

Кинетика и катализ, 2022, T. 63, № 6, стр. 845-857
Синтез в режиме низкотемпературного горения катализаторов на основе галлуазита для глубокого окисления углеводородов, СО и метанирования СО2
В. Н. Борщ a, *, И. М. Быстрова a, Е. В. Пугачева a, Е. М. Смирнова b, А. В. Ставицкая b, В. А. Винокуров b
a ФГБУН Институт структурной макрокинетики и проблем материаловедения им. А.Г. Мержанова РАН
142432 Московская область, Черноголовка, ул. Акад. Осипьяна, 8, Россия
b ФГАОУ ВО Российский государственный университет (национальный исследовательский университет) нефти
и газа им. И.М. Губкина
119991 Москва, Ленинский просп., 65, Россия
* E-mail: borsch@ism.ac.ru
Поступила в редакцию 24.02.2022
После доработки 11.07.2022
Принята к публикации 13.07.2022
- EDN: JMRCKO
- DOI: 10.31857/S0453881122060028
Аннотация
Синтез в режиме низкотемпературного горения в пористых средах был использован для получения нанесенных моно- и биметаллических катализаторов, содержащих 10 мас. % Ni (I), 10 мас. % Co (II) и 5 мас. % Со + 5 мас. % Ni (III) на галлуазитных нанотрубках. Образцы катализаторов исследованы методами РФА, СЭМ, рентгеноспектрального микроанализа, ТПВ, определена удельная поверхность методом БЭТ по физической адсорбции азота. Активность и селективность полученных катализаторов изучена на примере процессов глубокого окисления СО и пропана и гидрирования СО2 при атмосферном давлении. В глубоком окислении СО и пропана наиболее активными оказались Со-содержащие образцы, особенно биметаллический Со–Ni-катализатор. В процессе метанирования СО2 на исходных образцах ряд активности выглядел следующим образом: I > III > II. Восстановление катализаторов в токе водорода при 400°С в течение 1 ч привело к значительному росту их активности с максимумом при 350°С, ряд активности при этом сохранился. На восстановленном образце I была достигнута конверсия СО2 88.8% при селективности образования метана 97.7%.
ВВЕДЕНИЕ
Метод низкотемпературного горения в пористых средах или самораспространяющийся поверхностный термосинтез (self-propagating surface thermo synthesis, SSTS) [1–7] является перспективным подходом для получения нанесенных катализаторов. Суть метода SSTS заключается в пропитке носителя смесью растворов окислителя, чаще всего нитрата активного металла, и восстановителя (горючего). В качестве горючего могут использоваться различные органические соединения, растворимые в воде. После высушивания в образце путем нагрева инициируется реакция, протекающая в режиме волны горения или теплового взрыва. В результате реакции образуются высокодисперсные (в т.ч. наноразмерные) оксидные и/или металлические активные фазы (АФ) на поверхности пор носителя. Структура АФ зависит от использованного горючего и соотношения окислитель : горючее. Температуру в волне горения обычно поддерживают на уровне не выше 400°С, что облегчается высокой теплоотдачей в носитель. Такая достаточно низкая температура горения дает возможность сохранить структуру носителя при минимальном образовании побочных соединений АФ и носителя и предотвратить спекание частиц АФ. Метод SSTS позволяет получать широкий спектр нанесенных катализаторов с различными АФ на носителях, не подверженных окислительно-восстановительным превращениям в ходе реакции горения. Чаще всего в качестве носителей использовали стеклоткань [1–6] и γ-Al2O3 [1–5, 7], однако были синтезированы также катализаторы на основе пластин из сплавов Ti–Si, Ni–Al–Si, пористой стали и пористого титана [1], цеолитов NaХ, ZSM-5 и силикагеля [7]. Катализаторы с АФ на основе оксидов Mn, Co, Ni, Cu и Се в различных сочетаниях [1–3, 7] были испытаны в процессах глубокого окисления СО и углеводородов и метанирования СО2 [7] и продемонстрировали высокую активность. Pd- и Pd/Ag-cодержащие катализаторы, полученные путем SSTS, отличались высокой селективностью в процессе селективного гидрирования ацетилена в присутствии СО [4–6].
