

Кинетика и катализ, 2022, T. 63, № 4, стр. 405-417
Каталитическое разложение гидразина и его производных для получения водородсодержащих газовых смесей
В. А. Матышак a, О. Н. Сильченкова a, *
a ФГБУН Федеральный исследовательский центр химической физики им. Н.Н. Семенова РАН
119991 Москва, ул. Косыгина, 4, Россия
* E-mail: son1108@yandex.ru
Поступила в редакцию 10.01.2022
После доработки 25.03.2022
Принята к публикации 26.03.2022
- EDN: QPDZAH
- DOI: 10.31857/S0453881122040074
Аннотация
Гидразин и моногидрат гидразина широко используются в разнообразных областях науки и техники. Традиционно эти реагенты применяются в качестве восстановителей тяжелых металлов. В последнее время интерес к гидразину и моногидрату гидразина связан с задачей получения свободного от примесей водорода в результате каталитического разложения этих реагентов. В процессе разложения гидразина и моногидрата гидразина испытано большое количество разнообразных каталитических систем, проявляющих принципиально разную активность и селективность в образовании водорода. В обзоре приводится подробная систематизация этих данных и анализ причин различия свойств разных каталитических систем. На основе имеющейся информации можно полагать, что главную роль в определении направления процесса играет форма активации гидразина. Обсуждаются также результаты исследования механизма разложения гидразина при низких и высоких температурах. Рассмотренные в обзоре экспериментальные результаты показывают, что гидразин и моногидрат гидразина являются вполне перспективными и для запасания водорода, и в целях получения водорода, свободного от примесей, для эффективной работы топливных элементов.
ВВЕДЕНИЕ
Сокращение запасов ископаемого топлива наряду с ухудшающейся экологической обстановкой обуславливают необходимость поиска новых экологически чистых и экономически доступных источников энергии. Одним из наиболее перспективных возобновляемых энергоносителей считается водород.
При переходе на водородную энергетику основным направлением получения электроэнергии становится использование водорода в топливных элементах (ТЭ) [1, 2]. В водородных ТЭ протекает реакция окисления водорода кислородом в мембранном электрохимическом процессе с образованием электричества, тепла и воды. Энергетический КПД водородных ТЭ значительно выше, чем у традиционных энергоустановок, и может достигать 85%.
Вместе с тем весь водород, который производится в мире, не сможет покрыть потребность даже в автомобильном топливе. Это стимулирует поиск возможных путей улучшения существующих процессов получения водорода и разработку новых.
В качестве сырья для производства водорода рассматриваются как газы (метан, пропан), так и жидкие органические соединения (первичные спирты, муравьиная кислота) [3–6]. Очевидный недостаток этого подхода – образование побочных продуктов, в частности, оксидов углерода, являющихся ядом для платиновых электродов ТЭ. Избежать этого можно, если применять как исходное сырье реактивы, не имеющие в своем составе атомов углерода. В числе потенциальных реагентов для получения свободного от примесей водорода были предложены борогидриды металлов, аммиак и его производные, гидразин N2H4 и его производные и др. [6–14].
Кроме того, хранить Н2 можно либо как газ при высоком давлении (200–300 бар), либо в жидком виде при Т = –253°С [15]. И то, и другое экономически неэффективно. Возможный вариант решения проблемы безопасного хранения и транспортировки водорода – использование химических соединений с водородом с последующим получением водорода по месту требования [6–15].
В последние годы во многих публикациях рассматривается моногидрат гидразина, который считают не только источником водорода, но и перспективным жидкофазным реагентом для безопасного хранения и транспортировки водорода благодаря высокому содержанию водорода (8.0 вес. %), отсутствию побочных продуктов в случае полного разложения и стабильности в условиях окружающей среды [16–21].
В 70-х гг. прошлого столетия был большой интерес к каталитическому превращению гидразина N2H4 на катализаторе Ir/Al2O3 [22, 23]. За счет разложения гидразина можно создать импульс тяги, достаточный для работы рулевых двигателей космических аппаратов [24, 25].
Другой важной областью применения гидразина является химия и технология актиноидов [26]. Выраженные восстановительные свойства гидразина используются для восстановления актиноидов (U, Np, Pu) и их стабилизации в низших степенях окисления. Наиболее часто эти реакции ведут в присутствии катализатора Pt/SiO2. Восстановление ионов в таком случае сопровождается разложением гидразина [27, 28].
Подробно свойства гидразина и его производных рассмотрены в [29, 30]. Разложение гидразина описывается следующими уравнениями:
Маршрут реакции зависит от катализатора и температуры. Многие каталитические системы способны разлагать гидрат гидразина даже при комнатной температуре, при этом продуктами разложения являются нейтральные азот и водород [31–35].
Целью настоящего обзора является систематизация и анализ литературных данных об активности и селективности различных каталитических систем в процессе разложения гидразина и его производных. Большое внимание в обзоре уделяется рассмотрению данных о механизме образования водорода при низких и высоких температурах. Вопросы практического, технического, технологического использования получаемого при разложении гидразина водорода в обзоре не обсуждаются.
АКТИВНОСТЬ И СЕЛЕКТИВНОСТЬ РАЗЛИЧНЫХ КАТАЛИТИЧЕСКИХ СИСТЕМ
Классическим катализатором разложения гидразина является Shell 405 [36, 37]. В состав катализатора входит около 30 вес. % Ir, нанесенного на γ-Al2O3, с размером кластеров около 2 нм. Отметим, что испытывали разные варианты приготовления иридиевых катализаторов. В частности, катализаторы Ir/Al2O3, включающие от 2 до 35% Ir, приготовленные по альтернативной методике [38, 39], содержали в своем составе IrxOy. Кроме того, упоминаются поликристаллические и монокристаллические Ir(111)-образцы [40–42], а также частицы Ir, нанесенные на углеродные нанотрубки и нанонити [43, 44].
В большой степени результаты исследования иридиевых катализаторов обобщены в [45]. Было показано, что в атмосфере гидразина на Ir/Al2O3 возникают прочносвязанные поверхностные интермедиаты. Из них образуются продукты H2, N2 и NH3, которые обнаруживаются методом ТПД. Высокая прочность поверхностных комплексов требует повышения температуры десорбции. Азот в наиболее сильносвязанном состоянии десорбируется с Тмакс = 380°С. В области температур ниже 300°С образуются N2 и NH3, при Т > 500°С – H2 и N2, т.к. начинается разложение аммиака. Дисперсные кристаллиты Ir проявляют поверхностные свойства, аналогичные поликристаллическому Ir. Сравнение поверхностных свойств свежего и отработанного катализаторов показывает уменьшение активной поверхности после продолжительной выдержки гидразина как в лабораторном реакторе, так и в трастере. Это связано со снижением плотности поверхностных центров, доступных для адсорбции N2H4, без изменения плотности поверхностных состояний. Не было установлено, является ли сокращение площади поверхности результатом термического спекания кристаллитов или появления необратимо адсорбированных поверхностных частиц, которые образуются в атмосфере гидразина. Длительное воздействие атмосферы N2H4 способствует отравлению катализатора из-за примесей в N2H4 или из-за необратимо адсорбированных промежуточных частиц, таких как адсобированные атомы азота, что ведет к потере активности иридиевых центров.
