN: RMZNZT
- DOI: 10.31857/S0453881122030121
Аннотация
Изучена роль цеолита в Н-форме в тепло- и массообмене в гранулах композитного Со-катализатора синтеза Фишера–Тропша (СФТ). Показано, что введение в состав катализатора теплопроводящей добавки – металлического алюминия – повышает термическую стабильность гранул, однако не обеспечивает высокую производительность. Добавление в катализатор цеолита способствует интенсификации массообмена за счет снижения плотности и средней температуры кипения образующихся жидких углеводородов. При этом дополнительно улучшается теплообмен благодаря уносу тепла за счет теплоемкости отводящихся продуктов. На основании исследований по превращению жидких углеводородов в условиях СФТ предложен механизм их совместного протекания на Со-цеолитном катализаторе.
ВВЕДЕНИЕ
Синтез Фишера–Тропша (СФТ) – основная стадия технологий получения из углеродсодержащего сырья жидких углеводородов, которые служат альтернативным источником компонентов высококачественных моторных топлив, что определяет актуальность работ по созданию новых высокопроизводительных катализаторов [1–4]. Кроме того, эта стадия определяет аппаратурное оформление всей технологической цепочки: от соотношения Н2 : СО в исходном синтез-газе до количества итераций для достижения желаемого состава продуктов.
СФТ – сильно экзотермический процесс, в котором контроль температуры является критически важной задачей: энтальпия основной реакции составляет –165 кДж/мольCO, а молекулярно-массовое распределение (ММР) продуктов синтеза чувствительно к температуре катализатора. Местные перегревы на поверхности гранул приводят к смещению распределения в сторону более легких углеводородов, в первую очередь – метана, что крайне нежелательно. Все это повышает требования к управлению процессом и контролю за температурой, а, следовательно, и к организации тепломассообмена в каталитическом слое, в частности, в гранулах катализатора [5, 6].
В реализованных в промышленности технологиях, направленных на получение высокомолекулярных углеводородов, СФТ проводят при низких нагрузках на катализатор и малых конверсиях синтез-газа, чтобы избежать перегревов [2, 7]. Это приводит к ограничениям по производительности – не более 100 гC5+${\text{л}}_{{{\text{кт}}}}^{{ - 1}}$ ч–1 или 75–85 гC5+ ${\text{кг}}_{{{\text{кт}}}}^{{ - 1}}$ ч–1 [8, 9]. Отвод тепла от активных центров, на которых протекает взаимодействие СО и Н2, крайне незначителен в традиционных катализаторах на основе оксидов из-за низкой теплопроводности материала гранул, причем неизотермический режим эксплуатации гранул приводит к быстрой дезактивации катализатора [5, 10]. Следовательно, теплопроводность является одним из ключевых моментов в создании высокоэффективных гранулированных катализаторов СФТ.
Повышение термической устойчивости катализаторов СФТ может позволить интенсифицировать теплообмен в реакторе с неподвижным слоем. В литературе в качестве теплопроводящих компонентов предлагается использовать подложки из структурированных материалов [1, 11], углеродные материалы [12–14] или металлы [15, 16]. Таким образом, проблема теплоотвода может быть решена, но вопросы, связанные с массопереносом, останутся: высокомолекулярные продукты СФТ будут заполнять транспортные поры катализатора, снижая активность катализаторного слоя. В промышленности эта проблема решается путем периодической обработки катализатора в токе водорода при температуре, превышающей оптимальную для его эксплуатации.
Необходимость крекировать высокомолекулярные продукты СФТ – одно из препятствий на пути к совершенствованию промышленных технологий на его основе. Использование цеолитов в качестве компонентов кобальтовых катализаторов для получения из СО и Н2 компонентов моторных топлив уже на выходе из реактора СФТ является альтернативным направлением развития этого процесса [17–23]. Интенсификация массообмена in situ – в гранулах и в слое кобальтового катализатора – технологичное решение, позволяющее кроме массообмена интенсифицировать теплоперенос за счет снижения среднего молекулярного веса и плотности образующихся углеводородов С5+.
Цеолиты широко применяются в катализаторах нефтепереработки и нефтехимии, в том числе благодаря своей активности в крекинге и изомеризации углеводородов. При использовании этих свойств в качестве инструмента для интенсификации массообмена на поверхности гетерогенных катализаторов важно так сформировать каталитическую систему, чтобы крекинг не был чересчур глубоким и позволял получать жидкие, а не газообразные углеводороды [22, 23]. При разработке бифункциональных катализаторов СФТ чаще всего применяют HZSM-5, реже – HY и HBeta.
