

Кинетика и катализ, 2022, T. 63, № 4, стр. 418-432
Методы исследования реакций, протекающих на цеолитных катализаторах по механизму “hydrocarbon pool”
Е. С. Матвеенко a, М. В. Григорьев b, Т. А. Кремлева a, Е. В. Андрусенко a, b, Н. А. Косинов c, *
a ФГАОУ ВО Тюменский государственный университет, группа TsyfroCatLab
625003 Тюмень, ул. Володарского, 6, Россия
b Национальный исследовательский университет ИТМО, группа TheoMAT
191002 Санкт-Петербург, ул. Ломоносова, 9, Россия
c Eindhoven University of Technology, 5600 MB
PO Box 513 Eindhoven, Netherlands
* E-mail: N.A.Kosinov@tue.nl
Поступила в редакцию 03.11.2021
После доработки 15.03.2022
Принята к публикации 15.03.2022
- EDN: DVCQZP
- DOI: 10.31857/S0453881122040062
Аннотация
В настоящей работе рассмотрены особенности протекания каталитических реакций по механизму “hydrocarbon pool” на цеолитных катализаторах. Выделены и проанализированы основные инструментальные методы анализа, которые позволяют характеризовать изменения структуры катализаторов и выделять ключевые интермедиаты в ходе каталитического процесса. Отражены возможности и ограничения применения различных методов. В заключение обсуждены перспективные направления в изучении каталитических реакций данного типа.
ВВЕДЕНИЕ
Современная экологическая обстановка требует оптимизации технологических процессов с целью снижения выбросов в окружающую среду. Для эффективного решения этой проблемы можно выделить несколько основных направлений исследований: разработка альтернативных источников энергии, поиск оптимальных условий производства, совершенствование процессов с возможностью использования безотходных технологий и вторичного сырья. Данные тенденции являются актуальными и для нефтехимической отрасли в рамках модернизации добычи, переработки нефти и дальнейшей эксплуатации ее продуктов. Особое внимание сосредоточено на создании химических процессов, способных в будущем компенсировать недостаток нефти. Для решения химических аспектов большинства из представленных проблем широко применяются цеолитные катализаторы. Уникальные свойства цеолитов делают их незаменимыми для таких каталитических процессов как переработка нефти (крекинг, алкилирование, гидроизомеризация) и биомассы (лигниновые и целлюлозные фракции), а также депарафинизация тяжелых фракций нефти (топлива и масла) [1]. Помимо этого, особое внимание привлекают процессы, протекающие по механизму “hydrocarbon pool”: конверсия метанола и других органических веществ (фуран, метилгалогениды, метантиол) в высшие углеводороды, что обусловлено их практической значимостью (табл. 1).
Получаемые в результате олефины и ароматические соединения посредством окисления, дегидрирования, алкилирования могут служить источниками прекурсоров для фармацевтичекой, полимерной, текстильной промышленности [9]. Установки, предназначенные для конверсии природного газа в олефины через метанольный интермедиат, постепенно внедряются в эксплуатацию. Известно о функционировании установок следующих фирм: “ExxonMobil” (США), “UOP” (США), “Lurgi” (Германия), “Sinopec” (Китай) [10]. В связи с широким применением цеолитов в вышеупомянутых процессах и современным подходом к конверсии ряда органических веществ в углеводороды необходимо иметь возможность разрабатывать катализаторы с определенным набором свойств, отслеживать их селективность, эффективность и активность в ходе реакции, а также контролировать процесс деактивации, который является одной из главных проблем цеолитных катализаторов. Современные методы, такие как ультрафиолетовая и инфракрасная спектроскопия, спектроскопия комбинационного рассеяния и ядерного магнитного резонанса, хромато-масс-спектрометрия, а также термогравиметрия и рентгеновская дифракция, позволяют детально изучать не только изменение физико-химических свойств катализатора, но и определять детальные механизмы реакций. Тонкости применения этих методов и их возможности рассмотрены в настоящем обзоре с учетом специфики каталитических процессов, проходящих в присутствии цеолитных катализаторов.
Исключительные особенности цеолитов
На сегодняшний день цеолиты занимают главенствующую роль в процессах, лежащих в основе нефтехимического производства. Они относятся к тектосиликатам и имеют ряд полезных свойств, таких как распространенность в земной коре, нанопористость (размер пор 0.3–2 нм), термическая и химическая стабильность, широкий спектр структурных и химических особенностей, а также возможность к регенерации [11]. В настоящее время в базе данных международной цеолитной ассоциации (International Zeolite Association) [12] перечислено более 230 топологий цеолитов. Разнообразие цеолитных архитектур проявляется в различных размерах пор, размерности системы каналов (от 0D до 3D) и других структурных свойствах. Зачастую эти характеристики позволяют контролировать протекание химических реакций в микропористом пространстве цеолитов [13]. Контроль становится возможным благодаря селективности по форме. Протекание химической реакции внутри цеолитов зависит от того, насколько свободно реагенты, интермедиаты и продукты реакции могут диффундировать внутрь микропор и на поверхность катализатора [14]. Так, явление формаселективности было продемонстрировано при изучении конверсии метантиола в олефины (MtTO). Для этого проводили сравнение возможности применения цеолита H-ZSM-5 (топология MFI), имеющего трехмерные микропоры 10MR, и H-SSZ-13 (топология CHA), имеющего размерность 8MR, к процессу MtTO и конверсии метанола в олефины (МТО). Распределения продуктов превращения CH3OH и CH3SH на H-ZSM-5 сильно различались, использование же H-SSZ-13 обеспечивало схожую селективность по легким олефинам в обеих реакциях. Стоит отметить, что снижение конверсии CH3SH с течением времени для H-SSZ-13 привело к уменьшению выхода легких олефинов и более высокой общей селективности образования коксовых отложений. Несмотря на схожесть распределения продуктов, полученных в реакциях MTO и MtTO на H-SSZ-13, скорость его дезактивации в реакции MtTО выше, что связано с более быстрым образованием дезактивирующих частиц, которые блокируются внутри пор катализатора из-за их размера и не могут диффундировать на внешнюю поверхность [4, 15].