Поскольку Ir достаточно редкий и дорогой элемент, естественным желанием было разработать эффективные, но более дешевые катализаторы этого процесса. Первоначально внимание было обращено на металлы платиновой группы [46, 47]. Реакция эффективно протекает на массивном Rh [48] и нанесенном Rh/Al2O3 [18, 49].
Разложение гидразина также изучалось на металлических тонких пленках [50–52], фольге Ir [53] и на нанесенных системах [18, 19, 54–58].
Сравнительное исследование активности и селективности металлов VIII группы при Т < 180°С проведено в [59]. Обнаружено, что скорость реакции при 100°C меняется в ряду следующим образом (температурный интервал измерений 60–160°С):
Однако с точки зрения селективности по водороду этот ряд металлов выглядит иначе:
Только Pd, Pt, Rh имеют высокую (около 100%) селективность в разложении гидразина до азота и водорода, которая снижается при повышении температуры. На остальных катализаторах гидразин разлагается преимущественно до аммиака и азота и селективность по водороду не превышает 10%.
Во многих работах в реакции разложения гидразина были изучены свойства карбидов, нитридов, фосфидов переходных металлов – как массивных, так и нанесенных, как правило, на оксид алюминия [60–67].
В обзоре [68] отмечается, что за последние два десятилетия исследований гидрат гидразина показал свой большой потенциал в качестве жидкофазного материала для хранения водорода, и приводятся данные для большого числа нанокатализаторов разложения гидрата гидразина с высокой селективностью по водороду. Рассмотрены металлические наночастицы, металл–металлоксидные системы и катализаторы на носителях, а также стратегии повышения селективности по H2. Катализатор на основе Ni–Fe считается одним из идеальных кандидатов, не содержащих благородные металлы.
Многие металлические (Fe, Co, Ni, Cu, Ru, Rh, Ir, Pt и Pd) наночастицы проявляют вполне измеримую активность, но обладают низкой селективностью в образовании водорода при Т < 80°C [12]. Отмечается, что среди металлов наночастицы родия демонстрируют относительно высокую селективность по водороду (44%). Совсем другими свойствами в процессе разложения моногидрата гидразина обладают биметаллические наночастицы, особенно содержащие никель (например, Rh–Ni, Pt–Ni, Pd–Ni, Ni–Ir) [69–80].
Так, в [69] приведены результаты исследования Rh–Ni-наночастиц, приготовленных в виде черни. Выяснилось, что наибольшую селективность (100% при комнатной температуре) проявляют частицы с соотношением Rh : Ni = 4 : 1.
В [70] описываются биметаллические нанокатализаторы Ni1 –xPd, которые характеризуются высокой селективностью по водороду (>80%) при разложении гидразина в водном растворе при 323 K. В работе отмечается, что монометаллические Ni and Pd проявляют низкую активность или вовсе не активны в аналогичных условиях. Это означает, что каталитические свойства биметаллических наночастиц имеют эффект синергизма. Обнаружена корреляция между составом Ni–Pd-катализаторов и селективностью по водороду. Сравнительный анализ катализатора Ni0.6Pd0.4 и монометаллических компонентов (наночастиц Ni и Pd) методами EXAFS и XPS показал, что образование связей между атомами металлов является критическим фактором, определяющим высокую активность и селективности по водороду катализатора Ni0.6Pd0.4 в разложении гидрата гидразина. Присутствие в небольших количества Ir или Pt в биметаллических нанокатализаторах (Ni0.95Ir0.05 и Ni0.93Pt0.07) повышает селективность образования водорода даже при комнатной температуре [71, 72].
Приготовление высокодисперсных биметаллических наночастиц Ni0.6Pt0.4 с размером 2.4 нм описано в [74]. Показано, что присутствие NaOH в процессе приготовления положительно влияет на свойства катализатора, повышая его активность и селективность в разложении гидрата гидразина при комнатной температуре.
В работе [80] предлагается биметаллический катализатор Ni–Ir на основе CeO2, который продемонстрировал не только высокую каталитическую активность и селективность в образовании водорода из N2H4·H2O, но также и уменьшенную потребность в щелочном промоторе по сравнению с Ni–Pt-катализатором.
В [77] описывается недорогой катализатор на основе наночастиц сплава никеля и железа, обладающий 100% селективностью по водороду в реакции разложения гидрата гидразина в щелочном растворе при 343 K. Авторы считают, что созданный катализатор демонстрирует важность и перспективность использования комбинаций различных неблагородных металлов для создания активных нанокатализаторов, свойства поверхности которых (очевидно, из-за эффекта синергизма) будут в большей степени подходить для осуществления процесса разложения гидрата, чем свойства поверхности индивидуальных компонентов.
В последнее время проявляется тенденция к усложнению состава катализаторов. В работе [81] методом поэтапного нанесения были приготовлены триметаллические нанокатализаторы на основе меди с благородными металлами: Cu–Ag–Ir, Cu–Pd–Ir и Cu–Ag–Pd. Показано, что активность триметаллических катализаторов в разложении гидрата гидразина выше, чем биметаллических Cu–Ag, Cu–Ir и Cu–Pd из-за синергетического эффекта и электронных взаимодействий между тремя металлами. Для улавливания аммиака, образующегося в продуктах реакции вместе с водородом, использовался раствор Несслера.
В работе [17] также было продемонстрировано, что триметаллические наносистемы Ni/Fe/Pd, Ni/Fe/Ag, Ni/Fe/Cu более активны для получения водорода и азота в условиях окружающей среды, чем биметаллические Pd/Ag, Ag/Ni, Ni/Fe, тогда как монометаллические аналоги Pd, Ag, Ni, Fe и Cu не обладает реакционной способностью в этом процессе.
Нанокомпозит на основе графена CuNi/ La2O2CO3/rGO проявляет 100% селективность в образовании водорода при 70°C, что объясняют небольшим размером частиц CuNi (~3.2 нм), синергетическим эффектом между частицами металлов и сильным взаимодействием между металлом и носителем. Общая частота оборота катализатора (TOF) достигаeт 114.3 ч–1 [82].