Полимеризационный механизм СФТ оправдывает большой интерес к использованию кислотных свойств цеолитов в катализаторах СФТ для одностадийного (не подразумевающего стадию гидрокрекинга) получения компонентов моторных топлив [1, 18, 24–29]. Вторичные реакции, зависящие от длины цепи углеводородов, приводят к отклонениям от ММР, характерного для такого механизма, и могут быть применены для повышения селективности образования желаемых продуктов. Введение цеолитов в катализатор – способ вовлечь углеводороды, образовавшиеся из СО и Н2, во вторичные превращения, такие как крекинг и изомеризация.
Для совместного размещения в одной каталитической системе активных центров СФТ и превращений углеводородов используют различные способы, такие как физическое смешивание катализатора СФТ и цеолита, нанесение активного в СФТ металла на цеолит, его капсулирование в цеолит, компактирование в гранулы смеси активного металла и цеолита. Для промышленной реализации капсулированные катализаторы наименее перспективны, поскольку лимитирующей стадией процесса является транспорт исходных компонентов и продуктов синтеза. С нашей точки зрения наиболее перспективны пропиточные и компактированные (композитные) катализаторы [17, 30].
Цель настоящей работы – изучение взаимосвязи влияния теплопроводящей добавки и цеолита на показатели СФТ и состав продуктов синтеза при интенсификации тепло- и массообмена в гранулах многофункциональных кобальтовых катализаторов. Катализаторы смешения на основе скелетного кобальта были выбраны для выявления возможного синергетического действия добавок теплопроводящего компонента и цеолита, поскольку в этом случае активный металл не располагается на поверхности компонентов катализатора, как при нанесении из раствора.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Катализаторы, содержащие 20 мас. % скелетного Со, 10 мас. % цеолита (Beta, Y, морденит или ZSM-5) в Н-форме, 50 мас. % порошка металлического алюминия (с частицами в виде чешуек (ч) или сфер (сф)) и 20 мас. % бемита были приготовлены смешением компонентов катализатора с последующей экструзией и термообработкой. Для этого цеолит и бемит тщательно перемешивали и пептизировали азотной кислотой для получения связующего. В полученный гель вмешивали порошок металлического алюминия, увлажненный спиртом в весовом соотношении 3 : 1, а затем – порошок скелетного Со. Порошок скелетного Со получали из стехиометрического сплава Co2Al9 размером 100 меш. производства компании “Johnson Matthey” выщелачиванием раствором 10 М КОН при 60°С с последующим отмыванием сначала раствором щелочи, а затем 0.5 М HNO3. Готовый порошок соответствовал составу СоО0.75, содержал менее 0.3 мас. % Al и характеризовался размером частиц 1–15 мкм. Полученную пасту экструдировали через фильеру диаметром 2.5 мм. Экструдат сушили на воздухе в течение 10 ч, подвергали термической обработке, повышая температуру со скоростью 0.5°С/мин до 400°С, и выдерживали в изотермическом режиме 3 ч. Прокаленный экструдат измельчали до размера 2.5 × 2.5 мм. Обозначения и составы сравниваемых катализаторов приведены в табл. 1.
Таблица 1.
Исследуемые катализаторы, их состав, характеристики пористой системы и теплопроводящие свойства
| Катализатор | Состав*, мас. % | Объем макропор, см3/г | Объем мезопор, см3/г | Удельная поверхность, м2/г | Коэффициент теплопровод-ности, Вт м–1 К–1 | ||
|---|---|---|---|---|---|---|---|
| цеолит HBeta | тип Al** | Al2O3 | |||||
| R | – | – | 80 | 0.314 | 0.365 | 241 | 0.89 |
| RHB | 10 | – | 70 | 0.347 | 0.332 | 270 | 0.71 |
| Bсф | – | 50сф | 20 | 0.204 | 0.137 | 82 | 1.61 |
| Bч | – | 50ч | 20 | 0.552 | 0.184 | 91 | 4.99 |
| Eч | 10 | 50ч | 20 | 0.583 | 0.178 | 102 | 3.75 |
| Eч-сф | 10 | 38ч + 12сф | 20 | 0.436 | 0.153 | 114 | 2.00 |
На поверхности гранулы полученного таким образом катализатора хорошо видна крупная пористая система, доля макропор в которой составляет не менее 67%, и теплопроводящий каркас, образованный чешуйками алюминия (рис. 1а). На рис. 1б показан край крупной частицы кобальта скелетной поликристаллической структуры, обладающей развитой внутренней поверхностью. Кроме того, на микрофотографии отчетливо видно, что в краевой области этой частицы отдельные кристаллиты оксидов кобальта размером <20 нм выходят на поверхность.
Рис. 1.
СЭМ-изображение поверхности катализатора (а) и ПЭМ-изображение частицы скелетного Со (Со-Ra) на поверхности этого катализатора (б).