Галлуазит представляет собой природный глинистый минерал из группы ортосиликатов с общей формулой Al2Si2O5(OH)4. Он близок по формуле к каолиниту и представляет собой продукт дегидратации последнего в природе. В результате дегидратации слои каолинита сворачиваются в многослойные нанотрубки, которые являются характерной особенностью структуры галлуазита. Эти трубки (ГНТ) построены из чередующихся алюмокислородных и кремнекислородных слоев, связанных между собой гидроксильными группами. Наружная поверхность нанотрубок с диаметром 50–160 нм состоит из гидроксилированных тетраэдров [SiO4– х(ОН)х], в то время как внутренняя полость диаметром 20–30 нм покрыта гидроксилированными октаэдрами [AlO6– x(OH)x]. Длина нанотрубок достигает нескольких микрометров [8–12]. Вследствие такой структуры они обладают высокой адсорбционной емкостью, которая может быть увеличена при модификации поверхности органосиланами с различными функциональными группами [12–15], аминоспиртами [16], путем сульфонирования [17] и обработки кислотами [18]. На основе ГНТ можно получать ряд мезопористых носителей [19]. Очевидно, что вследствие специфической наноструктуры, нетипичной для всех известных ранее типов алюмосиликатов, как кристаллических, так и аморфных, ГНТ являются перспективными носителями для катализаторов, работающих в различных процессах. Структура ГНТ позволяет добиться равномерного распределения частиц АФ по размерам во внутренних полостях, препятствует укрупнению этих частиц и повышает устойчивость к их вымыванию оттуда. В связи с этим следует в первую очередь ожидать от катализаторов на основе ГНТ повышенной стабильности. В начале этого года появился наиболее полный на сегодняшний день обзор, посвященный разработкам и исследованию катализаторов различных процессов на основе ГНТ [19]. Однако нашлось лишь очень ограниченное число работ, в которых изучали катализаторы процессов глубокого окисления и гидрирования СО2. На биметаллическом катализаторе Pd–Ni/ГНТ, полученном пропиткой солями с последующим восстановлением, в процессе окисления СО достигнуто 100% конверсии уже при 128°С [20]. Введение в ГНТ предварительно полученных наночастиц Ru, позволило получить катализаторы с гораздо более высокой селективностью в процессе селективного окисления СО в присутствие Н2 (PROX), чем полученные путем пропитки солями с последующим восстановлением [21, 22]. Модификация катализатора CeO2–ZrO2/γ-Al2O3 добавками ГНТ существенно повысила его термическую стабильность и активность в процессах глубокого окисления углеводородов и СО, а также в селективном восстановлении NOx (TWC, Three Way Catalyst) [23]. В работе [24] получен высокоактивный палладиевый TWC на основе модифицированных основаниями Шиффа ГНТ, а дополнительное введение в эту композицию CeO2 привело к улучшению стабильности катализатора при повышенных температурах. Прокалка галлуазита с последующей гидрофобизацией внутренних полостей ГНТ цетилтриметиламмонийбромидом и нанесением палладия позволили снизить температуру 99% конверсии метана в процессе глубокого окисления с 620 до 425°С по сравнению с катализатором на основе немодифицированных ГНТ [25]. На оптимизированном по составу CuO–In2O3/ГНТ в процессе гидрирования СО2 до диметилового эфира достигнуто 7.6% конверсии СО2 с селективностью 65% при давлении 40 атм и температуре 250°С [26]. Однако работы по синтезу катализаторов с Co–Ni АФ на основе ГНТ, тем более с использованием процесса низкотемпературного горения, и исследованию их свойств в процессах глубокого окисления и гидрирования СО2, в литературе отсутствуют.
Ранее нами на основе предлагаемого метода были разработаны Co-, Mn- и Ni-содержащие нанесенные катализаторы на таких носителях, как γ-Al2O3, силикагель, цеолиты NaX и ZSM-5, и показана их высокая активность в процессах глубокого окисления и гидрирования СО2 [7]. В настоящей работе представлены результаты физико-химических и каталитических исследований катализаторов Co–Ni/ГНТ, полученных в процессе низкотемпературного горения, с целью выявления влияния особенностей носителя и метода приготовления на характеристики синтезированных систем. Никель является хорошим катализатором процессов гидрирования, а кобальт, как правило, более активен в процессах окисления, хотя известны также процессы гидрирования и переноса водорода с его участием. Поэтому катализаторы были испытаны в экологически важных процессах глубокого окисления и гидрирования СО2. Глубокое окисление лежит в основе процессов нейтрализации различных газовых выбросов техногенной природы, а гидрирование СО2 в литературе рассматривается в качестве варианта его функционализации как углеродсодержащего сырья и с целью уменьшения выбросов в атмосферу этого парникового газа.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Процесс получения катализаторов включал следующие стадии. Навеску галлуазита (6 г, “Sigma Aldrich”, 100–300 мкм) пропитывали по влагоемкости смесью растворов нитратов металлов (окислители) и мочевины (горючее), соотношение концентраций которых рассчитывали на полное восстановление нитратов металлов до свободных металлов. Образец высушивали при 90°С и помещали в трубчатый кварцевый реактор с плоским дном, закрытый сверху системой пылеулавливания. Реактор сверху продували аргоном. Включали подогреватель дна реактора, мощность которого не меняли в течение всего процесса. Температуру контролировали термопарой в середине слоя образца. Показания термопары через цифровой мультиметр Metex МЕ-31 (“Metex”, Корея) вводили в компьютер и строили термограммы процесса как зависимость температуры от времени. Реакция начиналась, как правило, у боковой поверхности реактора, проходила в режиме волны горения и визуально наблюдалась как волна потемнения, сопровождающаяся интенсивным газовыделением с выбросом пыли над слоем образца. После остывания образец в реакторе в среде аргона подвергали стабилизации 5% раствором Н2О2 для предохранения от самовоспламенения на воздухе высокодисперсных металлических фаз в составе АФ, а затем высушивали при 90°С. Из полученного катализатора удаляли фракцию <100 мкм. Следует отметить, что полное восстановление нитратов до металлов в процессе горения даже в инертной среде происходит далеко не всегда. Так, после синтеза и стабилизации катализатора 10% Co–5% Mn/γ-Al2O3 в составе АФ в ходе РФА обнаружили только фазу Co3O4, в то время как в составе приготовленного таким же методом катализатора 10% Co–10% Ni/силикагель наблюдались как фазы оксидов, так и металлов [7]. В любом случае непрореагировавшее горючее просто отмывалось от образца.
Рентгенофазовый анализ (РФА) образцов производили на дифрактометре ДРОН-3М (Россия), излучение FeKα. Анализ морфологии и элементного состава образцов осуществляли на сканирующем электронном микроскопе (СЭМ) Ultra plus (“Carl Zeiss”, Германия) с рентгеноспектральным микроанализатором JCXA-733 Superprobe (“JEOL”, Япония). Энергия луча при микроанализе составляла 20 кэВ, оценка глубины проникновения по [27, 28] дала величину около 4 мкм. Термопрограммируемое восстановление (ТПВ) проводили на установке AutoChem 2950 HP (“Micromeritics”, США). Образцы были предварительно прокалены при 500°С на воздухе в течение 3 ч. Перед началом измерений навески массой 0.1 г выдерживали в токе газовой смеси 7 об. % Н2 + + Ar с расходом 20 мл/мин до стабилизации базовой линии, затем производили нагрев со скоростью 10°С/мин с регистрацией сигнала детектором по теплопроводности.