Уникальные свойства никелевых катализаторов продемонстрированы в работах [55, 83–86]. Высокодисперсный никель, нанесенный на Al2O3, обеспечивает 100% конверсию и высокую селективность по водороду в разложении гидрата гидразина при комнатной температуре [84, 85].
В работе [55] катализатор с 13 вес. % Ni0.5Cu0.5/CeO2 показал 100% селективность по H2 в диапазоне температур 30–70°C и в 3.2 раза более высокую частоту оборотов при 50°C, равную 1450.0 ч–1, по сравнению с катализатором Ni/CeO2. Его реакционная способность превосходит таковую большинства известных катализаторов, не содержащих благородных металлов, и даже сопоставима с катализаторами на основе благородных металлов. Катализатор NiFe/Cr2O3 также обладал 100% селективностью по H2 при 70°C, значение TOF для него составило 893.5 ч–1 [86].
Таким образом, Ni – наиболее предпочтительный элемент среди переходных металлов VIII группы, обладающий достаточной активностью и высокой селективностью в образовании H2 из N2H4·H2O. Легирование Ni благородными металлами (такими как Pt, Ir, Rh) представляет собой эффективную стратегию улучшения каталитических свойств. Положительное влияние на селективность по водороду оказывает также взаимодействие металл–носитель и добавление щелочного промотора [68, 83].
Приведенные выше обзорные данные показывают, что в процессе разложения гидразина был исследован широкий круг катализаторов. Важной их характеристикой с точки зрения получения водорода является зависимость селективности процесса по водороду от температуры. По характеру этой зависимости катализаторы можно условно разделить на две группы.
Первый тип (массивный и нанесенный Ir, карбиды, нитриды, фосфиды переходных металлов и т.д.) характеризуется практически нулевой селективностью при низких температурах и возрастанием ее значения до 100% при 500–600°С (рис. 1). Пример такой зависимости можно найти в [62].
Рис. 1.
Селективность образования водорода в реакции разложения гидразина на катализаторе α-Mo2C/γ-Al2O3 с концентрацией Mo2C 8.7 (1), 15.4 (2) и 26.9% (3) и катализаторе 31.6% Ir/γ-Al2O3 (4) в зависимости от температуры и конверсии аммиака (X).

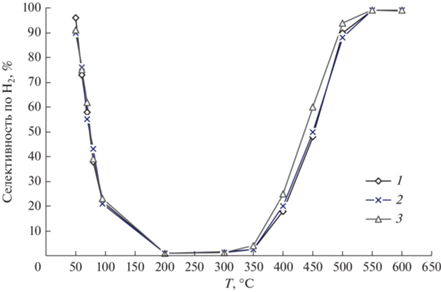
Для второго типа (упомянутые выше наночастицы металлов, такие как Ni, Pd, Pt) характерна высокая селективность при низких температурах, ее падение с повышением температуры и повторный подъем при Т > 300–400°С (рис. 2). В [14] приводятся результаты исследования катализаторов, демонстрирующих оба типа зависимости селективности от температуры. Образцы были приготовлены нанесением на SiO2 металлов VIII группы (Fe, Ru, Co, Rh, Ir, Ni, Pd, Pt) для изучения в реакции разложения гидразина. Измерение активности и селективности проводили в проточном микрореакторе. Все испытанные катализаторы были способны производить водород и азот при Т > 300°С (первый тип катализаторов). Однако образцы, содержащие Ni, Pd и Pt, производили водород и азот и при низких температурах, проявляя значительную селективность по водороду в интервале 30–60°С. Лучшим из них оказался Ni/SiO2, обладавший при высокой активности селективностью более 90% при температурах 30–40 и 500–700°C. Авторы, в частности, выяснили, что увеличение концентрации никеля приводит к росту и активности, и селективности. Этот факт означает, что с повышением содержания никеля в катализаторе механизм реакции не изменяется.
Рис. 2.
Селективность образования H2 на различных катализаторах: 1 − 21% Ni/SiO2 (восстановлен при 500°С); 2 − 21% Ni/Al2O3; 3 − 21% Ni/AC. Конверсия гидразина при 40°С составляет более 90%.
Кроме того, на каталитические параметры влияет также метод синтеза и свойства носителя. В [38] описывается иридиевый катализатор, полученный методом, включающим многократные пропитки с перемешиванием раствора IrCl3 и носителя Al2O3 с последующей сушкой и прокаливанием. Было показано, что увеличение количества пропиток может повлиять на размер и распределение наночастиц Ir, а именно, большее количество пропиток приводило к меньшему размеру частиц. Такой катализатор, в отличие от традиционно приготовленных, проявляет высокую селективность (>90%) по водороду при низких температурах благодаря малому размеру наночастиц Ir.
Отметим, что главный недостаток первого типа катализаторов – слишком высокая температура достижения значительной селективности по водороду. Общим недостатком каталитических систем второго типа является падение активности и селективности при Т > 100–150°С. Чересчур узкий температурный интервал активности характерен для разложения и гидразина, и гидрата гидразина.
Приведенные примеры показывают, что разделение катализаторов на два класса вполне очевидно. По-видимому, на различных катализаторах (или на одном и том же, но по-разному приготовленном) реализуется разный механизм разложения гидразина.
МЕХАНИЗМ ПРОЦЕССА РАЗЛОЖЕНИЯ ГИДРАЗИНА
Активное исследование механизма разложения гидразина проводилось в 70-х гг. прошлого века [22, 23, 87–90], однако ясного представления о механизме сформулировать не удалось. По одной версии считается, что образование продуктов происходит в результате тримолекулярной реакции [87]. По другой – процесс протекает с разрывом связи N–N в молекуле гидразина. На следующем этапе из атомов N и H появляются азот, водород и аммиак. В итоге в [23] была предложена следующая схема процесса, которая демонстрирует одновременное образование водорода, азота и аммиака [23]:
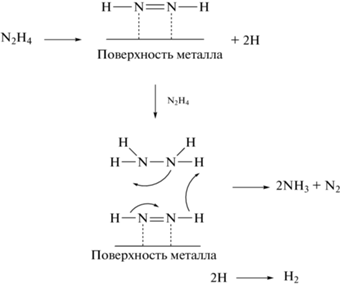
Схема 1 . Предполагаемый механизм образования продуктов каталитического разложения гидразина.