В качестве образца сравнения по такой же методике был приготовлен катализатор, который вместо цеолита содержал дополнительное количество связующего. Катализатор загружали в стальной проточной реактор с внутренним диаметром 10 мм. Температуру в реакторе поддерживали подаваемой в рубашку реактора пароводяной смесью под давлением на границе насыщения.
Для характеристики катализаторов были использованы методы низкотемпературной сорбции азота, определения влагоемкости, измерения теплопроводности, РФА, сканирующей и просвечивающей электронной микроскопии.
Объем мезопор и удельную площадь поверхности катализаторов находили по низкотемпературной сорбции азота на приборе NOVAWin (“Quantachrome Instruments”, США) с использованием фирменных программных продуктов.
Объем макропор катализатора определяли по влагоемкости гранул (ГОСТ 24160-2014). Для этого навеску массой не менее 2 г сушили при 110°С в течение 4 ч, после чего помещали в эксикатор с хлористым кальцием и охлаждали до комнатной температуры. Затем навеску взвешивали и заливали дистиллированной водой комнатной температуры объемом не менее 3 объемов навески. Если через 10 мин выдерживания с периодическим перемешиванием наблюдались выходящие из пор пузырьки газа, то навеску выдерживали под слоем жидкости еще 10 мин, и так до прекращения появления пузырьков. Затем жидкость сливали, стряхивая остатки жидкости на фильтровальную бумагу, бюкс с влажной навеской взвешивали. Количество поглощенной воды рассчитывали по формуле:
где m1 – масса сухой навески, г; m2 – масса влажной навески, г; m2– m1 – количество поглощенной жидкости, г. Влагоемкость (см3/г) рассчитывали, принимая плотность воды равной 0.9971 г/см3.Коэффициент теплопроводности катализаторов вычисляли на основании данных по теплоемкости, полученных на приборе DSC8000 (“PerkinElmer”, США), и температуропроводности, полученных на приборе LFA 457/2/G MicroFlash analyzer (“NETZSCH”, Германия), по формуле:
где ρ – кажущаяся плотность, рассчитанная из геометрических параметров гранул катализатора, кг/м3, α – температуропроводность, м2/с; Cp – теплоемкость, Дж кг–1 K–1. Метод лазерной вспышки относится к динамическим методам измерения коэффициента теплопроводности разнообразных материалов (пластмасс, пористых композитов, керамик и др.). Сущность метода состоит в определении скорости прохождения фронта изменения температуры в образце, сформованном в плоскопараллельную таблетку с известными геометрическими характеристиками. Короткий импульс лучистой энергии поглощается в тонком слое фронтальной поверхности плоского образца. Вызванное этим возмущение температуры регистрируется на тыльной поверхности образца [31].Дифракционный анализ проводили при комнатной температуре на порошковом дифрактометре Stoe Stadi P (“STOE Cie GmbH”, Германия). Были использованы CuKα-излучение, монохроматор на вторичном пучке, точечный сцинтилляционный детектор.
Микрофотографии поверхности кобальт–алюминий-цеолитных систем и кристаллитов кобальта получены методом растровой и просвечивающей электронной микроскопии с помощью электронных микроскопов TESCAN VEGA 3 SEM (“TESCAN”, Чехия) и JEM-2010 (“JEOL”, Япония) соответственно.
Характеристики пористой системы и теплопроводящих свойств катализаторов, использованных для выявления отдельных вкладов цеолита на примере HBeta и теплопроводящей добавки в виде порошка металлического алюминия и совместного взаимного влияния этих компонентов катализатора, представлены в табл. 1. Из данных табл. 1 видно, что введение в состав катализатора металлического алюминия приводит к увеличению коэффициента теплопроводности в 1.8–5.6 раз, причем в большей степени при использовании порошка алюминия в виде чешуек – с 0.89 до 4.99 Вт м–1 К–1. При добавлении или замене чешуйчатого алюминия на сферический объем макропор катализатора снижается в 1.5 раза, а при добавлении цеолита – возрастает в 1.1 раза. Удельная поверхность катализаторов при введении в их состав цеолита также растет. Высокие значения величин поверхности и объема мезопор катализаторов без теплопроводящей добавки связаны, вероятно, с бóльшим содержанием связующего.
Перед синтезом катализаторы активировали в токе водорода, подаваемого со скоростью 3000 ч–1 при давлении 0.1 МПа и температуре 400°С в течение 1 ч.