Структура типичных цеолитов в основном состоит из тетраэдрических блоков [SiO4]. Путем внедрения в нее катионов трехвалентных элементов (Al3+, B3+, Fe3+) вместо тетравалентного кремния возможно создавать отрицательный заряд, который требует наличия компенсирующего катиона, а также обеспечивает бифункциональность катализатора [16]. Отрицательный заряд цеолитного каркаса может быть компенсирован протоном, что приводит к образованию кислых мостиковых гидроксильных групп (Si–(OH)–Al) [11, 14]. Таким образом, данная структурная особенность цеолитов позволяет варьировать их кислотные свойства. Примером может служить работа Р.И. Кузьминой с соавт. [17], где отмечается влияние внедрения различных типов металлов и неметаллов на силу кислотных центров цеолита. Так, введение в структуру цеолита кобальта и меди уменьшает кислотность активных центров и делает невозможным протекание конверсии этанола в жидкие углеводороды, а внедрение фосфора и циркония приводит к росту каталитической активности, конверсии и селективности образования пропилена [17].
Другим преимуществом цеолитов является относительно высокая (гидро-)термическая стабильность, которая позволяет проводить реакции в агрессивных условиях как в газовой, так и в жидкой фазах [18, 19]. Благодаря этим свойствам цеолиты играют важную роль во многих процессах, включающих адсорбцию, разделение, ионный обмен и катализ [20]. Сочетание селективности по форме, сильной кислотности, возможности стабилизировать активные центры делают цеолиты практичными каталитическими материалами, играющими определяющую роль в процессе НСР.
Механизм “hydrocarbon pool”
Уникальная реакционная способность цеолитов наблюдается в процессах, идущих по механизму “hydrocarbon pool” (HCP). HCP лежит в основе превращения метанола в углеводороды (МТН) и олефины (МТО) [2, 21, 22]. Аналогичные механизмы установлены и для протекающих на цеолитах превращениях метилгалогенидов [3] и метантиола [4] в высшие углеводороды, конверсии этилена в пропилен [5], ароматизации фуранов [6] и легких алканов [7, 8]. Общие особенности химизма реакций, идущих по механизму НСР, заключаются в появлении сокаталитических углеводородных интермедиатов внутри микропористой структуры цеолитов, наличии индукционного периода, когда происходит образование этих интермедиатов, а также сильной зависимости стабильности интермедиатов и продуктов от топологии пор катализатора [6]. В исследованиях отмечается, что основными реакционноспособными катионными интермедиатами являются полизамещенные ароматические соединения (например, метилированные циклопентенильные и гексаметилбензольные катионы) [11, 23]. Фактически данные структуры могут выступать в качестве органических сокатализатаров, в чем и состоит их определяющая роль [5]. При этом наблюдается “confinement effect” (эффект ограничения), суть которого заключается в возможности влияния на стабильность промежуточных продуктов и, соответственно, на селективность всей реакции путем варьирования размеров пор и кислотности цеолитного катализатора [24]. Стоит отметить, что нецеолитные катализаторы не проявляют активности в вышеприведенных реакциях. Таким образом, процессы, идущие по механизму HCP, могут осуществляться только на цеолитах.
Тонкости механизма HCP изучаются на протяжении последних 30-ти лет. В исследованиях подчеркивается главенствующая роль свойств цеолитных катализаторов в активации исходных веществ и косвенное каталитическое воздействие цеолитов [2]. При этом сама реакция протекает между молекулами, стабилизированными в порах цеолита (органическими сокатализаторами) и находящимися в газовой фазе. Процесс НСР проходит через следующие основные стадии: короткий индукционный период, сопровождающийся возникновением первых С–С-связей, стационарное образование углеводородов в концепции двойного цикла, включающего олигомеризацию, метилирование, деалкилирование, дегидрирование и дегидратацию инкапсулированных молекул карбокатионных/радикальных интермедиатов, представляющих собой активные центры, обуславливающие автокаталитический характер процесса и определяющие путь появления целевых продуктов [25].
Рассмотрим принцип механизма HCP на примере процесса конверсии метанола в углеводороды, в котором можно выделить два параллельных каталитических цикла. Алкены образуются как одни из первичных продуктов С–С-сочетания, затем метилируются метанолом и впоследствии подвергаются крекингу в одном цикле (алкеновый цикл), в то время как ароматические углеводороды метилируются и впоследствии деалкилируются в другом (ареновый цикл), подвергаясь изомеризации и трансалкилированию [26, 27]. В олефиновом цикле наблюдается рост цепи с образованием высших алкенов, которые способны переносить водород, обуславливая появление алканов и ароматических углеводородов (схема 1 ) [28].

Схема 1 . Механизм HCP с двойным циклом, предложенный для процесса превращения метанола в углеводороды катализируемого цеолитами. Пунктирная линия указывает на процесс переноса водорода.
Таким образом, взаимосвязь двух циклов осуществляется посредством реакций гидридного переноса с образованием алканов.
Влияние коксообразования на активность катализаторов
Помимо проблемы контроля активности и селективности каталитических реакций, протекающих по механизму HCP, такие процессы осложнены появлением кокса, который вызывает дезактивацию катализаторов [29]. Термин “кокс” используется для описания смеси высококипящих ароматических, алифатических и графитоподобных углеродистых соединений, формирующихся как внутри пор цеолита, так и на внешней поверхности [30]. Образование продуктов уплотнения в цеолитных катализаторах описывается моделью “горящей сигары” и подразумевает неоднородную дезактивацию, которая протекает от входа к выходу из слоя катализатора. Наиболее ярко признаки дезактивации выражены во входной части реактора (наблюдается окрашивание в черный цвет); средняя зона, в которой протекает конверсия метанола отличается серой окраской; наименее дезактивированная зона, имеющая белый цвет, находится в выходной части реактора [31]. Более стабильная работа катализатора при протекании процессов с образованием коксовых отложений в ходе реакций на сегодняшний день достигается за счет использования реакторов с кипящим слоем, что обеспечивает равномерное закоксование катализатора по всему объему и лучший отвод тепла [32, 33]. Помимо этого, с процессом дезактивации цеолитных катализаторов в промышленности борются посредством регенерации, например, путем сжигания кокса во время циклического процесса, в ходе которого катализатор непрерывно циркулирует между реактором и регенератором [34]. Для разработки новых подходов к решению проблемы дезактивации необходимо понимание химических тонкостей механизмов, определяющих активность и селективность реакций HCP. При этом следует иметь представления не только о природе промежуточных и конечных продуктов реакции, но и о состоянии активных центров катализатора и изменениях их структуры во времени. Ниже мы обсудим основные способы изучения реакций, протекающих по механизму “hydrocarbon pool”.