Интересные данные получены при изучении процесса разложения гидразина с использованием меченого гидразина. Так, в [23] определяли состава продуктов при превращении смеси обычного гидразина и изотопа гидразина, меченого по азоту. Опыты проводили в статических условиях в присутствии нанесенных на α-Al2O3 металлов (0.5% Ir, 0.5% Rh, 5% Ni, 1% Pt, 5% Co, 0.5% Ru, 5% Pd, 5% Cu). Выяснилось, что в продуктах реакции отсутствует азот N14N15, а есть только ${\text{N}}_{2}^{{14}}$ и ${\text{N}}_{2}^{{15}}$. Этот факт означает, что в исследованном температурном интервале (65–210°С) в процессе превращения гидразина связь N−N действительно не разрывается, а осуществляется процесс внутримолекулярного дегидрирования гидразина. Возможной причиной этого явления авторы считают потерю поверхностной подвижности реакционных интермедиатов NН2, NН, N. Отсутствие подвижности препятствует перемешиванию изотопов.
Аналогичный результат получен в [87] при изучении разложения гидразина N15 на Fe/MgO в интервале температур 43–365°C. Частицы с массой 28 (14N14N), 29 (14N15N) и 30 (15N15N) определяли масс-спектрометрически. В образце 15N2H4 присутствовало 5.1% 14N15NH4.
В продуктах в качестве возможных интермедиатов разложения гидразина до водорода и азота не обнаружены амид-, имид- и нитрид-частицы. Это означает, что связь N−N в молекуле гидразина остается стабильной. Авторы считают, что аммиак в системе получается в результате протекания вторичной реакции между образовавшимися ранее молекулами водорода и азота.
Экспериментальные результаты, приведенные в [91–95], также свидетельствуют в пользу внутримолекулярного дегидрирования N2H4, приводящего к появлению N2 и Н2. Важным для подтверждения такой точки зрения является обнаружение в термодесорбционных спектрах на поверхности Ni(100) в качестве интермедиата в разложении гидразина частиц диимида N2H2 [91].
Дополнительным подтверждением являются данные, полученные при исследовании взаимодействия гидразина с поверхностью Pt(111) [92]. Авторы предположили, что при низких температурах разрыв связи N−H и сохранение связи N−N связаны с тем, что хемосорбция N2H4 осуществляется через оба атома азота (мостиковая форма адсорбции). Кроме того, на Pt(111) прочность связи водород–металл выше, чем прочность связи азот–металл. В результате барьер для разрыва связи N−H ниже барьера для разрыва связи N−N. Десорбция водорода зависит от заполнения поверхности и температуры и лимитируется как скоростью рекомбинации атомов, так и скоростью разрыва связи N−H.
Приведенные данные показывают, что механизм внутримолекулярного дегидрирования гидразина с образованием водорода и азота действительно существует. Однако детальное стадийное его описание отсутствует. Обычно предлагаются многостадийные схемы (до 20 стадий) [96], основанные на термодесорбционных и кинетических измерениях.
Следует отметить работы, в которых приводятся результаты изучения механизма с помощью ИК-спектроскопии [22, 66, 97–105]. Немногочисленность таких исследований связана с тем, что большинство перечисленных выше катализаторов не обладают подходящими спектральными характеристиками. Так, в [22] ИК-спектральные измерения адсорбции гидразина на Ir/γ-Al2O3 проведены в ограниченном диапазоне волновых чисел (1000–2000 см–1). При комнатной температуре адсорбция гидразина приводит к появлению полос поглощения (п. п.) при 1665, 1620, 1460, 1270 см–1. Полосы при 1665 см–1 соответствуют колебаниям в адсорбированном гидразине, остальные могут быть отнесены к колебаниям как в гидразине, так и в адсорбированном аммиаке. При понижении давления и повышении температуры до 180°С в спектре адсорбированного гидразина наблюдаются те же п. п., что и в спектре адсорбированного аммиака. Такой же результат был получен в [97, 104] при изучении взаимодействия гидразина с поверхностью оксидных систем TiO2 и Cu/TiO2. В итоге был сделан вывод о том, что разложение гидразина на этих системах проходит через промежуточное образование аммиака. Очевидно, что для построения достоверного стадийного механизма процесса необходимо изучение природы и свойств промежуточных комплексов (интермедиатов) с помощью спектрокинетического метода [100], который заключается в одновременном измерении скорости превращения поверхностных комплексов (ИК-спектроскопия) и скорости образования продуктов.
Основные характеристики адсорбции гидразина и моногидрата гидразина суммированы в табл. 1–3.
Таблица 2.
Спектральные характеристики адсорбционных комплексов гидразина на разных носителях
| Катали-затор | Частота п. п., см–1 | Ссылка | ||||||
|---|---|---|---|---|---|---|---|---|
| N−H валентное |
NH2 ножничное |
NH2 ножничное |
NH2 веерное |
NH2 веерное |
N−N валентное |
NH2 маятниковое |
||
| TiO2 | − | − | 1610 | − | − | 1225 | 1180 | [103] |
| TiO2 | 3334 3300 3268 |
1614 | 1591 | 1323 | 1305 | 1143 | 1084 | [97] |
| Ir/Al2O3 | − | 1665 | 1620 | 1460 | 1270 | − | − | [22, 98] |
| CuO/TiO2 | − | 1611 | 1560 | 1350 | 1280 | 1180 | − | [104] |
| MoC | − | 1615 | − | 1460 | − | 1260 | − | [66] |
Таблица 3.
Спектральные характеристики моногидрата гидразина и адсорбционных комплексов моногидрата гидразина на разных носителях
| Катализатор | Частота п. п., см–1 | Ссылка | ||||||
|---|---|---|---|---|---|---|---|---|
| N−H валентное |
NH2 ножнич-ное |
NH2 ножнич-ное |
NH2 веерное |
NH2 веерное |
N−N валент-ное |
NH2 маятни-ковое |
||
| N2H4 гидрат | 3333 3190 |
− | 1609 | − | − | 1282 | 1083 | [105] |
| N2H4 гидрат | 3335 | − | 1587 | 1465 | 1352 | 1142 | − | Настоящая работа |
| N2H4 гидрат Al2O3 |
− | − | − | 1445 | − | 1270 | − | |
| N2H4 гидрат Pd/Al2O3 |
− | 1690 | − | 1460 | − | 1270 | − | |
| N2H4 гидрат 5%Cu/Ce0.8ZrO2 |
− | − | 1530 | 1405 | 1315 | 1270 | − | |
Приведенные данные говорят о том, что при низких температурах в зависимости от типа катализатора реализуются два разных механизма разложения гидразина: через промежуточное образование аммиака и внутримолекулярное дегидрирование, однако их детальное стадийное описание отсутствует.
Механизм образования водорода и азота
Анализ литературы показывает, что для описания разложения гидразина используется набор гипотетических стадий. В качестве примера приведем набор стадий из ранней работы [47]:
Схема построена по результатам исследования кинетики разложения паров гидразина на палладии. Катализатор – палладиевая фольга при 120–165°С, полупроницаемая палладиевая мембрана при 220°С. При 140°С образуется преимущественно азот и водород. Количество гидразина, разлагающегося с появлением аммиака, составляет несколько процентов.