Каталитические свойства образцов исследовали в синтезе Фишера–Тропша и в превращении смеси жидких углеводородов (СЖУ). Для СФТ применяли смесь Н2 и СО с мольным соотношением 2 : 1, которую подавали с объемной скоростью 1000–6000 ч–1, синтез проводили при давлении 2 МПа, варьируя температуру в диапазоне 170–260°С. Длительность эксперимента при постоянных условиях составляла 6 ч, постоянство каталитических показателей достигалось через 3 ч. Каталитические показатели сравнивали при оптимальной температуре, которой соответствовала наибольшая производительность по целевому продукту – смеси жидких углеводородов. Из двух близких значений этого показателя выбирали то, при котором селективность образования углеводородов С5+ была выше. Превращения СЖУ исследовали в том же диапазоне температур 170–260°С при давлении 0.1 МПа, СЖУ подавали шприцевым насосом в поток водорода со скоростью 1.2 мл/ч. В качестве сырья использовали углеводороды следующего состава (мас. %): олефины С5+ – 6.3, парафины С5+ линейные – 82.3 и разветвленные – 11.4; фракции С5–С10 – 50.3, С11–С18 – 38.3 и С19+ – 11.4. Анализ газа и жидких углеводородов проводили через 1 ч выдерживания при постоянных условиях.
Состав исходного синтез-газа и газообразных продуктов синтеза определяли газо-адсорбционной хроматографией на приборе Кристаллюкс-4000м (НПФ “Мета-хром”). Колонку с молекулярными ситами 5А (3 м × 3 мм) использовали для разделения CO и СН4, а с HayeSep (3 м × 3 мм) – СО2 и углеводородов С2–С4. Детектор – катарометр, газ-носитель – гелий (20 мл/мин). Режим – температурно-программированный (60–200°С, скорость нагрева – 10°С/мин). Состав СЖУ анализировали методом газожидкостной хроматографии на приборе Кристаллюкс-4000м (НПФ “Мета-хром”). Колонка – капиллярная 50 м, неподвижная фаза – DB-Petro. Детектор – ПИД, газ-носитель – гелий (30 мл/мин). Режим – температурно-программированный (50–270°С, скорость нагрева – 4°С/мин).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В табл. 2 представлены результаты СФТ, полученные при скорости синтез-газа 3000 и 5000 ч–1. Можно видеть, что введение теплопроводящей добавки в состав катализатора расширяет диапазон условий его эксплуатации, причем повышение скорости газа в большинстве случаев приводит к росту производительности. Совместное введение теплопроводящей добавки и цеолита способствует дальнейшему увеличению производительности, а селективность образования метана снижается. Наблюдаемый синергетический эффект от сочетания цеолита и металлического алюминия объясняется интенсификацией одновременно тепло- и массообмена на поверхности гранул катализатора. Интересно отметить, что оптимальная температура синтеза при этом снижается.
Таблица 2.
Показатели СФТ и состав образующихся углеводородов С5+ в зависимости от скорости подачи синтез-газа (3000/5000 ч–1)
| Катализатор | Т, °С | Конверсия СО, % | Производительность, г кг–1 ч–1 | Селективность, мол. % | |
|---|---|---|---|---|---|
| С5+ | СН4 | ||||
| R | 231/– | 68/– | 154/– | 37/– | 33/– |
| RHB | 242/– | 49/– | 120/– | 39/– | 28/– |
| Bсф | 225/233 | 61/64 | 151/242 | 48/43 | 30/33 |
| Bч | 249/255 | 68/62 | 304/426 | 50/46 | 29/31 |
| Eч | 237/246 | 72/44 | 396/312 | 56/44 | 17/19 |
| Eч-сф | 234/240 | 77/69 | 384/493 | 56/48 | 17/23 |
Введение в состав катализатора цеолита ведет к изменению состава углеводородов С5+ (табл. 3): доля н-парафинов снижается в результате протекания реакций изомеризации, а высокомолекулярных соединений – благодаря крекингу, при этом содержание фракции С5–С10 повышается. Рост доли олефинов свидетельствует о бимолекулярном механизме этих превращений [32, 33]. Увеличение коэффициента теплопроводности способствует повышению расчетной плотности СЖУ, образовавшихся при 3000 ч–1, с 0.707 до 0.712–0.713 г/см3 и средней температуры их кипения со 149 до 159–162°С, что может объясняться уменьшением вероятности местных перегревов, а, следовательно, снижением температуры внутри гранул. Одновременное введение теплопроводящей добавки и цеолита, как и одного только цеолита, приводит к уменьшению этих показателей до 0.692–0.695 г/см3 и 124–129°С соответственно. В результате удаление продуктов С5+ с поверхности катализатора облегчается, способствуя как массообмену, так и отводу тепла. Таким образом, добавление в состав Со-катализатора цеолита в Н-форме интенсифицирует массообмен в гранулах катализатора за счет снижения плотности и температуры кипения образующихся жидких углеводородов.
Таблица 3.