ИССЛЕДОВАНИЕ ПРОЦЕССОВ HCP
Спектроскопия (УФ, КР, ИК, ЯМР)
Различные виды спектроскопии достаточно часто применяют для изучения каталитических реакций [35]. Среди них можно выделить ультрафиолетовую (УФ) спектроскопию, в основе которой лежит использование фотонов в диапазоне ультрафиолетового и видимого света, возбуждающих электронные переходы в атомах и молекулах. Типичные функциональные группы, которые могут быть идентифицированы УФ-спектроскопией, включают сопряженные двойные связи, ароматические кольца, карбокатионы [36]. Исследования с помощью УФ-спектроскопии позволяют выявить полициклические ароматические вещества, такие как полиметилантрацены, диенильные и триенильные карбокатионы, диены и полиалкилароматические вещества [37, 38]. Например, применяя данный метод при изучении реакции МТО в режиме операндо, Qian Qingyun и др. [38] наблюдали изменения в составе промежуточных продуктов при повышении температуры. Авторы пришли к выводу о том, что метилирование активных центров боковой цепи при реализации механизма HCP способствует дезактивации цеолитного катализатора и ведет к образованию прекурсоров кокса, а не к получению олефинов (рис. 1) [38].
Рис. 1.
УФ-спектры катализатора H-SAPO-34, полученные в режиме операндо в течение периода запрограммированной по температуре реакции MTO.

Высокая чувствительность метода УФ-спектроскопии к олефиновым и ароматическим соединениям ограничивает возможность его использования на начальных стадиях реакции [39, 40].
Решить эту проблему можно с помощью инфракрасной (ИК) спектроскопии, которая широко применяется для исследования структурных особенностей соединений, образующихся при начальном контакте метанола с цеолитами, таких как метоксильные группы диметилового эфира [41, 42]. ИК-спектроскопия относится к абсорбционным спектроскопическим методам и предусматривает использование излучения в инфракрасной области для анализа молекулярных колебаний, существующих в образце. ИК-спектроскопия обычно служит для определения содержания органических веществ, дает более интенсивные сигналы поверхностных функциональных групп (CO, CH3О, пиридин и т.д.) и поверхностных гидроксилов (M–OH) [42]. Также этот метод применяется для изучения химических свойств каталитически активных центров (окислительно-восстановительных характеристик, проявление свойств оснований и кислот Льюиса или Бренстеда) [43, 44]. Использование синхротронной инфракрасной микроскопии для идентификации изменений, происходящих на начальной стадии реакции МТН, продемонстрировано в работе Minova и др. [45]. Исследование было посвящено изучению образования первых углерод-углеродных связей, алкенов, а также ключевых олигомерных и диметилциклопентенильных катионов – основных интермедиатов НСР (рис. 2a). ИК-спектроскопия позволила зафиксировать начальный этап возникновения С–С-связей из метанола, а также депротонирование первоначально сформировавшихся поверхностных метоксильных групп. Анализ ИК-спектров показал, что олигомеры олефинов и диметилциклопентенильные катионы можно рассматривать как первые интермедиаты НСР, получение из которых алкенов и метилзамещенных ароматических соединений протекает более эффективно, чем в результате прямого процесса образования С–С-связей, наблюдаемого на начальных стадиях реакции [45]. Основной проблемой при использовании метода ИК-спектроскопии является появление кокса, сильно поглощающего ИК-излучение. В результате анализ катализатора на более поздней стадии реакции становится затруднительным [22, 46, 47].
Рис. 2.
а – ИК-спектры, записанные с интервалом 2 с (показаны первые 6 сканов) после импульсного ввода 2 мл пропена в поток N2 над кристаллом HZSM-5 при 523 К. Черная кривая представляет собой окончательный спектр в серии через 400 с после пульса пропена. На вставках: расширение области CH-колебаний (слева) и изменение интенсивности полос 2960, 2870 и 1510 см–1 в зависимости от времени (справа). б – Спектры комбинационного рассеяния катализатора 0.5Pt1Sn/SBA-100 при различном времени протекания реакции.

В то время как с помощью ИК-спектроскопии возможно обнаружить колебания, обусловленные изменением дипольного момента, спектроскопия комбинационного рассеяния (КР) чувствительна к колебаниям, связанным с изменением поляризуемости [48]. Достоинство данного метода заключается в возможности анализировать оптически непрозрачные образцы, а также фиксировать нужные колебания в водных фазах. Спектроскопия комбинационного рассеяния позволяет получать более интенсивные сигналы каталитически активных центров (М–О–М-, М–О-колебания) в широком диапазоне реакционных условий при различных температурах и давлениях [49–51]. Jose P. Ruelas-Leyva и др. в работе [52], посвященной изучению влияния образования кокса на активность катализатора, утверждали, что применение КР-спектроскопии дает возможность получить информацию не только о структуре (размерах частиц) образующегося кокса, но и о его положении на носителе (рис. 2б). Был исследован процесс коксообразования, идентифицированы два типа кокса и условия их формирования, а также изучено влияние коксообразования на конверсию и селективность, отмечены закономерности изменения размеров частиц кокса при различных временах реакции и объяснено явление “миграции кокса” – спекание частиц и движение их к носителю с постепенным переходом от мягкого кокса к твердому [52].
Так как КР-спектроскопия обладает сравнительно низкой чувствительностью, ее необходимо использовать в совокупности с методом ИК-спектроскопии в качестве источника дополнительной информации [42].
Твердотельная спектроскопия ядерного магнитного резонанса (ЯМР), которая дает как структурную, так и динамическую информацию о каталитических системах, рассматривается как один из наиболее релевантных методов для исследований механизмов НСР [29]. Высокое разрешение и чувствительность делают твердотельный ЯМР мощным инструментом для изучения гетерогенных катализаторов [53]. Данный метод обычно применяется для идентификации молекул, измерения тонких электронных эффектов, определения структуры, исследования интермедиатов в химических реакциях [29, 42, 54]. Например, в работе Zhang Wenna и др. [55] с помощью твердотельной ЯМР-спектроскопии изучали реакционноспособные и стабильные промежуточные продукты реакции МТО. В основе наблюдений лежало взаимодействие между образующимися интермедиатами и порами цеолита. Были идентифицированы основные промежуточные ионы карбения и установлено, что механизм метилирования их боковой цепи является энергетически выгодным путем для образования олефинов. В работе подчеркивается, что селективность процесса МТО контролируется порами цеолита за счет их размера и стерического фактора (рис. 3) [55].