В работе [106] приводится обобщенный набор стадий. Последовательность (I) описывает как раз процесс низкотемпературного образования азота и водорода, в то время как схема (II) отображает образование аммиака и азота.
(I)
$\begin{gathered} {{{\text{N}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}\left( {\text{a}} \right) \to {{{\text{N}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{3}}}}\left( {\text{a}} \right) + {\text{H}}\left( {\text{a}} \right) \to \\ \to {{{\text{N}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{2}}}}\left( {\text{a}} \right) + {\text{2H}}\left( {\text{a}} \right) \to \\ \to {{{\text{N}}}_{{\text{2}}}}{\text{H}}\left( {\text{a}} \right) + 3{\text{H}}\left( {\text{a}} \right) \to {{{\text{N}}}_{{\text{2}}}}\left( {\text{a}} \right) + {\text{4H}}\left( {\text{a}} \right).~~~~~~~ \\ \end{gathered} $(II)
$\begin{gathered} ~{{{\text{N}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}\left( {\text{a}} \right) \to 2{\text{N}}{{{\text{H}}}_{2}}\left( {\text{a}} \right), \\ {\text{N}}{{{\text{H}}}_{{\text{2}}}}\left( {\text{a}} \right) \to {\text{NH}}\left( {\text{a}} \right) + {\text{H}}\left( {\text{a}} \right) \to {\text{N}}\left( {\text{a}} \right) + {\text{2H}}\left( {\text{a}} \right), \\ {\text{N}}{{{\text{H}}}_{{\text{2}}}}\left( {\text{a}} \right) + {\text{H}}\left( {\text{a}} \right) \to {\text{N}}{{{\text{H}}}_{{\text{3}}}}\left( {\text{a}} \right), \\ {{{\text{N}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}\left( {\text{a}} \right) \to {\text{N}}{{{\text{H}}}_{{\text{3}}}}\left( {\text{a}} \right) + {\text{NH}}\left( {\text{a}} \right), \\ {{{\text{N}}}_{2}}{{{\text{H}}}_{4}}\left( {\text{a}} \right) + 2{\text{H}}\left( {\text{a}} \right) \to {{{\text{N}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{5}}}}\left( {\text{a}} \right) + {\text{H}}\left( {\text{a}} \right) \to \\ \to {{{\text{N}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{6}}}}\left( {\text{a}} \right) \to 2{\text{N}}{{{\text{H}}}_{{\text{3}}}}\left( {\text{a}} \right), \\ {{{\text{N}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{2}}}}\left( {\text{a}} \right) + {{{\text{N}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}{\text{(г}}) \to {{{\text{N}}}_{{\text{4}}}}{{{\text{H}}}_{{\text{6}}}}\left( {\text{a}} \right) \to \\ \to 2{\text{N}}{{{\text{H}}}_{{\text{3}}}}\left( {\text{a}} \right) + {{{\text{N}}}_{{\text{2}}}}\left( {\text{a}} \right). \\ \end{gathered} $Здесь индексами (a) и (г) обозначены адсорбированные и газообразные комплексы соответственно.
Получается, что экспериментальные исследования обеспечили информационную базу данных о реакциях образования продуктов каталитического разложения гидразина. Как полагают авторы [106], теоретические исследования необходимы для достижения прогресса в понимании механизма каталитического разложения гидразина. Из расчетных работ можно упомянуть кинетический анализ каталитического разложения N2H4 [107], расчеты DFT адсорбции гидразина на поверхности Ni(100) [108] и Ir(111) [109, 110].
В [109] DFT-расчеты были выполнены для изучения разложения N2H4 на поверхности Ir(111). При этом рассматривался процесс внутримолекулярного и межмолекулярного дегидрирования N2H4. Расчет соответствующих энергетических характеристик показал, что на поверхности Ir(111) крайне маловероятно получение больших количеств N2 через внутримолекулярное дегидрирование при низких температурах вследствие относительно высоких активационных барьеров. Вместе с тем, внутримолекулярный разрыв связи N–N в гидразине протекает легче, чем разрыв связи N–H, в результате может появляться комплекс NH2. Предлагается расчетная последовательность стадий образования азота и водорода (схема (III) ).
| N2H4(a) + N2H4(газ) + Ir6/кластер адсорбция гидразина | 0 ккал/моль | |
| N2H4(a) + N2H4/Ir6/кластер | –42.5 ккал/моль | |
| Седловая точка 1,2-H2 элиминирования | 17.6 ккал/моль | |
| N2H4(a) + H2(газ) + цис-HNNH/Ir6/кластер | –21.0 ккал/моль | (III) |
| Седловая точка 1,2-H2 элиминирования | 32.2 ккал/моль | |
| N2H4(a) + 2H2(газ) + N2/Ir6/кластер десорбция N2 | –33.2 ккал/моль | |
| N2H4(a) + 2H2(газ)+N2(газ) + Ir6/кластер | –17.2 ккал/моль |
Вспомогательную роль в стадиях этой схемы играет молекула N2H4(a). Недостатком механизма является его построение для высоких температур. Нужно отметить, что убедительно обоснованной экспериментально или теоретически стадийной схемы образования азота и водорода при низких температурах к настоящему времени нет.
Механизм образования азота и аммиака
Гораздо более продвинутой выглядит ситуация с разработкой представлений о механизме образования азота и аммиака. Практически единодушно считается, что начальной стадией является разрыв связи N–N с образованием ключевого интермедиата NH2 [110–112].
В [111] методом DFT изучено каталитическое действие относительно нового материала – N4 графена, модифицированного ванадием. Разложение гидразина исследовалось по двумя направлениям: расщепление связи N–N в стадии N2H4 → NH2 + NH2 и расщепление связи N–H в стадии N2H4 → N2H3 + H. Показано, что разрыв связи N–N на модифицированной ванадием поверхности имеет меньшую энергию активации, чем расщепление связи N–H.
Похожее исследование выполнено в работе [112] для катализатора Ni1/C31. Расчеты электронной структуры, основанные на геометрии адсорбции, подтвердили, что активация связи N–N происходит легче, чем активация связей N–H, главным образом из-за более сильного взаимодействия между 2p- и 3d-орбиталями атомов азота и никеля, чем между 1s- и 3d-орбиталями атомов водорода и никеля соответственно.