Влияние типа цеолита в составе катализатора на состав образующихся углеводородов С5+ в зависимости от скорости подачи синтез-газа (3000/5000 ч–1)
| Катализатор | СnH2n | н-СnH2n+2 | изо-СnH2n+2 | С5–С10 | С11–С18 | С19+ | Расчетная плотность, г/см3 | Средняя температура кипения, °С |
|---|---|---|---|---|---|---|---|---|
| R | 8/– | 78/– | 14/– | 59/– | 32/– | 9/– | 0.707/– | 149/– |
| RHB | 41/– | 27/– | 32/– | 81/– | 19/– | 0/– | 0.696/– | 131/– |
| Bсф | 2/5 | 86/82 | 12/13 | 52/58 | 39/35 | 9/7 | 0.713/0.708 | 162/151 |
| Bч | 10/17 | 80/73 | 10/10 | 55/60 | 36/34 | 9/6 | 0.712/0.710 | 159/155 |
| Eч | 32/40 | 46/43 | 22/17 | 75/79 | 23/20 | 2/1 | 0.695/0.697 | 129/133 |
| Eч-сф | 35/37 | 42/44 | 23/19 | 76/80 | 23/19 | 1/1 | 0.692/0.690 | 124/121 |
Влияние типа цеолита на свойства катализатора было изучено на примере наиболее распространенных в нефтехимии фожазита, морденита и пентасила и несколько менее популярного цеолита бета (табл. 4). Использованные цеолиты отличались кислотностью, определенной методом ТПД аммиака, десорбировавшегося в диапазоне 300–500°С, а катализаторы на их основе – объемом транспортных пор, зависящим от размеров кристаллитов и их конгломератов, характерных для исходных порошков цеолитов (табл. 5). При сравнении показателей СФТ, полученных при 3000 и 5000 ч–1, видно, что большого объема макропор недостаточно для интенсивного массообмена: наибольший объем макропор характеризует катализатор на основе цеолита HMor – 0.54 см3/г, однако в его присутствии увеличение скорости синтез-газа не приводит к повышению производительности, которая составляет 320 г кг–1 ч–1 при 3000 ч–1 и 313 г кг–1 ч–1 при 5000 ч–1. Кроме того, катализаторы на основе цеолитов с низкой кислотностью отличаются невысокой селективностью уже в синтезе при 3000 ч–1. Тогда как наибольшая кислотность 561 ${{{\text{мкмол}}{{{\text{ь}}}_{{{\text{N}}{{{\text{H}}}_{{\text{3}}}}}}}} \mathord{\left/ {\vphantom {{{\text{мкмол}}{{{\text{ь}}}_{{{\text{N}}{{{\text{H}}}_{{\text{3}}}}}}}} {\text{г}}}} \right. \kern-0em} {\text{г}}}{\text{,}}$ характерная для цеолита HZSM-5, способствует повышению производительности катализатора на его основе, которая достигает 453 г кг–1 ч–1 при 3000 ч–1. Однако увеличение скорости синтез-газа до 5000 ч–1 оказывает незначительное влияния на этот показатель, вероятно, из-за малого объема макропор, не обеспечивающего доступность этих кислотных центров. Наибольшая производительность в синтезе при 5000 ч–1 – 493 г кг–1 ч–1 – зафиксирована при использовании катализатора на основе цеолита HBeta. Данный цеолит характеризуется довольно высокой кислотностью (394 ${{{\text{мкмол}}{{{\text{ь}}}_{{{\text{N}}{{{\text{H}}}_{{\text{3}}}}}}}} \mathord{\left/ {\vphantom {{{\text{мкмол}}{{{\text{ь}}}_{{{\text{N}}{{{\text{H}}}_{{\text{3}}}}}}}} {\text{г}}}} \right. \kern-0em} {\text{г}}}$), а содержащий его катализатор – средним объемом макропор (0.44 см3/г). Также в присутствии этого катализатора наблюдались меньшие потери в конверсии СО при увеличении скорости подачи синтез-газа. Кроме того, увеличение силы кислотных центров приводит к сокращению доли высокомолекулярных продуктов и возрастанию содержания изомеров (табл. 5). Таким образом, для превращений углеводородов доступность кислотных центров цеолитов важнее их силы. Стоит отметить, что повышение кислотности входящего в состав катализатора цеолита способствует снижению оптимальной температуры синтеза, вероятно, за счет уменьшения среднего молекулярного веса углеводородов С5+, что облегчает удаление продуктов с поверхности катализатора, интенсифицируя массообмен. Так, при 3000 ч–1 оптимальная температура синтеза составляла 231, 234, 238 и 243°С в присутствии цеолитов HZSM-5, HBeta, HMor и HY, а при 5000 ч–1 – 237, 240, 244 и 249°С соответственно (табл. 3).
Таблица 4.