Удельную поверхность измеряли путем физической адсорбции азота по БЭТ на установке, созданной по схеме ГОСТ 23401-90 и по изложенной там методике.
Полученные образцы испытывали на установке с проточным кварцевым реактором с неподвижным слоем катализатора (1 см3, фракция 100–300 мкм, без добавок кварца). Исходную газовую смесь и продукты реакции гидрирования СО2 анализировали на компьютеризованном хроматографе 3700 (“Хроматограф”, Россия) с двумя колонками (наполнители – цеолит 5А и Porapak Q, газ-носитель – гелий, расход – 30 мл/мин, температура колонок – 70°С, детектор ДТП). Для анализа исходной газовой смеси и продуктов реакции глубокого окисления применяли пятикомпонентный газоанализатор Автотест 02.03П (“Мета”, Россия) нулевого класса точности. В реакциях глубокого окисления СО и пропана газовая смесь имела состав (об. %) 0.2С3Н8, 0.6СО и 2.0О2, остальное – азот. Объемная скорость подачи газа составляла 120 000 ч–1. Для гидрирования СО2 использовали смесь состава (об. %) 5.4СО2, 21.6Н2 (СО2 : Н2 = 1 : 4), остальное – гелий. Объемная скорость подачи газа в этом процессе – 3600 ч–1. Восстановление образцов катализаторов осуществляли в том же реакторе в токе водорода (20 мл/мин) при температуре 400°С в течение 1 ч.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В настоящей работе были получены образцы катализаторов с содержанием (в пересчете на металлы) 10 мас. % Ni (I), 10 мас. % Co (II) и 5 мас. % Co + 5 мас. % Ni (III). Термограммы процесса синтеза катализаторов приведены на рис. 1.
Рис. 1.
Термограммы процесса синтеза катализаторов: a – I, б – II, в – III (I – 10 мас. % Ni, II – 10 мас. % Co, III – 5 мас. % Co + 5 мас. % Ni).
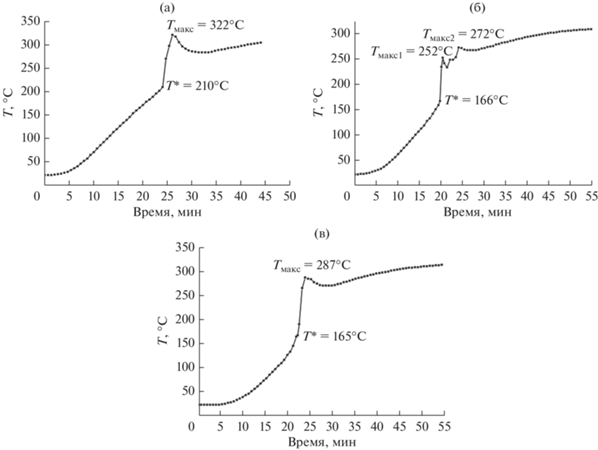
Величинами T* обозначены точки перегиба на кривых, когда тепловыделение за счет химической реакции в волне горения начинает превосходит теплопоступление от внешнего источника нагрева. Эта величина близка к понятию температуры самовоспламенения. Отмечены также максимальные температуры во фронте волны горения Tмакс. Обращают на себя внимание существенно более низкие значения Т* для катализаторов II и III по сравнению с образцом I. По-видимому, такой эффект связан с явлением автокатализа, когда образующиеся в начале горения оксометаллические кобальтсодержащие АФ образцов II и III начинают катализировать этот процесс. Чисто никелевые АФ образца I гораздо менее активны в окислении. Кроме того, на термограмме синтеза катализатора II наблюдается расщепление пика горения в максимуме. Нами ранее наблюдалось это явление в ходе синтеза катализатора 10% Co–5% Mn/γ-Al2O3 [7], что было объяснено результатом прохождения двух волн – горения и догорания, связанных со сложным автокаталитическим механизмом горения, включающим несколько стадий разложения нитрата кобальта и последующего окисления мочевины.
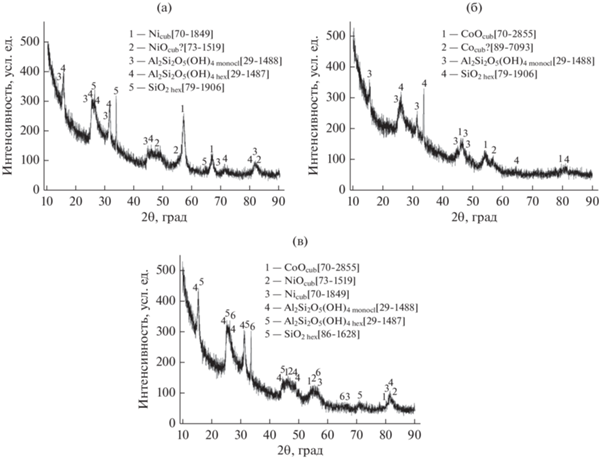
Фазовый состав полученных образцов был исследован методом РФА. Рентгенограммы приведены на рис. 2.
Рис. 2.
Рентгенограммы образцов катализаторов: a – I, б – II, в – III (I – 10 мас. % Ni, II – 10 мас. % Co, III ‒ 5 мас. % Co + + 5 мас. % Ni).