В упоминавшейся выше теоретической работе [109] расчеты на основе функционала плотности показали, что разложение гидразина на Ir происходит в основном за счет межмолекулярных взаимодействий, причем главным является радикал NH2, который последовательно отрывает атомы водорода от гидразина с образованием аммиака и водорода. Последовательность стадий представлена на схеме (IV) .
| 2Ir/кластер + 3N2H4(газ) адсорбция трех N2H4, первый разрыв NN и миграция | 0.0 ккал/моль | |
| 2(NH2/N2H4/Ir6/кластер) | –139.5 ккал/моль | |
| Седловая точка первого отщепления с помощью NH2 | –140.5 ккал/моль | |
| NH3/N2H3/Ir6/кластер + NH2/N2H4/Ir6/кластер десорбция NH3 и миграция | –148.0 ккал/моль | |
| NH2/N2H3/Ir6/кластер + N2H4/Ir6/кластер + NH3(газ) | –122.8 ккал/моль | |
| Седловая точка второго отщепления с помощью NH2 | –84.2 ккал/моль | |
| NH3/HNNH/Ir6/кластер + N2H4/Ir6/кластер + NH3(газ) десорбция NH3, второй разрыв NN и миграция |
–84.7 ккал/моль | (IV) |
| NH2/HNNH/Ir6/кластер + NH2/Ir6/кластер + 2NH3(газ) | –86.4 ккал/моль | |
| Седловая точка третьего отщепления с помощью NH2 | –43.1 ккал/моль | |
| NH3/NNH/Ir6/кластер + NH2/Ir6/кластер + 2NH3(газ) десорбция NH3, второй разрыв NN и миграция |
–107.8 ккал/моль | |
| NH2/NNH/Ir6/кластер + Ir6/кластер + 3NH3(газ) | –115.4 ккал/моль | |
| Седловая точка четвертого отщепления с помощью NH2 | –48.9 ккал/моль | |
| NH3/N2/Ir6/кластер + Ir6/кластер + 3NH3(газ) десорбция NH3 и N2 | –142.0 ккал/моль | |
| 2Ir6/кластер + N2(газ) + 4NH3(газ) | –108.8 ккал/моль |
В работе [106] похожий механизм реализуется при низких температурах:
При высокой температуре к нему добавляется канал разрыва связи N–H с последующим образованием H2. Сложившиеся представления, как видно, связаны с тем, что при разложении гидразина на Ir(111) маршрут с наименьшей энергией включает в себя N−N-расщепление (прямой барьер ≤ 10 ккал/моль) с появлением поверхностного комплекса NH2, который последовательно отрывает четыре атома H от N2H4, проходя весь путь до N2. Первый отрыв, по существу, не имеет барьера, а у остальных трех седловая точка энергии ниже, чем у газофазных реагентов. Кроме того, барьер разрыва связи N−N в адсорбированном состоянии значительно меньше по сравнению с энергией связи (65 ккал/моль) в газовой фазе. В итоге, сомнений в способности катализатора подавлять поверхностно-радикальный характер процесса не остается. Последовательное отщепление атома H от четырех радикалов NH2 прекрасно объясняет низкотемпературный маршрут 3N2H4 → 4NH3 + N2.
Высокотемпературный маршрут N2H4→ N2 + + 2H2 более неоднозначный. Установлено, что начальный разрыв связи N−H сначала в гидразине, а затем в N2H3 с образованием поверхностных атомов H имеют суммарный барьер немного ниже, чем у реагентов газовой фазы. Четыре таких N−H-разрыва могли бы произвести два H2 и N2. Однако в таком случае число требуемых стадий диссоциации, скачков и рекомбинаций велико. Был предложен более короткий молекулярный маршрут через вспомогательные 6-центровые седловые точки. Эти два процесса вполне могут конкурировать в высокотемпературном интервале, и они не являются взаимоисключающими. Первые два атома водорода, безусловно, легко удаляются из гидразина путем отщепления от NH поверхностных атомов H с последующей рекомбинацией H + H. Последние два водорода в адсорбированном цис-HNNH направлены наружу от поверхности, и их удаление с помощью вспомогательных молекулярных образований конкурирует с радикальным отщеплением последних двух атомов водорода.
ЗАКЛЮЧЕНИЕ
В процессе разложения гидразина испытано большое количество разнообразных каталитических систем. Исследователи руководствовались желанием создать катализатор эффективный, но менее дорогой, чем иридиевый.
Важной характеристикой катализатора с точки зрения получения водорода является зависимость селективности процесса по водороду от температуры. По характеру этой зависимости катализаторы делятся на две группы.
Причина такого различия свойств разных катализаторов в одной реакции к настоящему времени неизвестна. Можно полагать, что главную роль в определении направлении процесса играет форма активации гидразина. Если поверхность катализатора устроена так, что адсорбция гидразина происходит в мостиковой (через оба атома азота) форме, следует ожидать ослабления связи N−N и ее разрыва. При этом образуется комплекс NH2, играющий ключевую роль в низкотемпературном образовании азота и аммиака.
Если активная поверхность устроена так, что гидразин адсорбируется в линейной форме, в системе протекает внутримолекулярное дегидрирование с образованием водорода и азота при низкой температуре.
Теоретические и экспериментальные результаты показывают, что гидразин и моногидрат гидразина являются вполне перспективными и для запасания водорода, и для получения свободного от примесей водорода для эффективной работы топливных элементов. Нужно заметить, что широкое экономическое применение процесса станет возможным только после решения проблемы активации инертного азота для использования в различных химических процессах [113, 114].
Список литературы
Hydrogen and other Alternative Fuels for Air and Ground Transportation. Ed. Pohl H.W. UK. Wiley, 1995. 206 p.
Deng Z.-Y., Ferreira J.M.F., Sakka Y. // J. Am. Ceram. Soc. 2008. V. 91. P. 3825.
Nikolaidis P., Poullikkas A. // Renew. Sustain. Energy Rev. 2017. V. 67. P. 597.
Чесноков В.В., Лисицын А.С., Соболев В.И., Герасимов Е.Ю., Просвирин И.П., Чесалов Ю.А., Чичкань А.С., Подъячева О.Ю. // Кинетика и катализ. 2021. Т. 62. С. 472.
Khan Z. // Int. J. Hydrogen Energy. 2019. V. 44. P. 1150.
Lang C., Jia Y., Yao X. // Energy Storage Mater. 2020. V. 26. P. 290.
Ekinci A. // Кинетика и катализ. 2020. Т. 61. С. 540.
Al-Thabaiti S.A., Khan Z., Malik M.A. // Int. J. Hydrogen Energy. 2019. V. 44. P. 16452.
Jiang H.L., Singh S.K., Yan J.M., Zhang X.B., Xu Q. // Chem. Sus. Chem. 2010. V. 3. P. 541.
Yadav M., Xu Q. // Energy Env. Sci. 2012. P. 9698.
Lan R., Irvine J.T.S., Tao S. // Int. J. Hydrog. Energy. 2012. V. 37. P. 1482.
Singh S.K., Xu Q. // Catal. Sci. Technol. 2013. V. 3. P. 1889.