Влияние типа цеолита в составе катализатора на показатели СФТ в зависимости от скорости подачи синтез-газа (3000/5000 ч–1)
| Катализатор | Цеолит | Топт, °С | Конверсия СО, % | Производи-тельность, г кг–1 ч–1 |
Селек-тивность, мол. % | [изо-СnH2n+2], мас. % | [C19+], мас. % |
|---|---|---|---|---|---|---|---|
| Еч-сфHZSM5 | HZSM-5 | 231/237 | 75/50 | 453/462 | 62/56 | 26/24 | 2/1 |
| Еч-сфHBet | HBeta | 234/240 | 77/65 | 384/493 | 56/48 | 23/19 | 1/1 |
| Еч-сфHMor | HMor | 238/244 | 74/48 | 320/313 | 47/42 | 17/15 | 2/2 |
| Еч-сфHY | HY | 243/249 | 73/60 | 453/462 | 62/56 | 15/10 | 3/3 |
Таблица 5.
Влияние размера частиц исходного порошка цеолита на кислотность и объем макропор катализатора
| Катализатор | Цеолит | Кислотность (300–550°С), ${\text{мкмол}}{{{\text{ь}}}_{{{\text{N}}{{{\text{H}}}_{{\text{3}}}}}}}{\text{/г}}$ | Объем макропор, см3/г | Размер частиц порошка цеолитов | |
|---|---|---|---|---|---|
| кристаллиты, нм (СЭМ) | поликристаллы, мкм (РФА) | ||||
| Еч-сфHZSM5 | HZSM-5 | 561 | 0.38 | 41 ± 2 | 0.1–0.3 |
| Еч-сфHBetа | HBeta | 394 | 0.44 | 12 ± 0.5 | 0.1–1 |
| Еч-сфHMor | HMor | 160 | 0.54 | 122 ± 6 | 0.1–1.5 |
| Еч-сфHY | HY | 89 | 0.52 | 94 ± 4 | 0.5–1 |
Итак, для термической устойчивости гранул в условиях повышенной нагрузки по сырью, необходимой для достижения высокой производительности, требуется одновременная интенсификация тепло- и массообмена как в катализаторном слое, так и в отдельных гранулах катализатора: повышенная теплопроводность гранул способствует их устойчивости к местным перегревам, а достаточный объем макропор и наличие цеолита в Н-форме обеспечивают интенсивный массообмен на их поверхности.
Ранее нами было проведено сравнительное исследование влияния реакционной среды на превращение СЖУ в присутствии цеолитсодержащих Со-катализаторов в диапазоне температур 170–260°C [34], которое показало, что в восстановительной среде вклад продуктов изомеризации был наибольшим, как и доля метана в газообразных продуктах. Кроме того, было сделано наблюдение, что зависимость состава продуктов от температуры имеет различный характер в диапазонах 170–210 и 220–260°C. Этот эффект в дальнейшем был объяснен с помощью термогравиметрического исследования, которое продемонстрировало, что состав поверхностных соединений, образовавшихся при проведении синтеза в диапазоне 170–260 и 170–210°С в присутствии цеолитсодержащего и бесцеолитного катализаторов заметно отличается [35].
Проведенное в настоящей работе сравнение закономерностей, выявленных при изучении превращений жидких углеводородов, позволило установить, что конверсия жидких углеводородов в газообразные продукты при 170–200°С на кобальтовом и кобальт-цеолитном катализаторах имеет близкие значения (рис. 2а), а при 210–260°С этот показатель выше в присутствии цеолитного примерно в 2 раза. Интересно отметить, что в более низком интервале температур доля метана в газе, образовавшемся на цеолитном катализаторе, больше, чем на бесцеолитном, а при повышении температуры реакции в случае цеолитного катализатора этот показатель снижается в 2 раза, составляя 10% независимо от температуры, а в случае бесцеолитного – линейно возрастал, достигая 45% при 260°С.
Рис. 2.
Влияние цеолита в составе катализатора на превращения жидких углеводородов: конверсия в газообразные углеводороды (K) и доля метана в образовавшемся газе (СН4) (а): изменения в групповом (б) и фракционном (в) составе жидких углеводородов. Метка B указывает на бесцеолитный катализатор, Е – на цеолитсодержащий.