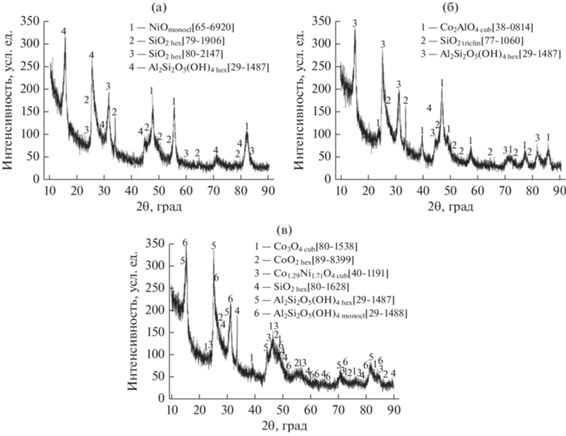
Как можно видеть, АФ катализаторов представлены оксидными и сложнооксидными фазами (образцы I и III) и фазой алюмината кобальта Co2AlO4 для образца II. Заметим, что катализатор 5% Co/γ-Al2O3, полученный по такой же методике с той же композицией горючее/окислитель, но в присутствие воздуха, содержал только оксиды кобальта CoO и Co3O4 [1]. Во всех образцах помимо галлуазита (согласно данным РФА в образце III он находится в двух модификациях – кроме обычной гексагональной также и в моноклинной) обнаруживается также SiO2, содержащийся в виде примеси в исходном галлуазите.
Результаты измерения удельной поверхности исходного галлуазита и катализаторов на его основе приведены в табл. 1.
Таблица 1.
Удельная поверхность образцов исходного галлуазита и катализаторов на его основе
| Образец | Sуд, м2/г |
|---|---|
| Галлуазит | 58.3 |
| I | 63.1 |
| II | 64.1 |
| III | 70.0 |
| Примечание: I – 10 мас. % Ni, II – 10 мас. % Co, III – 5 мас. % Co + 5 мас. % Ni. | |
Несколько бóльшую удельную поверхность образцов катализаторов можно объяснить добавкой более высокодисперсной, по сравнению с галлуазитом, АФ, что подтверждается результатами исследования морфологии образцов катализаторов (рис. 3).
Рис. 3.
Морфология образцов катализаторов по данным СЭМ: a – I, б – II, в – III (I – 10 мас. % Ni, II – 10 мас. % Co, III – 5 мас. % Co + 5 мас. % Ni).

На всех микрофотографиях на внешней поверхности образцов наблюдаются зерна АФ, сравнимые или более мелкие по размерам, чем ГНТ. Структура нанотрубок галлуазита сохраняется практически полностью за исключением образца I, где ГНТ в значительной мере укорочены и раздроблены, по-видимому, вследствие более высокой температуры горения при синтезе этого катализатора. Результаты элементного анализа зерен катализаторов в областях, отмеченных на рис. 3, приведены в табл. 2
Таблица 2.
Элементный состав катализаторов по данным рентгеноспектрального микроанализа в точках, указанных на рис. 3
| № точки | Рис. 3а | Рис. 3б | Рис. 3в | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| O | Al | Si | Ni | O | Al | Si | Co | O | Al | Si | Co | Ni | |
| мас. % | |||||||||||||
| 1 | 59.78 | 14.24 | 14.72 | 11.26 | 51.83 | 13.8 | 12.49 | 21.88 | 61.50 | 16.10 | 16.29 | 3.17 | 2.94 |
| 2 | 50.80 | 12.40 | 12.67 | 24.13 | 59.23 | 15.85 | 15.51 | 9.41 | 62.05 | 14.70 | 16.23 | 3.70 | 3.3 |
| 3 | 53.27 | 13.10 | 14.87 | 18.76 | 63.00 | 14.67 | 15.12 | 7.21 | 59.27 | 14.03 | 14.53 | 6.05 | 6.12 |
| 4 | 63.08 | 13.74 | 15.12 | 8.06 | 55.16 | 14.69 | 14.08 | 16.07 | 55.71 | 16.05 | 16.91 | 5.53 | 5.80 |
| 5 | 60.86 | 14.40 | 15.24 | 9.50 | 52.69 | 13.92 | 13.65 | 19.74 | 64.73 | 13.37 | 14.11 | 3.75 | 4.04 |
| 6 | 61.25 | 14.63 | 15.23 | 8.89 | 61.38 | 14.27 | 14.23 | 10.12 | 65.25 | 12.95 | 12.90 | 4.24 | 4.66 |
| 7 | 64.44 | 14.40 | 14.92 | 6.24 | 49.00 | 14.63 | 13.91 | 22.46 | 59.80 | 14.22 | 16.34 | 4.90 | 4.74 |
| 8 | 61.89 | 15.03 | 16.13 | 6.95 | 64.84 | 13.58 | 13.82 | 7.76 | 58.39 | 15.28 | 16.80 | 4.60 | 4.93 |
| 9 | 46.98 | 10.52 | 10.62 | 31.88 | 61.70 | 13.19 | 13.93 | 11.18 | 64.34 | 14.50 | 14.65 | 3.26 | 3.25 |
| 10 | 66.96 | 13.98 | 14.37 | 4.69 | 53.13 | 12.82 | 13.09 | 20.96 | 59.58 | 14.17 | 16.98 | 4.52 | 4.75 |
| 11 | 59.39 | 14.96 | 16.05 | 9.6 | 57.37 | 14.60 | 15.20 | 12.83 | 52.75 | 14.48 | 14.00 | 9.41 | 9.36 |
| 12 | 62.56 | 14.50 | 15.31 | 7.63 | 51.89 | 13.51 | 12.44 | 22.16 | 49.05 | 11.06 | 10.55 | 15.11 | 14.23 |
| 13 | 54.81 | 15.23 | 15.80 | 14.16 | 63.70 | 13.54 | 16.68 | 6.07 | 60.84 | 14.52 | 14.86 | 4.77 | 5.01 |
| 14 | 60.23 | 14.99 | 15.60 | 9.18 | 60.02 | 14.81 | 14.98 | 10.19 | 62.83 | 14.84 | 15.46 | 3.45 | 3.42 |