Song J., Ran R., Shao Z. // Int. J. Hydrogen Energy. 2010. V. 35. P. 7919.
Zheng M., Cheng R., Chen X., Li N., Li L., Wang X., Zhang T. // Int. J. Hydrogen Energy. 2005. V. 30. P. 1081.
Schlapbach L., Züttel A. // Nature. 2002. V. 414. P. 353.
Qiu Y.-P., Wang W.-Z., Chen M.-H., Shi Q., Yang Z.-Q., Wang P. // J. Mater. Chem. A. 2021. V. 9. P. 18385.
Al-Thubaiti K.S., Khan Z. // Int. J. Hydrogen Energy. 2020. V. 45. P. 13960.
Firdous N., Janjua N.K., Wattoo M.H.S. // Int. J. Hydrogen Energy. 2020. V. 45. P. 21573.
Motta D., Barlocco I., Bellomi S., Villa A., Dimitratos N. // Nanomater. 2021. V. 11. P. 1340.
Huang W., Liu X. // Fuel. 2022. V. 315. Article 123210.
Qiu Y.-P., Cao G.-X., Wen H., Shi Q., Dai H., Wang P. // Int. J. Hydrogen Energy. 2019. V. 44. P. 15110.
Contour J.P., Pannetier G. // J. Catal. 1972. V. 24. P. 434.
Maurel R., Menezo J.C. // J. Catal. 1978. V. 51. P. 293.
Janjua N.K., Firdous N., Bhatti A.S., Khan Z.S. // Appl. Catal. A: Gen. 2014. V. 479. P. 9.
Vieira R., Bastos-Netto D., Ledoux M.-J., Pham-Huu C. // Appl. Catal. A: Gen. 2005. V. 279. P. 35.
Колтунов В.С. Кинетика реакций актиноидов. Москва: Атомиздат, 1974. 312 с.
Anan’ev A.V., Broudic J.-C., Brossard Ph. // Appl. Catal. A: Gen. 2003. V. 242. P. 1.
Ананьев А.И., Тананаев И.Г., Шилов В.П. // Успехи химии. 2005. Т. 74. С. 1132.
Hydrazine and its Derivatives, 2nd edition. Ed. Schmidt E.W. New York: Wiley Interscience, 2001. 2232 p.
Ewald E. // Int. J. Hydrogen Energy. 1998. V. 23. P. 803.
Manukyan K.V., Cross A., Rouvimov S., Miller J., Mukasyan A.S., Wolf E.E. // Appl. Catal. A: Gen. 2014. V. 476. P. 47.
Singh S.K., Zhang X.-B., Xu Q. // J. Am. Chem. Soc. 2009. V. 131. P. 9894.
Singh S.K., Zhang X.-B., Xu Q. // Europ. J. Inorg. Chem. 2011. V. 14. P. 2232.
Jain P., Anila K.A., Vinod C.P. // Chem. Select. 2019. V. 4. № 9. P. 2734.
Tong D.G. // J. Mater. Chem. A. 2019. V. 7. P. 20442.
Schulz-Ekloff G., Hoppe R. // Catal. Lett. 1990. V. 6. P. 383.
Soares Neto T.G., Cobo A.J.G., Cruz G.M. // Appl. Catal. A: Gen. 2003. V. 250. № 2. P. 331.
Cho S.J., Lee J., Lee Y.S., Kim D.P. // Catal. Lett. 2006. V. 109. P. 181.
Jang Y.B., Kim T.H., Sun M.H., Lee J., Cho S.J. // Catal. Today. 2009. V. 146. P. 196.
Sawin H.H., Merrill R.P. // J. Chem. Phys. 1980. V. 73. P. 996.
Smith O.I., Solomon W.C. // Ind. Eng. Chem. Fundam. 1982. V. 21. P. 374.
Papapolymerou G.A.-P., Botis A.G., Papargyris A.D., Spiliotis X.D., Kasidakis D. // React. Kinet. Catal. Lett. 1993. V. 49. P. 339.
Prasad V., Vasanthkumar M.S. // J. Nanoparticle Res. 2015. V. 17. P. 1.
Vieira R., Pham-Huu C., Keller N., Ledoux M.J. // Chem. Commun. 2002. V. 9. P. 954.
Falconer J.L., Wise H. // J. Catal. 1976. V. 43. P. 220.
Block J. // Z. Phys. Chem. N. F. 1972. V. 82. P. 1.
Хоменко А.А., Аппельбаум Л.О. // Кинетика и катализ. 1976. Т. 17. С. 691.
Prasad J., Gland J.L. // Langmuir. 1991. V. 7. P. 722.
Davis K.M.C., Sayer C.F. // J. Chem. Soc. Faraday Trans. 1972. V. 68. P. 1884.
Contaminard R.C.A., Tompkins F.C. // J. Chem. Soc. Faraday Trans. 1971. V. 67. P. 545.
Cosser R.C., Tompkins F.C. // J. Chem. Soc. Faraday Trans. 1971. V. 67. P. 526.
Al-Haydari Y.K., Saleh J.M., Matloob M.H. // J. Phys. Chem. 1985. V. 89. P. 3285.
Wood B.J., Wise H. // J. Catal. 1975. V. 39. P. 471.
Kang W., Varma A. // Appl. Catal. B: Env. 2018. V. 220. P. 409.
Kang W., Varma A. // Appl. Catal. B: Env. 2019. V. 249. P. 54.
Dai H.B., Zhong Y.J., Kang Q., Sun L.X., Wang P. // Int. J. Hydrogen Energy. 2017. V. 42. P. 5684.
Qiu Y.-P., Wang W.-Z., Chen M.-H., Shi Q., Yang Z., Wang P. // J. Mater. Chem. A. 2021. V. 9. P. 18385.
Liu X., Liu Y., Wang J., Ma J. // Ind. Eng. Chem. Res. 2022. V. 61. P. 1636.
Maurel F., Menezo J.C., Barrault J. // J. Chim. Phys. 1973. V. 70. P. 1221.
Chen X., Zhang T., Ying P., Zheng M., Wu W., Xia L., Li T., Wanga X., Li C. // Chem. Commun. 2002. V. 8. P. 288.
Chen X., Zhang T., Zheng M., Wu Z., Wu W., Li C. // J. Catal. 2004. V. 224. P. 473.
Chen X., Zhang T., Zheng M., Xia L., Li T., Wu W., Wang X., Li C. // Ind. Eng. Chem. Res. 2004. V. 43. P. 6040.
Liang C., Ding L., Wang A., Zhiqiang M., Qiu J., Zhang T. // Ind. Eng. Chem. Res. 2009. V. 48. P. 3244.