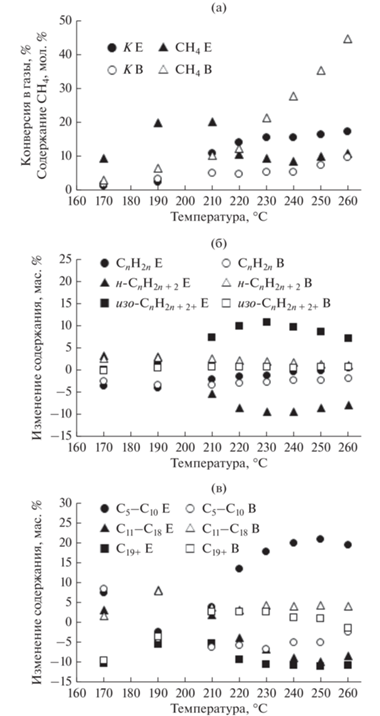
Изменения в групповом составе жидких углеводородов ниже 210°С мало зависят от состава катализатора и вызваны в основном гидрированием олефинов (рис. 2б), тогда как выше 210°С на цеолитном катализаторе активно протекает изомеризация н-парафинов, причем при Т < 240°С олефины являются реагентами, а при Т > 240°С – продуктами реакций. На поверхности бесцеолитного катализатора при повышении температуры продолжает протекать гидрирование олефинов, хотя можно отметить и небольшой вклад продуктов изомеризации.
Изменения во фракционном составе, наблюдаемые выше 210°С, также зависят от наличия цеолита в катализаторе: в его присутствии углеводороды С11 и выше превращаются в С5–С10, а в его отсутствие, наоборот, доля более тяжелых компонентов несколько увеличивается за счет снижения доли легких компонентов (рис. 2в). Полученные данные позволяют предположить, что ниже 210°С превращения жидких углеводородов протекают в основном на активных центрах, содержащих Со, а выше 210°С – цеолит. При этом механизмы реакций и состав промежуточных поверхностных соединений могут отличаться, как было предположено в [35].
Как отмечено выше, повышение содержания изомеров при превращении СЖУ наблюдали и в присутствии бесцеолитного катализатора, что хорошо объясняется механизмом, предложенным А.Л. Лапидусом в работе [36]. Было показано, что процесс изомеризации может протекать и на тех же кобальтовых центрах, на которых протекает синтез углеводородов из CO и H2, причем атомы водорода имеют одинаковую возможность оторваться с образованием цис–транс-изомеров в соотношении близком к единице. Механизм А.Л. Лапидуса, в свою очередь, выдвинут на основании механизма превращения α-олефинов во “внутренние” олефины [37].
ЗАКЛЮЧЕНИЕ
Таким образом, определены условия СФТ, при которых кобальт-цеолитные катализаторы проявляют бифункциональные свойства. Наиболее вероятен следующий механизм образования углеводородов: активные кобальтсодержащие центры катализируют синтез углеводородов из CO и H2, которые участвуют в кислотно-катализируемых реакциях на цеолитных центрах в основном по карбкатионному механизму, поскольку превращение смеси углеводородов в присутствии цеолитсодержащего катализатора сопровождается увеличением доли олефинов С5–С10 [35, 36]. Предположительно, на поверхности кобальт-цеолитного катализатора одновременно протекает ряд последовательных и параллельных реакций, причем образовавшиеся углеводороды могут десорбироваться как продукты или реадсорбироваться на кобальтовом или бренстедовском кислотном центре. Количество актов реадсорбции зависит от свойств катализатора и условий синтеза и определяет конечный состав продуктов.
Введение в состав Со-катализатора теплопроводящей добавки приводит к интенсификации теплообмена на его поверхности, что способствует увеличению производительности и селективности в образовании жидких углеводородов, особенно в условиях повышенной нагрузки по синтез-газу, а роль цеолита заключается в создании дополнительных возможностей для интенсификации тепло- и массообмена на поверхности катализатора за счет снижения среднего молекулярного веса образующихся жидких углеводородов, что дополнительно повышает производительность и селективность по жидким углеводородам.
Список литературы
Синева Л.В., Мордкович В.З. // Научный журнал Российского газового общества. 2019. Т. 20. № 1. С. 42.
Greener Fischer–Tropsch Processes. Eds. Maitlis P.M., Klerk A. Weinheim: Wiley-VCH. 2013. 372 p.
Sousa-Aguiar E.F., Noronha F.B., Faro A., Jr. // Catal. Sci. Technol. 2011. V. 1. P. 698.
Brosius R., Kooyman P.J., Fletcher J.C.Q. / Nanotechnology in Catalysis. Eds. Sels B., Van de Voorde M. Wiley-VCH, 2017. P. 449.
Fratalocchi L., Visconti C.G., Groppi G., Lietti L., Tronconi E. // Chem. Eng. J. 2018. V. 349. P. 829.
Яковенко Р.Е., Нарочный Г.Б., Савостьянов А.П., Кирсанов В.А. // Химическое и нефтегазовое машиностроение. 2015. № 3. С. 11.
Соромотин В.Н., Яковенко Р.Е., Медведев А.В., Митченко С.А. // Кинетика и катализ. 2021. Т. 62. № 6. С. 811.
Хасин А.А. // Газохимия. 2008. № 2. С. 28.