| 15 | 60.00 | 14.87 | 15.73 | 9.40 | 59.74 | 15.02 | 15.90 | 9.34 | 55.83 | 15.09 | 16.18 | 6.50 | 6.40 |
| 16 | – | – | – | – | 49.73 | 11.49 | 11.21 | 27.57 | 61.10 | 14.16 | 14.23 | 5.30 | 5.21 |
| 17 | – | – | – | – | 62.09 | 14.20 | 16.10 | 7.61 | 57.94 | 16.09 | 15.75 | 5.84 | 4.38 |
| 18 | – | – | – | – | 60.13 | 12.64 | 13.50 | 13.73 | 62.16 | 13.18 | 13.07 | 6.08 | 5.51 |
| 19 | – | – | – | – | 61.71 | 15.38 | 15.97 | 6.94 | 54.90 | 14.78 | 15.27 | 7.73 | 7.32 |
| 20 | – | – | – | – | – | – | – | – | 59.20 | 15.54 | 16.18 | 4.64 | 4.44 |
| Среднее | 59.09 | 14.08 | 14.83 | 11.99 | 57.28 | 13.90 | 14.18 | 13.56 | 59.36 | 14.49 | 15.12 | 5.59 | 5.45 |
Из данных табл. 2 следует, что средняя поверхностная концентрация активных металлов достаточно хорошо воспроизводит их расчетное объемное содержание, хотя разброс поверхностных концентраций между зернами образцов достаточно велик (примерно в три раза). Возможно, это объясняется различной ориентацией ГНТ в зернах, представляющих собой достаточны рыхлые конгломераты. При исследовании морфологии поверхности катализаторов оказалось, что гранулы АФ преимущественно тяготеют к изломам ГНТ, выходам их внутренних каналов. По-видимому, при пропитке образцов раствор вследствие капиллярного эффекта заполняет прежде всего внутренние каналы ГНТ, и в процессе горения АФ образуются внутри и на торцах ГНТ.
На первом этапе каталитических исследований образцы полученных катализаторов были испытаны в процессе глубокого окисления. Результаты представлены на рис. 4.
Рис. 4.
Зависимости конверсий СО (1) и пропана (2) от температуры в присутствии образцов катализаторов: a – I, б – II, в – III (I – 10 мас. % Ni, II – 10 мас. % Co, III – 5 мас. % Co + 5 мас. % Ni).
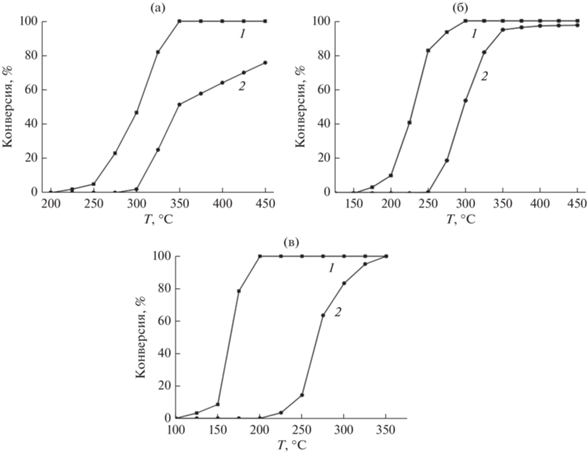
Как следует из рис. 4, наиболее низкую активность в этом процессе ожидаемо проявил образец катализатора I. Полная конверсия СО достигается только при температуре 350°С, а конверсия пропана даже при 450°С не превышает и 80%. При сравнении образцов II и III можно заметить, что активность катализатора III заметно превосходит таковую для катализатора II, несмотря на то, что содержание более активного в окислении элемента, кобальта, в образце II в два раза больше. Превышение температуры 100%-ной конверсии СО для катализатора II по сравнению с III соcтавляет 100°C, а температуры 90%-ной конверсии пропана – около 30°С, при том что на образце II 100%-ная конверсия пропана не достигается вплоть до 450°С. Причина, очевидно, заключается в разном химическом составе АФ образцов. У катализатора III, как было отмечено выше, АФ состоит из оксидов кобальта, не считая смешанного оксида кобальта-никеля, гораздо более активных в процессе окисления, чем алюминат кобальта в образце II. Факт низкой активности алюминатов металлов, в частности, кобальта, по сравнению с оксидами этих металлов был отмечен нами ранее [29]. Отметим, что результаты, зафиксированные в присутствии образца III, находятся на уровне показателей специально разработанного для процесса глубокого окисления и полученного таким же методом катализатора на основе γ-Al2O3 с гораздо более активной в этом процессе фазой Co10%–Mn5%, имеющего удельную поверхность 155 м2/г [7]. На катализаторе, содержащем 10 мас. % (Ni + Co), нанесенных на наноструктурированный CeO2, в процессе окисления СО температура 90%-ной конверсии T90 = 196°C, что практически совпадает с нашими результатами, а в глубоком окислении пропана Т50 = 322°С, что примерно на 50°С выше таковой для образца III (рис. 4в) [35]. Более того, даже при 600°С конверсия пропана на этом катализаторе составляет всего 88.2%. Таким образом, в процессах глубокого окисления СО и пропана полученные катализаторы продемонстрировали очень высокую активность.