Santos J.B.O., Valeca G.P., Rodrigues R.J.A.J. // J. Catal. 2002. V. 210. P. 1.
Zheng M., Chen X., Cheng R., Li N., Sun J., Wang X., Zhang T. // Catal. Commun. 2006. V. 7. P. 187.
Cheng R., Shu Y., Zheng M., Li L., Sun J., Wang X., Zhang T. // J. Catal. 2007. V. 249. P. 397. https://doi.org/10.1016/j.jcat.2007.04.007
Mnatsakanyan R., Zhurnachyan A.R., Matyshak V.A., Manukyan K.V., Mukasyan A S. // J. Phys. Chem. Solids. 2016. V. 96–97. P. 115.
Cheng Y., Wu X., Xu H. // Sustain Energy Fuel. 2019. V. 3. P. 343.
Singh S.K., Xu Q. // J. Am. Chem. Soc. 2009. V. 131. № 50. P. 18032.
Singh S.K., Iizuka Y., Xu Q. // Int. J. Hydrog. Energy. 2011. V. 36. P. 11794.
Singh S.K., Xu Q. // Chem. Commun. 2010. V. 46. № 35. P. 6545.
Singh S.K., Xu Q. // Inorg. Chem. 2010. V. 49. P. 6148.
Wang J., Zhang X.-B., Wang Z.-L., Wang L.-M., Zhang Y. // Energy Env. Sci. 2012. V. 5. P. 6885.
Singh A.K., Xu Q. // Int. J. Hydrog. Energy. 2014. V. 39. № 17. P. 9128.
Du Y., Su J., Luo W., Cheng G. // ACS Appl. Mater. Interfaces. 2015. V. 7. P. 1031.
Jiang Y., Kang Q., Zhang J., Dai H.-B., Wang P. // J. Power Sources. 2015. V. 273. P. 554.
Singh S.K., Singh A.K., Aranishi K., Xu Q. // J. Am. Chem. Soc. 2011. V. 133. P. 19638.
Zhou L., Luo X., Xu L., Wan C., Ye M. // Catalysts. 2020. V. 10. P. 930.
Qiu Y.-P., Shi Q., Zhou L.-L., Chen M.-H., Chen C., Tang P.-P., Walker G.S., Wang P. // ACS Appl. Mater. Interfaces. 2020. V. 12. № 16. P. 18617.
Dai H., Zhong Y., Wang P. // Progress in Natural Sci: Mater. Int. 2017. V. 27. P. 121.
Khan Z., AL-Thabaiti S.A., Rafiq M.Z.A. // Int. J. Hydrogen energy. 2021. V. 46. P. 39754.
Hong X., Yao Q., Long J., Li X., Chen X., Lu Z.-H. // Ind. Eng. Chem. Res. 2021. V. 60. P. 16224.
He L., Liang B., Huang Y., Zhang T. // Natl. Sci. Rev. 2018. V. 5. P. 356.
He L., Huang Y., Wang A., Wang X., Chen X., Delgado J.J., Zhang T. // Angew. Chem. – Int. Ed. 2012. V. 51. P. 6191.
He L., Huang Y., Wang A, Liu Y., Liu X., Chen X., Delgado J.J., Wang X., Tao Zhang // J. Catal. 2013. V. 298. P. 1.
Chen J., Zou H., Minghong Q.Y., Li L.X., Lu Z.-H. // Appl. Surf. Sci. 2020. V. 501. ID 144247.
Block J., Schulz-Ekloff G. // J. Catal. 1973. V. 30. P. 327.
Aika K.I., Ohhata T., Ozaki A. // J. Catal. 1970. V. 19. P. 140.
Gosser R.C., Tompkins F.C. // Trans. Faraday Soc. 1971. V. 67. P. 545.
Davis K.M.C., Sayer S.F. // Trans. Faraday Soc. 1972. V. 68. P. 1884.
Huang S.X., Rufael T.S., Gland J.L. // Surf. Sci. Lett. 1993. V. 290. P. L673–L676.
Alberas D.J., Kiss J., Liu Z.-M., White J.M. // Surf. Sci. 1992. V. 278. P. 51.
Wagner M.L., Schmidt L.D. // Surf. Sci. 1991. V. 257. P. 113.
Bu Y., Shinn D.W., Lin M.C. // Surf. Sci. 1992. V. 276. P. 184.
Prasad J., Gland J.L. // Surf. Sci. 1991. V. 258. P. 67.
Kolinko P.A., Kozlov D.V., Vorontsov A.V., Preis S.V.A. // Catal. Today. 2007. V. 122. P. 178.
Chuang C.-C., Shiu J.-S., Lin J.-L. // Phys. Chem. Chem. Phys. 2000. V. 2. P. 2629.
Contour J.P., Pannetier G. // Bull. Soc. Chim. France. 1970. P. 4260.
Matyshak V.A., Krylov O.V. // Catal. Today. 1995. V. 25. P. 1.
Sacconi L., Sabatini A. // J. Inorg. Nucl. Chem. 1963. V. 25. P. 1389.
Durig J.R., Bush S.F., Mercer E.E. // J. Chem. Phys. 1965. V. 44. P. 4238.
Sathyanarayana D.N., Nicholls D. // Spectrochim. Acta. Part A. 1978. V. 34. P. 263.
Amores J.M.G., Escribano V.S., Ramis G., Busca G. // Appl.Catal. B: Env. 1997. V. 13 P. 45.
Ramis G., Li Yi, Busca G. // Catal. Today. 1996. V. 28. P. 373.
Firdous N., Janjua N.K., Qazi I., Wattoo M.H.S. // Int. J. Hydrogen Energy. 2016. V. 41. P. 984.
Zhang P.-X., Wang Y.-G., Huang Y.-Q., Zhang T., Wu G.-S., Li J. // Catal. Today. 2011. V. 165. P. 80.
de Medeiros J.E., Valenc G.P. // Braz. J. Chem. Eng. 1998. V. 15. P. 126.
Agusta M.K., David M., Nakanishi H., Kasai H. // Surf. Sci. 2010. V. 604. P. 245.
Schmid M.W., Gordon M.S. // Z. Phys. Chem. 2013. V. 227. P. 1301.
Lu X., Francis S., Motta D., Dimitratos N., Roldan A. // Phys. Chem. Chem. Phys. 2020. V. 22. P. 3883.
Küçük H., Akca A. // Comput. Theor. Chem. 2022. V. 1208. Article 113539.
Genç A.E., Küçük H., Alp I.O., Akça A. // Int. J. Hydrogen Energy. 2020. V. 58. P. 33407.
Shilov A.E. // Pure Appl. Chem. 1992. V. 64. № 10. P. 1409.
Матышак В.А., Караваев М.М. // Химическая промышленность. 1998. № 9. С. 537.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