Punt A. / Proceedings of the 10th Natural Gas Conversion Symposium (NGCS 10). March 2–7, 2013. Doha, Qatar. KN5.2.
Merino D., Sanz O., Montes M. // Chem. Eng. J. 2017. V. 327. P. 1033.
Merino D., Sanz O., Montes M. // Fuel. 2017. V. 210. P. 49.
Kim J., Nese V., Joos J., Jeske K., Duyckaerts N., Pfänder N., Prieto G. // J. Mater. Chem. A. 2018. V. 6. № 44. P. 21978.
Asalieva E., Sineva L., Sinichkina S., Solomonik I., Gryaznov K., Pushina E., Kulchakovskaya E., Kulnitskiy B., Ovsyannikov D., Mordkovich V. // Appl. Catal. A: Gen. 2020. V. 601. P. 117639.
Kim J., Nese V., Joos J., Jeske K., Duyckaerts N., Pfänderc N., Prieto G. // J. Mats Chem. A. 2018. V. 6. № 44. P. 21978.
Wang D., Wang Z., Li G., Li X., Hou B. // Ind. Eng. Chem. Res. 2018. V. 57. № 38. P. 12756.
Asalieva E., Gryaznov K., Kulchakovskaya E., Ermolaev I., Sineva L., Mordkovich V. // Appl. Catal. A: Gen. 2015. V. 505. P. 260.
Синева Л.В., Асалиева Е.Ю., Мордкович В.З. // Успехи химии. 2015. Т. 84. С. 1176.
Яковенко Р.Е., Зубков И.Н., Савостьянов А.П., Соромотин В.Н., Краснякова Т.В., Папета О.П., Митченко С.А. // Кинетика и катализ. 2021. Т. 62. № 1. С. 109.
Kibby C., Jothimurugesan K., Das T., Lacheen H. S., Rea T., Saxton R. J. // Catal. Today. 2013. V. 215. P. 131.
Wang H., Pei Y., Qiao M., Zong B. Design of Bifunctional Solid Catalysts for Conversion of Biomass-Derived Syngas into Biofuels / Production of Biofuels and Chemicals with Bifunctional Catalysts. Eds. Fang Z., Smith Jr. R., Li H. Springer: Singapore, 2017. V. 8. P. 137.
Sartipi S., Makkee M., Kapteijn F., Gascon J. // Catal. Sci. Technol. 2014. V. 4. № 4. P. 893.
Мурзин Д.Ю. // Кинетика и катализ. 2020. Т. 61. № 1. С. 85.
Zhu C., Bollas G.M. // Appl. Catal. B: Env. 2018. V. 235. P. 92.
Sun B., Qiao M., Fan K., Ulrich J., Tao F. // ChemCatChem. 2011. V. 3. P. 542.
Brosius R., Fletcher J.C.Q. // J. Catal. 2014. V. 317. P. 318.
Li W., He Y., Li H., Shen D., Xing C., Yang R. // Catal. Comm. 2017. V. 98. P. 98.
Kang S., Ryu J., Kim J., Kim H., Lee C., Lee Y., Jun K. // Modern Res. Catal. 2014. V. 3. P. 99.
Subramanian V., Zholobenko V.L., Cheng K., Lancelot C., Heyte S., Thuriot J., Paul S., Ordomsky V.V., Khodakov A.Y. // ChemCatChem. 2015. V. 8. № 2. P. 380.
Xing C., Yang G., Yang M.W.R., Yang R., Tan L., Zhu P., Wei Q., Li J., Mao J., Yoneyama Y., Tsubaki N. // Fuel. 2015. V. 148. P. 48.
Яковенко Р.Е., Бакун В.Г., Зубков И.Н., Нарочный Г.Б., Папета О.П., Савостьянов А.П. // Катализ в промышленности. 2020. Т. 20. № 4. С. 275.
ГОСТ Р 57943-2017 (ИСО 22007-4:2008).
Jentoft F.C., Gates B.C. // Top. Catal. 1997. V. 4. P. 1.
Anderson B.G., Schumacher R.R., van Duren R., Singh A.P., van Santen R.A. // J. Mol. Catal. A: Chem. 2002. V. 181. P. 291.
Sineva L.V., Gorokhova E.O., Pushina E.A., Kulchakovskaya E.V., Mordkovich V.Z. // Mendeleev Commun. 2020. V. 30. P. 362.
Sineva L.V., Gorokhova E.O., Gryaznov K.O., Ermolaev I.S., Mordkovich V.Z. // Catal. Today. 2021. V. 378. P. 140.
Лапидус А.Л. // Химия твердого топлива. 2013. № 6. С. 5.
Egiebor N.O., Cooper W.C. // Appl. Catal. 1985. V. 17. P. 47.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