Рис. 5.
Зависимости концентраций СО2 (1, 4), СН4 (2, 5) и СО (3, 6) от температуры в процессе гидрирования СО2 на исходных (1, 2, 3) и восстановленных (4, 5, 6) образцах катализаторов: a – I, б – II, в – III (I – 10 мас. % Ni, II – 10 мас. % Co, III – 5 мас. % Co + 5 мас. % Ni).
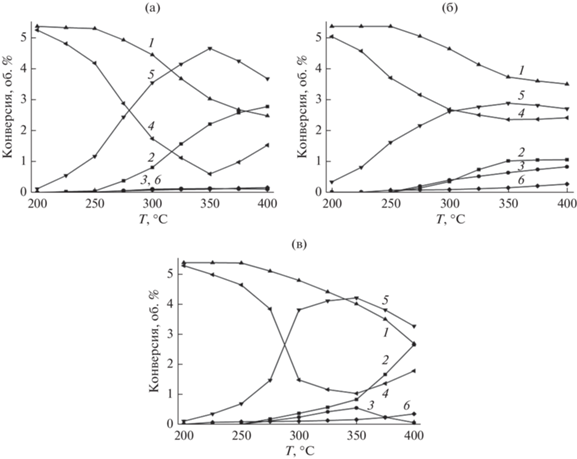
Рис. 6.
Рентгенограммы восстановленных образцов катализаторов: a – I, б – II, в – III (I – 10 мас. % Ni, II – 10 мас. % Co, III – 5 мас. % Co + 5 мас. % Ni).
Следующим этапом исследований было испытание катализаторов в реакции гидрирования СО2 при атмосферном давлении. Вначале гидрирование проводили на исходных синтезированных образцах. Затем образцы подвергали восстановлению, после чего процесс гидрирования повторяли. Результаты приведены на рис. 5.
Основными продуктами процесса, как можно видеть, являются метан и СО. Конверсия СО2 и выход метана монотонно растут с повышением температуры. Наиболее активны среди исходных образцов никельсодержащие катализаторы – I, а затем III. Конверсия СО2 на образце I при 400°С достигает 53.8% при селективности по метану 96.2%, а на образце III – 50.1 и 98.1% соответственно. После восстановления активность всех образцов (а в случае образца II – и селективность по метану) кардинально возрастают, однако ряд активности I > III > II сохраняется. Максимальная конверсия СО2 на восстановленном образце I теперь уже составляет 88.8% при селективности по метану 97.7%, а на образце III – 81.0 и 96.6% соответственно. Следует указать, что эти показатели зафиксированы при 350°С, а при дальнейшем повышении температуры они падают. Экстремальные зависимости конверсии СО2 и выхода метана с максимумами в области 300–350°С нами были отмечены ранее для полиметаллических катализаторов, полученных методом самораспространяющегося высокотемпературного синтеза (СВС) [30]. Мы объяснили это явление как обратимую дезактивацию катализаторов в этом процессе в области Т > 350°С, поскольку такая зависимость повторялась неоднократно. И тогда, и в настоящем исследовании такой ее характер сохранялся после 3–4 повторных экспериментов без заметного изменения активности и селективности катализаторов. Механизм этого явления пока не вполне ясен. Отметим, что похожая экстремальная зависимость выхода метана на катализаторе была также обнаружена в работе [36] на никелевых катализаторах (10 мас. % высокодисперсного Ni на различных оксидах) в интервале температур 300–400°С, когда максимальная конверсия СО2 составила ≤80% при селективности по метану около 93%. На катализаторе 15 мас. % Ni/γ-Al2O3, приготовленном методом упаривания и прокалки кислой смеси растворов солей Al и Ni и обладающем удельной поверхностью 242 м2/г, конверсия СО2 при 350°С была 45%, селективность по метану – 91% [37]. В наших экспериментах выход метана в расчете на пропущенный СО2 в присутствии образца I был несколько выше (87 против 85%), чем на ранее полученном нами этим же методом катализаторе Co10%–Ni10%/ZSM-5 с удельной поверхностью 236 м2/г [7]. В работе [38] на образце 2% Co/ZrO2 c самой высокой дисперсностью кобальта, приготовленном пропиткой из растворов органических кислот, при 400°С конверсия СО2 составила 85%, а селективность по метану – 99%. Отметим, что такие показатели, сравнимые с нашими, были достигнуты при давлении 3 МПа.
Для понимания причины столь сильного повышения активности катализаторов после восстановления был проведен РФА восстановленных образцов. Рентгенограммы приведены на рис. 6.
Как следует из полученных результатов, после восстановления в составе АФ образцов I и III появляются фазы металлического никеля, который, как известно, обладает высокой активностью в реакциях гидрирования. На образце II имеет место восстановление исходной фазы алюмината кобальта до оксида Co(II) и, возможно, до металлического кобальта (под вопросом). Судя по составу исходного катализатора III, в котором присутствуют простые и сложные оксиды Со, и по его активности, оксиды кобальта гораздо более активны в процессе гидрирования, чем алюминат кобальта в составе исходного образца II.
С целью исследования процесса восстановления катализаторов были сняты кривые термопрограммирумого восстановления (ТПВ) образцов (рис. 7).
Рис. 7.
Кривые ТПВ образцов катализаторов: a – I, б – II, в – III (I – 10 мас. % Ni, II – 10 мас. % Co, III – 5 мас. % Co + + 5 мас. % Ni).