Рис. 3.
ЯМР-спектр стабильных органических веществ в цеолите H-RUB-50 после непрерывной реакции с метанолом 13C при 300°C в течение 25 мин. Оптимизированные структуры катионов (триметилциклопентенил, триметилбензиниум, тетраметилбензиниум), содержащихся в H-RUB-50, приведены с рассчитанными химическими сдвигами 13C. Звездочкой обозначены вращающиеся боковые полосы. Фиолетовая линия – участок обозначенного черной линией спектра в увеличенном масштабе.
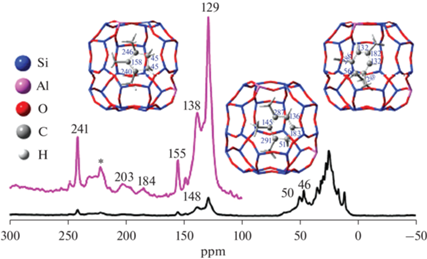
Как и любой физико-химический метод анализа, твердотельная ЯМР-спектроскопия имеет определенные недостатки. Во-первых, это зависимость чувствительности метода от силы поляризации ядерных спинов и исключительная возможность анализа изотопов, имеющих ненулевой ядерный спин (1H, 13C, и т.д.). Во-вторых – отсутствие возможности на сегодняшний день проведения регистрации ЯМР-спектров в режиме операндо, обусловленная технологическими сложностями и особенностями метода [55].
Таким образом, универсального спектроскопического метода изучения HCP процессов HCP не существует, однако сочетание разных вариантов анализа позволяет получить достаточно глубокое понимание механизмов реакций. Так, Wei Wang и др. [29, 56] использовали комбинацию методов ЯМР и УФ-спектроскопии для исследования механизма НСР и каталитической роли алкильных групп, а также реакционной способности поверхностных метоксигрупп, в то время как ЯМР-спектроскопия дала возможность более подробно идентифицировать алкильные сигналы, а УФ-спектроскопия – обнаружить низкие количества ароматических соединений и ионов карбения [29, 56]. Эта методика была применена для изучения образования углеводородов при конверсии метанола на слабодеалюминированном цеолите H-ZSM-5 (Si/Al = 22) [56]. Так, комбинированный метод ЯМР–УФ является подходящим инструментом для исследования тонкостей реакций, протекающих по механизму HCP. Ниже мы обсудим неспектроскопические методы анализа реакций HCP.
Газовая хроматография–масс- спектрометрия (ГХ–МС)
Газовая хроматография–масс-спектрометрия (ГХ–МС) – это универсальный аналитический метод, который позволяет высокоспецифично характеризовать вещества по газохроматографическим индексам удерживания и масс-спектрам. Вещества с перекрывающимися хроматографическими пиками различают по их масс-спектрам. Изомеры с похожими или идентичными масс-спектрами различают по индексам удерживания. Таким образом, ГХ и МС дополняют друг друга при анализе смесей [57–59].
Сфера применения метода ГХ–МС определяется списком аналитов, которые могут быть разделены методом газовой хроматографии. Это относительно низкомолекулярные и термически стойкие аналиты, несущественно распадающиеся при нагреве в инжекторе хроматографа и имеющие среднюю или низкую полярность. Наиболее распространенным вариантом ГХ–МС является анализ с применением ионизации электронным ударом (ЭУ) [60]. При проведении скрининга спектры, полученные при нормированных условиях ионизации (70 эВ), сравнивают с библиотечными масс-спектрами. Данный метод анализа находит широкое применение в различных областях катализа и позволяет получать информацию о том, какие молекулы и в каком количестве присутствуют не только на поверхности, но и в порах катализатора во время реакции.
С помощью ГХ–МС была выявлена зависимость между составами продуктов и структурой катализаторов. Например, в работе Shewangizaw Teketel [61] использовались катализаторы ZSM-22, ZSM-23, EU-1 и ZSM-48 для процесса MTH. Обнаружено, что небольшие различия в системе каналов оказывают влияние на распределение продуктов реакции МТН в цеолитах с 10MR. Катализаторы ZSM-22, ZSM-23 и ZSM-48 обеспечивали высокую селективность по углеводородам C5+, однако фракция C5+ без ароматических соединений получалась только над цеолитами ZSM-22 и ZSM-23.
В дальнейшем уже в работах Jinzhe Li [62] были изучены реакции, происходящие в процессе MTO с образцами катализатора типа SAPO c 8MR, имеющего различные размеры пустот. Установлено, что вид пустот определяет размер молекул и реакционную способность и, как следствие, приводит к различиям в протекании процессов MTO.
С помощью метода ГХ–МС был более детально исследован механизм процесса MTO при мягких условиях на катализаторах SAPO и раскрыты причины их довольно быстрой дезактивации. В ходе анализа обнаружен новый вид продуктов уплотнений – алмазоидные углеводороды, которые формируются и скапливаются в катализаторе SAPO-34 [63]. Накопление этих циклических и насыщенных углеводородов сводило на нет образование HCP и могло объяснить быструю дезактивацию SAPO-34 при низких температурах реакции. Стоит уделить внимание и подготовке образца к проведению ГХ–МС-анализу коксовых отложений. Как правило, используется следующая стандартная методика [64]. Первоначально осуществляют последовательные экстракции с помощью CCl4 и CH2Cl2 для удаления продуктов уплотнения из пор. Затем проводят обработку 40% плавиковой кислотой, чтобы разрушить каркас цеолита и извлечь оставшиеся продукты. После всех экстракций к полученной вытяжке добавляют внутренний стандарт (например, аценафтен) и выполняют анализ. Полученные площади пиков по данным ГХ–МС для каждого компонента затем сопоставляют с внутренним стандартом.
Как и любой другой метод анализа ГХ–МС имеет свои недостатки, а именно:
– необходимость растворения катализатора плавиковой кислотой;
– невозможность анализировать многие нелетучие полиароматические органические вещества.
Для подтверждения спектроскопических данных, а также в целях получения недостающей информации, которую не может дать метод ГХ–МС, обычно необходимо использовать другие методы физико-химического анализа.
Порошковая рентгеновская дифракция
Метод порошковой рентгеновской дифракции основан на изучении дифракционной картины, применим для материалов с дальним порядком и не подходит для аморфных соединений. С его помощью можно определять структуру и параметры элементарной ячейки цеолитных материалов.