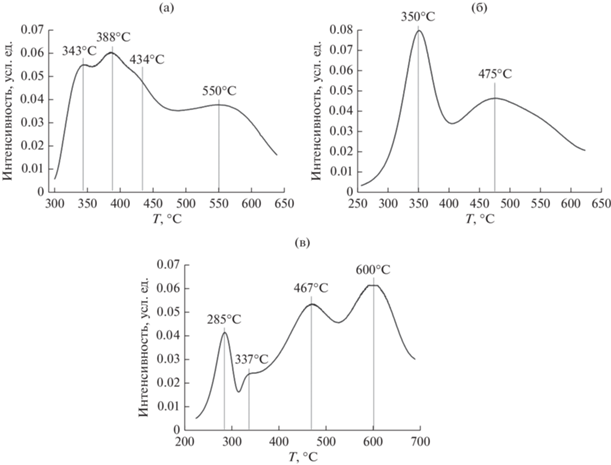
В интервале температур восстановления до 400°С на кривой ТПВ катализатора I идентифицируются два близких пика с максимумами при 343 и 388°С и слабый пик в районе перегиба при 434°С. Сравнивая РФА исходных и восстановленных образцов (рис. 2a и 6a), можно констатировать, что происходит практически полное восстановление оксида никеля (содержание NiO под вопросом) до металла. В работе [31] на кривой ТПВ катализатора Ni–Ce–La–O в этой области температур был обнаружен широкий пик с максимумами при 340, 390°С и слабым максимумом на нисходящей ветви в районе 430–440°С. Авторы приписывают этот пик восстановлению катионов никеля в разных структурах – как в составе частиц и кластеров оксида никеля, так и в твердых растворах. По данным рис. 2б и 6б, схема восстановления АФ катализатора II выглядит следующим образом: Co2AlO4 = Co(II)Co(III)AlO4 → CoO (+ Al2O3). Дальнейшее восстановление СоО если и протекает, то лишь в малой степени (обнаружение металлического кобальта, как уже отмечалось, под вопросом). В работе [32] было проведено ТПВ шпинелей Co3O4 и Co2MgO4. При восстановлении Co3O4 на кривой ТПВ обнаружено два пика при 390 и 485°С. Первый пик идентифицирован как соответствующий стадии восстановления Со3+ → → Со2+, второй – Со2+ → Со0. В случае Co2MgO4 два пика, отнесенные к этим же стадиям, смещены в область более низких температур – 379 и 455°С соответственно. Резонно предположить, что пики на рис. 7б также отвечают двум последовательным стадиям восстановления Co2AlO4, несколько смещенным по сравнению с Co2MgO4. Заметим, что температуры последовательного восстановления Co3O4 сильно зависят от морфологии этого оксида. Для наностержней Co3O4 они лежат в интервале 375–308 и 374–418°С [33], для других наноформ эти стадии протекают при 200–270 и 270–370°С соответственно [34]. Наконец, согласно данным РФА (рис. 2в и 6в) восстановление образца III представляет собой достаточно сложный процесс. Металлический никель может появляться только при восстановлении сложного оксида Co1.29Ni1.71O4 одновременно с выделением CoO. Кроме того, низший оксид кобальта образуется также при восстановлении высшего его оксида в исходном образце. ТПВ сложного оксида NixCo3– xO4 (x ≤ 1) [34] продемонстрировало наличие двух пиков в области 200–400°С, приписываемых процессу одновременного восстановления ионов кобальта и никеля. Если, по данным [34], ионный состав сложного оксида NiCo2O4 выглядит как ${\text{N}}{{{\text{i}}}^{{2 + }}}{\text{Co}}_{2}^{{3 + }}{\text{O}}_{4}^{{2 - }},$ то оксид Co1.29Ni1.71O4 можно записать в виде ${\text{Ni}}_{{{\text{0}}{\text{.71}}}}^{{{\text{3 + }}}}{\text{N}}{{{\text{i}}}^{{{\text{2 + }}}}}{\text{Co}}_{{{\text{1}}{\text{.29}}}}^{{{\text{3 + }}}}{\text{O}}_{4}^{{2 - }}{\text{.}}$ В таком случае первый пик при 285°С соответствует первой стадии процесса восстановления высших оксидов кобальта и никеля, а пик при 337°С (по аналогии с первым пиком ТПВ образца I) – частичному восстановлению NiO до металла.
Вероятно, процессы восстановления катализаторов под влиянием реакционной среды могут иметь место и в результат многократного повторения опытов, однако таких данных у нас пока нет. Как было отмечено выше, после 3–4 экспериментов заметных изменений в активности и характере ее зависимости от температуры выявлено не было.
ЗАКЛЮЧЕНИЕ
Методом низкотемпературного горения смесей нитратов металлов и мочевины (температуры во фронте волны горения при синтезе образцов составляют 272–332°С) получены образцы нанесенных катализаторов на основе галлуазитных нанотрубок (ГНТ) с содержанием (по металлу) 10 мас. % Ni (I), 10 мас. % Co (II) и 5 мас. % Co + + 5 мас. % Ni (III). Согласно данным РФА, активные фазы катализаторов содержат простые и сложные оксиды металлов. Значения удельной поверхности образцов лежат в интервале 63.1–70.0 м2/г, что несколько превышает поверхность галлуазита (58.3 м2/г) и говорит о высокой дисперсности частиц АФ с характерными размерами, меньшими, чем таковые ГНТ (20–30 нм по внутренней полости). По данным сканирующей электронной микроскопии ГНТ в основном сохраняют свою форму после синтеза, но становятся существенно короче, что наиболее заметно для образца I с наивысшей температурой горения. В процессе глубокого окисления при объемной скорости 120 000 ч–1 наиболее активными оказались кобальтсодержащие катализаторы, особенно образец III, в присутствии которого полная конверсия СО достигается при 200°С, а пропана – при 350°С. Ряд активности исходных образцов катализаторов в процессе гидрирования СО2 выглядит как I > III > II. Конверсия СО2 на образце I при 400°С составляет 53.8%, селективность по метану – 96.2%. Восстановление катализаторов в токе водорода при 400°С в течение 1 ч приводит к резкому возрастанию их активности, а в случае образца II – и селективности образования метана. Ряд активности при этом сохраняется, но максимальная конверсия СО2, равная 88.8% при селективности по метану 97.7%, получена на образце I уже при температуре 350°С. На основании данных РФА восстановленных катализаторов и ТПВ высказаны некоторые предположения о возможных причинах роста активности и схемах восстановления АФ.