Большинство цеолитов имеют строго регулярные кристаллические структуры (рис. 4). В координационной сфере алюминия и кремния находятся 4 атома кислорода, таким образом формируются тетраэдрические координационные полиэдры (КЧ = 4). Каркас состоит из тетраэдров ${\text{SiO}}_{4}^{{4 - }}$ и ${\text{AlO}}_{4}^{{5 - }}$, данные элементы соединяются друг с другом через мостиковые атомы кислорода, в результате чего возникает трехмерная каркасная структура, включающая в себя микропоры.

В интересующих нас реакциях HCP происходит образование углеводородов внутри цеолитных пор. Зачастую наличие этих углеводородов способствует снижению активности катализатора и может приводить к полной его дезактивации. Процесс дезактивации цеолитных катализаторов в реакциях MTH был изучен в работе [30] с помощью метода рентгеновской дифракции. В работе использовались катализаторы типа MFI. Реакцию MTH проводили до полной дезактивации катализатора, а затем разделяли его на слои. При сравнении рентгенограмм дезактивированных слоев и свежего катализатора наблюдались явные изменения интенсивности характеристических дублетов при 23.5° и 45° (рис. 5). Данные дублеты присущи катализаторам ZSM-5 орторомбической сингонии и представляют особый интерес, поскольку при дезактивации их интенсивность снижается, и на рентгенограмме появляется единичный пик, который свидетельствует об изменении категории симметрии с низшей орторомбической до средней тетрагональной. Дескриптором этого перехода служит разница между векторами элементарной ячейки a и b. Сигналы на рентгенограммах верхнего и среднего слоев (наиболее дезактивированных) заменяют дублеты на ренгенограмме свежего катализатора. Исчезновение дифракционных дублетов происходит за счет увеличения числа молекул кокса, расположенных в верхнем и среднем слоях цеолитного катализатора. На рис. 5 четко прослеживается изменение рефлексов для всех образцов.
Рис. 5.
Дифрактограммы дезактивированных слоев катализаторов ZSM-5. Сплошная линия – свежий катализатор; пунктирная и точечная линии – верхний и средний слои катализатора соответственно. а, б – Образцы, дезактивированные при 400°C и Pатм; в, г –образцы ZSM-5, дезактивированные при Р = 20 бар и 370°C.

В качестве параметра дескриптора для описания дезактивации использовалась разница между параметрами элементарной ячейки (a – b). Например, для свежего катализатора MFI-27 значение (a – b) составляло 0.172 Å, а для дезактивированного – 0.0 Å. Более подробно зависимость изменения параметра (а – b) от степени дезактивации описана в работе [65]. Для других катализаторов ZSM-5 также характерен аналогичный переход, но разница между параметрами (a – b) элементарной ячейки дезактивированных цеолитов не всегда была нулевая. В действительности такая тенденция наблюдалась только у MFI-27. В случае коммерческого катализатора CBV-8014 в ходе дезактивации величина (a – b) уже составляла 0.14 Å.
Стоит отметить, что по разнице параметров (a – b) можно достаточно точно фиксировать степень дезактивации, которая определяется общим содержанием кокса, в режиме операндо (рис. 6).
Рис. 6.
Зависимость разницы между параметрами элементарной ячейки (a – b) от содержания кокса в катализаторе.

Согласно литературным данным дезактивация действительно прогрессирует от входа к выходу (сверху вниз) согласно модели Haw [66]. В экспериментах, проводимых при атмосферном давлении, верхние слои катализатора были довольно сильно дезактивированы, как видно из высокого общего содержание в них кокса. При проведении реакции под высоким давлением наблюдалась иная картина. Верхние и средние слои были полностью дезактивированы, а нижние – гораздо меньше. Однако для некоторых катализаторов в среднем слое было больше кокса, чем верхнем. Причина этого заключается в высокой линейной скорости газа, которая достигается в узких трубчатых реакторах. Когда линейная скорость газа высока, необходимо, чтобы количество вещества в реакторе было достаточным для запуска, автокаталитического процесса реакции MTH. Верхний слой катализатора, находящийся в верхней части реактора, играет роль инициатора автокаталитических частиц. Этот слой не участвует в преобразованиях MTH и остается менее дезактивированным, в отличии от среднего слоя.
Рентгеновская дифракция находит применение и в изучении процессов MTO на мелкопористых катализаторах [67]. Авторами было установлено, что при конверсии метанола в олефины дескриптором хода реакции и накопления кокса является параметр элементарной ячейки c в ромбоэдрической ячейке катализатора SAPO-34. При сравнении процентных изменений параметров элементарной ячейки у SAPO-34 и ZSM-5 было предположено, что структура SAPO-34 более подвержена деформации, чем ZSM-5.
В дальнейшем D. Wragg предпринял попытку изучить более детально процесс MTO на SAPO-34 [68]. Для этого он использовал дифракцию рентгеновских лучей высоких энергий с разрешением по времени и пространству в режиме операндо. В результате было выявлено, что:
– зарождение первой С–С-связи в рамках механизма HCP зависит от скорости потока и количества каталитических центров;
– образование веществ на первых этапах HCP первоначально идет в противоположном направлении от потока реагентов;
– появление конгломератов кокса зависит от скорости потока и количества каталитических центров;
– дезактивация происходит из-за формирования тяжелого кокса.
Термический анализ
С тоски зрения каталитической химии особый интерес среди методов термического анализа представляют термогравиметрический анализ (ТГА) и дифференциальная сканирующая калориметрия (ДСК). Термогравиметрический анализ дает возможность регистрировать изменение массы образца в зависимости от температуры [69]. Дифференциальная сканирующая калориметрия – это аналитический метод, в котором измеряют скорость теплового потока к образцу или от него в то время когда он подвергается нагреву/охлаждению согласно контролируемой температурной программе в контролируемой атмосфере. Полученные данные позволяют проводить анализ количества (или обнаруживать образование) органических соединений на катализаторе. Кроме этого, на основании данных ТГА и ДСК можно судить о появлении на катализаторе ”мягкого” (на поверхности катализатора) и “твердого” (внутри пор катализатора) кокса, и о влиянии условий на протекание этих процессов. Например, в работе R.B. Rostami с коллегами [70] была изучена дезактивация катализатора SAPO-34 при разных температурах протекания процесса МТО. Обнаружено, что в зависимости от температуры менялось соотношение между “мягким” и “твердым” коксом. Разложение кокса, образовавшегося в результате проведении реакции при 673 и 733 К, начиналось при 550 и 700 К соответственно. Согласно данным ТГА повышение температуры в ходе реакции приводило к увеличению общего содержания кокса на SAPO-34 в процессе MTO.
Хотелось бы отметить, что не всегда изменение теплового эффекта на кривых ДТА обусловлено варьированием локализации коксовых отложений. Сдвиг положения пиков может быть связан и с природой коксовых отложений, формирующихся при различных условиях протекания процесса. Так, например, при низких температурах на катализаторах SAPO-34 в процессе MTO образуются адамантаноподобные отложения, которые в свою очередь будут приводить к смещению пика на термограмме [71].
Методы ТГА и ДСК нашли применение и для исследования других процессов. Например, в работе [72] рассматривалось влияние Mo (катализатор Mo/ZSM-5) на количество образовавшегося кокса на поверхности (“мягкого”) и внутри (“твердого”) катализатора. Обнаружено, что процесс образования “твердого” кокса мало зависит от содержания Мо. Так, при содержании 1–5% Мо поры катализаторов блокируются частицами углерода в равной степени независимо от количества молибдена.
При увеличении доли Мо в катализаторе содержание “мягкого” кокса (450–550°C) возрастает, а процесс горения смещается от 550–600°C в более низкотемпературную область (рис. 7).
Рис. 7.
Дифференциальные термогравиметрические кривые для катализаторов Mo/ZSM-5 (1–5% Mo) с различным содержанием кокса.
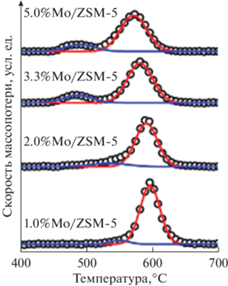
Причиной столь различного температурного поведения “мягкого” и “твердого” кокса авторы [72] считают блокирование диффузии кислорода в поры цеолита “мягким” коксом, а также каталитический характер его горения на поверхности. В работе [73] была изучена скорость коксования и происхождение кокса на морденитовых катализаторах в реакциях получения пропилена при различных температурах (393, 473, 623 и 723 К). Результаты температурного окисления коксовых отложений, образовавшихся при различных температурах после 240 мин работы катализатора, свидетельствуют о том, что с повышением температуры коксования полное окисление кокса затрудняется, его ароматичность увеличивается.
На рис. 7 приведены кривые ДТГ (дифференциальная термогравиметрия) продуктов дезактивации. Установлено, что пики, соответствующие коксовым отложениям, полученным при 393 К, имели максимумы при 570 и 778 К. Кокс, образовавшийся при 473 К, характеризовался пиками, смещенными в сторону более высоких температур – 589 и 780 К соответственно. Первый пик, связанный с более активными компонентами, постепенно исчезал по мере повышения температуры коксования, в то время как интенсивность второго пика снижалась. При температуре реакции 623 К на кривой ДСК появлялся третий пик с максимумом при 830–840 K. Этот пик становился единственным для сформировавшегося при 723 K кокса. Два пика, наблюдаемые в случае коксов, образовавшихся при 393 и 473 K, соответствуют горению разветвленных парафинов, олефинов и полиолефинов. Следовательно, при повышении температуры коксования эти соединения (парафины и олефины) замещаются менее реакционноспособными ароматическими углеводородами, что приводит к резкому снижению интенсивности второго пика и росту интенсивности третьего, максимум которого смещается в более высокотемпературную область по мере увеличения ароматичности кокса.
Изотопное мечение
Метод изотопных меток традиционно находит широкое применение для изучения механизмов реакций. Он позволяет оценивать реакционную способность молекул в порах катализатора и газовой фазе, степень конверсии исходных реагентов, селективность реакции по определенному веществу, образование и каталитическую роль интермедиатов [74–76]. Это становится возможным благодаря анализу соотношения меченных и немеченных исходных веществ или распределения углерода 13C и 12C в продуктах реакции. Одним из главных достоинств применения метода изотопных меток является возможность оценки соотношения вкладов алкенового и аренового циклов в механизм НСР. На этом принципе основана работа S. Svelle и соавт. [77] по определению каталитической роли олефинов и органических ароматических соединений в рамках механизма НСР. На катализатор H-ZSM-5 подавали импульс 12СН3ОН на протяжении 18 мин с последующим переключением на метанол 13СН3ОН. Для определения содержания 13С в образующихся соединениях в зависимости от времени эксперимента был использован метод ГХ–МС [77]. Основная идея заключалась в сопоставлении содержания 13С в газовой фазе и хорошо адсорбированной органике для понимания их механистической функции в каталитическом процессе (рис. 8). Экспериментальные данные показали, что более высокое содержание 13С в алкенах C3–C6 по сравнению с этеном и ароматическими соединениями обусловлено включением изотопа в эти группы молекул по разным механизмам. В ходе работы авторы пришли к выводам, что в присутствии цеолита H-ZSM-5 этен образуется из низших метилбензолов, а пропен и высшие олефины – в ходе метилирования и взаимопревращений алкенов. Таким образом, результаты свидетельствуют о потенциале совместного использования метод изотопных меток в комбинации с рассмотренными ранее изотопчувствительными спектроскопическими методами (ИК, КР, ЯМР) или ГХ–МС, а также подтверждают эффективность применения изотопного мечения для исследования каталитических процессов, протекающих по механизму HCP.

ЗАКЛЮЧЕНИЕ
Настоящая работа демонстрирует релевантность применения цеолитных катализаторов в современных каталитических процессах и необходимость улучшения их характеристик. Использование не только метанола, но и других потенциально возобновляемых реагентов (алкилгалогенидов, алкилтиолов, высших спиртов, фуранов) – многообещающий способ получения ценных углеводородов. Рассмотренные примеры подчеркивают, что конверсия углеводородов на цеолитах идет по сложным механизмам. Совместное применение описанных выше спектроскопических, хроматографических, диффракционных, термогравиметрических и изотопных методов позволяет достичь глубокого понимания реакционных путей изучаемых процессов и особенно – этапов реакций, идущих по механизму НСР. На сегодняшний день процессы МТН, проходящие в соответствии с НСР, постепенно становятся коммерциализированными, что обуславливает растущий интерес к данной области исследований. Таким образом, расширение знаний о возможностях протекания каталитических процессов в соответствии с НСР является важной задачей химии.
На наш взгляд дальнейший прогресс в области исследования HCP реакций возможен, в первую очередь, при комбинации методов характеризации (рис. 9). Например, изотопное мечение совместно с изотоп-чувствительной спектроскопией позволяет получать глубочайшее понимание реакционных процессов в порах цеолитов. Применение ин ситу и операндо методов это еще одно потенциально важное направление исследований. Реакции в порах цеолитов крайне чувствительны к условиям и поэтому важно удостовериться, что спектроскопическая информация получена в релевантных условиях. Наконец, синтез цеолитных катализаторов с заданными свойствами (топология, размер кристаллов, текстурные свойства, присутствие каталитически активных металлических центров), которые позволят однозначно связывать спектроскопические данные со структурой катализатора, будет играть важную роль в установлении точных механизмов HCP реакций.

Список литературы
Li Y., Li L., Yu J. // Chem. 2017. V. 3. P. 928.
Olsbye U., Svelle S., Bjrgen M., Beato, P., Janssens T.V.W., Joensen F., Bordiga S., Lillerud K.P. // Angew. Chem. 2012. V. 51. P. 5810.
Wei Y., Zhang D., Xu L., Chang F., He Y., Meng S., Su B., Liu Z. // Catal. Today. V. 131. 2008. P. 262.
Yu M., Tormene N., Bolshakov A., Mezari B., Liutkova A., Kosinov N., Hensen E.J.M. // Chem. Commun. 2021. V. 57. P. 3323.
Lee K., Hong S.B. // ACS Catal. 2019. V. 9. P. 10640.
Uslamin E.A., Kosinov N., Filonenko G.A., Mezari B., Pidko E., Hensen E.J.M. // ACS Catal. 2019. V. 9. P. 8547.
Uslamin E.A., Saito H., Sekine Y., Hensen E.M.J., Kosinov N. // Catal. Today. 2021. V. 369. P. 184.
Kosinov N., Wijpkema A.S.G., Uslamin E., Rohling R., Coumans F.J.A.G., Mezari B., Parastaev A., Poryvaev A.S., Fedin M.V., Pidko E.A., Hensen E.M.J. // Angew. Chem. Int. Ed. 2017. V. 57. P. 1016.
Иванова И., Понаморева О., Андриако Е., Нестеренко Н. // Общественно-деловой журнал энергетическая политика. 2021. № 6. С. 68.
Хаджиев С.Н., Магомедова М.В., Пересыпкина Е.Г. // Нефтехимия. 2015. Т. 55. № 5. С. 355.
Smeets S., Zou X. / Zeolites Catal. Prop. Appl. London: The Royal Society of Chemistry, 2017. P. 37.
Database of Zeolite Structures [electronic resource]: International Zeolite Association. - Access mode: http://www.iza-online.org/ (date of appeal: 15.08.2021).
Guisnet M., Gilson J. // Focus Catal. 2003. P. 8.
Zhan B.Z., Iglesia E. // Angew. Chem. 2007. V. 46. P. 3697.
Родионова Л.И., Князева Е.Е., Коннов С.В., Иванова И.И. // Нефтехимия. 2019. Т. 59. № 3. С. 333.
Чистяков А.В., Жарова П.А., Губанов М.А., Николаев С.А., Егорова Т.Б., Гехман А.Е., Цодиков М.В. // Кинетика и катализ. 2017. Т. 58. № 6. С. 726.
Кузьмина Р.И., Пилипенко Ю.А., Хорошилов И.И., Фролов М.П. // Изв. Сарат. ун-та. Нов. сер. Сер. Химия. Биология. Экология. 2015. Т. 15. № 4. С. 30.
Kosinov N., Liu C., Hensen E.J.M., Pidko E.A. // Chem. Mater. 2018. V. 30. P. 3177.
Blakeman P.G., Burkholder E.M., Chen H.Y., Collier J., Fedeyko J.M., Jobson H., Rajaram R.R. // Catal. Today. 2014. V. 231. P. 56.
Prodinger S., Shi H., Eckstein S., Hu. Z.H., Olarte M.V., Camaioni D.M., Derewinski M.A., Lercher J.A. // Chem. Mater. 2017. V. 29. P. 7255.
Vogt E.T.C., Whiting G.T., Chowdhury A.D., Weckhuysen B.M. // Adv. Catal. 2015. V. 58. P. 143.
Collett C.H., Mcgregor J. // Catal. Sci. Technol. 2016. V. 6. P. 363.
Wang W., Jiang Y., Hunger M. // Catal. Today. 2006. V. 113. P. 102.
Mynsbrugge J.V., Bell A.T. // J. Catal. 2021. V. 401. P. 89.
Plessow P.N., Studt F. // Catal. Lett. 2018. № 148. P. 1246.
Olsbye U., Bjørgen M., Svelle S., Lillerud K.P., Kolboe S. // Catal. Today. 2005. V. 106. P. 108.
Blasco T. // Chem. Soc. Rev. 2010. V. 39. P. 4685.
Parker S.F., Kombanal A.J. // Catalysts. 2021. V. 11(10). P. 1204.
Brogaard R.Y., Henry R., Schuurman Y., Medford A.J., Moses P.G., Beato P., Svelle S., Nørskov J.K., Olsbye U. // J. Catal. 2014. V. 314. P. 159.
Rojo-Gama D., Nielsen M., Wragg D.S., Dyballa M., Holzinger J., Falsig H., Lundegaard L.F., Beato P., Brogaard R.Y., Lillerud K.P., Olsbye U., Svelle S. // ACS Catal. 2017. V. 7. P. 8235.
Guisnet M., Magnoux P. // Appl. Catal. 2001. V. 212. P. 83.
Yarulina I., Chowdhury A.D., Meirer F., Weckhuysen B.M., Gascon J. // Nature Catal. 2018. V. 1. P. 398.
Tian P., Wei Y., Ye M., Liu Z. // ACS Catal. 2015. V. 5. № 3. P. 1922.
Del Campo P., Slawinski W.A., Henry R., Erichsen M.W., Svelle S., Beato P., Wragg D., Olsbye U. // Surf. Sci. 2016. V. 648. P. 141.
Jiang Y., Huang J., Reddy Marthala V.R., Ooi Y.S., Weitkamp J., Hunger M. // Micropor. Mesopor. Mater. 2007. V. 105. P. 132.
Hunger M., Weitkamp J. // Angew. Chem. Int. Ed. 2001. V. 40. P. 2954.
Jiang Y., Huang J., Weitkamp J., Hunger M. // Stud. Surf. Sci. Catal. 2007. V. 170. P. 1137.
Qingyun Q., Vogt C., Mohamed M., Asiri A.M., Al-Thabaiti S.A., Basahel S.N., Ruiz-Martinez J., Weckhuysen B.M. // ChemCatChem. 2014. V. 6. P. 3396.
Weckhuysen B.M. // Phys. Chem. Chem. Phys. 2003. V. 5. P. 4351.
Mores D., Stavitski E., Kox M.H.F., Kornatowski J., Olsbye U., Bert M., Weckhuysen B.M. // Chem. A. Eur. J. 2008. V. 14. P. 11320.
Yamazaki H., Shima H., Imai H., Yokoi T., Tatsumi T., Kondo J.N. // Angew. Chem. Int. 2011. V. 50. P. 1853.
Li J., Wei Z., Chen Y., Jing B., He Y., Dong M., Jiao H., Li X., Qin Z., Wang J., Fan W. // J. Catal. 2014. V. 317. P. 277.
Millet J.M.M. // Adv. Catal. 2007. V. 51. P. 309.
Wachs I.E., Roberts C.A. // Chem. Soc. Rev. 2010. V. 39. P. 5002.
Minova I., Matam S., Greenaway A., Catlov R.A., Frogley M.D., Cinque G., Wright P.A., Howe R.F. // ACS Catal. 2019. V. 9. P. 6564.
Palumbo L., Bonino F., Beato P., Bjørgen M., Zecchina A., Bordiga S. // J. Phys. Chem. 2008. V. 112. P. 9710.
Howe R.F., Russell F., McGregor J. // Catal. Lett. 2016. V. 146. P. 1242.
Wachs I.E. // Catal. Today. 2005. V. 100. P. 79.
Banares M.A., Mestl G. // Adv. Catal. 2009. V. 52. P. 43.
Bordiga S., Lamberti C., Bonino F., Travertd A., Thibault-Starzyk F. // Chem. Soc. Rev. 2015. V. 44. P. 7262.
Bard J.A., Debad J.D. Encyclopedia of Analytical Chemistry. Chichester: Wiley, 2006. 9842 p.
Ruelas-Leyva J.P., Maldonado-Garcia L.F., Talavera-Lopez A., Santos-López I.A., Picos-Corrales L.A., Santolalla-Vargas C.E., Gómez Torres S.A., Fuentes G.A. // Catalysts. 2021. V. 11. P. 128.
Ivanova I.I., Kolyagin Y.G. // Chem. Soc. Rev. 2010. V. 39. P. 5018.
Aerts A., Kirschhock C.E.A., Martens J.A. // Chem. Soc. Rev. 2010. V. 39. P. 4626.
Zhang W., Chen J., Xu S., Yueying C., Wei Y., Zhi Y., Huang J., Zheng A., Wu X., Meng X., Xiao F., Deng F., Liu Z. // ACS Catal. 2018. V. 8(12). P. 10950.
Hunger M., Wang W. // Chem. Commun. 2004. P. 584.
Niessen W.M. // Rev. J. Am. Soc. Mass Spectrom. 2001. V. 12. P. 1348.
Jaegers N.R., Mueller K.T., Wang Y., Hu J.Z. // Acc. Chem. Res. 2020. V. 53(3). P. 611.
Gas chromatography and mass spectroscopy. Practical guide. Eds. Sparkman D.O., Penton Z.E., Kitson F.G. Oxford: Elsevier, 2011. P 3.
Гладилович В.Д., Подольская Е.П. // Научное приборостроение. 2010. Т. 20. № 4. С. 36
Teketel S., Skistad W., Benard S., Olsbye U., Lillerud K.P., Beato P., Svelle S. // ACS Catal. 2011. V. 2(1). P. 26.
Li J., Wei Y., Chen J., Xu S., Tian P., Yang X., Liu Z. // ACS Catal. 2014. V. 5(2). P. 661.
Wei Y., Li J., Yuan C., Xu S., Zhou Y., Chen J., Liu Z. // Chem. Commun. 2012. V. 48(25). P. 3082.
Holmes S.M., Garforth A., Maunders B., Dwyer J. // Appl. Catal. A: Gen. 1997. V. 151(2). P. 355.
Alvarez A.G., Viturro H., Bonetto R.D. // Mater. Chem. Phys. 1992. V. 32. P. 135.
Haw J.F., Marcus D.M. // Top. Catal. 2005. V. 34. P. 41.
Wragg D.S., Johnsen R.E., Balasundaram M., Norby P., Fjellvag H., Grønvold A., Fuglerud T., Hafizovic J., Vistad O.B., Akporiaye D. // J. Catal. 2009. V. 268. P. 290.
Wragg D.S., O’Brien M.G., Bleken F.L., Di Michiel, M., Olsbye, U., Fjellvag H. // Heterogen. Catal. 2012. V. 51. P. 1.
Ивлев В.И., Фомин Н.Е., Юдин В.А., Окин М.А., Панькин Н.А. Термический анализ. Учебное пособие. Саранск: Изд-во Мордовcкого ун-та, 2017. С. 2.
Rostami R.B., Ghavipour M., Di Z., Wang Y., Behbahani R.M. // RSC Adv. 2015. V. 5(100). P. 81965.
Wei Y., Li J., Yuan C., Xu S., Zhou Y., Chen J., Liu Z. // Chem. Commun. 2012. V. 48(25). P. 3082.
Kosinov N., Uslamin E.A., Coumans F.J.A.G., Wijpkema A.S.G., Rohling R.Y., Hensen E.J.M. // ACS Catal. 2018. V. 8(9). P. 8459.
Henriques C.A., Afonso J.C., Magnoux P., Guisnet M., Monteiro J.L.F. Progress in zeolite and microporous materials studies in surface science and catalysis. Penselvania: Materials Research Society, 1999. 980 p.
Svelle S., Joensen F., Nerlov J., Olsbye U., Lillerud K.P., Kolboe S., Bjørgen M. // J Am. Chem. Soc. 2006. V. 128(46). P. 14770.
Lin S., Zhi Y., Chen W., Li H., Zhang W., Lou C. // J. Am. Chem. Soc. 2021. V. 143(31). P. 12038.
Zhang B., Zhong Z., Zhang J., Ruand R. // J. Anal. Appl. Pyrolysis. 2018. P. 1.
Svelle S., Joensen F., Nerlov J., Olsbye U., Lillerud K.-P., Kolboe S., Bjørgen M. // J. Am. Chem. Soc. 2006. V. 128(46). P. 14770.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