Список литературы
Завьялова У.Ф., Третьяков В.Ф., Бурдейная Т.Н., Лунин В.В., Шитова Н.Б., Рыжова Н.Д., Шмаков А.Н., Низовский А.И., Цырульников П.Г. // Кинетика и катализ. 2005. Т. 46. № 5. С. 795. (Zav’yalova U.F., Tret’yakov V.F., Burdeinaya T.N., Lunin V.V., Shitova N.B., Ryzhova N.D., Shmakov A.N., Nizovskii A.I., Tsyrul’nikov P.G. // Kinet. Catal. 2005. V. 46. P. 752.)
Zavyalova U., Scholz P., Ondruschka B. // Appl. Catal. A: Gen. 2007. V. 323. P. 226.
Афонасенко Т.Н., Шляпин Д.А., Леонтьева Н.Н., Гуляева Т.И., Буяльская К.С., Тренихин М.В., Цырульников П.Г. // Кинетика и катализ. 2011. Т. 52. № 6. P. 864. (Afonasenko T.N., Shlyapin D.A., Leont’eva N.N., Gulyaeva T.I., Buyal’skaya K.S., Trenikhin M.V., Tsyrul’nikov P.G. // Kinet. Catal. 2011. V. 52. P. 843.)
Mironenko O.O., Shitova N.B., Kotolevich Y.S., Sharafutdinov M.R., Struikhina N.O, Smirnova N.S., Kochubey D.I., Protasova O.V., Trenikhin M.V., Stonkus O.A., Zaikovskii V.I., Goncharov V.B., Tsyrul’nikov P.G. // Int. J. SHS. 2012. V. 21. № 2. P. 139.
Kotolevich Y.S., Khramov E.V., Mironenko O.O., Zubavichus Ya.V., Murzin V.Yu., Frey D.I., Metelev S.E., Shitova N.B., Tsyrulnikov P.G. // Int. J. SHS. 2014. V. 23. № 1. P. 9.
Kotolevich Y.S., Mamontov G.V., Vodyankina O.V., Petrova N.I., Smirnova N.S., Tsyryul’nikov P.G., Trenikhin M.V., Nizovskii A.I., Kalinkin A.V., Smirnov M.Y. Goncharov V.B. // Int. J. SHS. 2017. V. 26. № 4. P. 234.
Borshch V.N., Dement’eva I.M., Khomenko N.Yu. // Int. J. SHS. 2019. V. 28. № 1. P. 45.
Guimarães L., Enyashin A.N., Seifert G., Duarte H.A. // J. Phys. Chem. C. 2010. V. 114. P. 11358.
Yuan P., Tan D., Annabi-Bergaya F. // Appl. Clay Sci. 2015. V. 112–113. P. 75.
Hillier S., Brydson R., Delbos E., Fraser T., Gray N., Pendlowski H., Phillips I., Robertson J., Wilson I. // Clay Miner. 2016. V. 51. № 3. P. 325.
Kamble R., Ghag M., Gaikawad S., Panda B.K. // J. Adv. Sci. Res. 2012. V. 3. № 2. P. 25.
Peixoto A.F., Fernandes A.C., Pereira C., Pires J., Freire C. // Micropor. Mesopor. Mater. 2016. V. 219. P. 145.
Barrientos-Ramírez S., Oca-Ramírez G.M., Ramos-Fernández E.V., Sepúlveda-Escribano A., Pastor-Blas M.M.M., González-Montiel A. // Appl. Catal. A: Gen. 2011. V. 406. P. 22.
Zou M., Dub M., Zhan M., Yang T., Zhu H., Wang P., Bao S. // Mater. Res. Bull. 2015. V. 61. P. 375.
Jiang J., Zhang Y., Yan L., Jiang P. // Appl. Surf. Sci. 2012. V. 258. P. 6637.
Matusik J., Wścisło A. // Appl. Clay Sci. 2014. V. 100. P. 50.
Silva S.M., Peixoto A.F., Freire C. // Appl. Catal. A: Gen. 2018. V. 568. P. 221.
Abbasov V.M., Ibrahimov H.C., Mukhtarova G.S., Abdullayev E. // Fuel. 2016. V. 184. P. 555.
Massaro M., Noto R., Riela S. // Catalysts. 2022. V. 12. № 2. P. 149.
Ahmad Y.H., Mohamed A.T., Hassan W.M., Soliman A., Mahmoud K.A., Aljaber A.S., Al-Qaradawi S.Y. // Appl. Surf. Sci. 2019. V. 493. P. 70.
Wang L., Chen J., Rudolph V., Zhu Z. // Adv. Powder Tech. 2012. V. 23. № 4. P. 465.
Wang L., Chen J., Ge L., Rudolph V., Zhu Z. // J. Phys. Chem. C. 2013. V. 117. P. 4141.
Ouyang J., Zhao Z., Yang H., Zhang Y., Tang A. // Appl. Clay Sci. 2018. V. 152. P. 221.
Li W.-J., Wey M.-Y. // Sci. Total Env. 2019. V. 675. P. 397.
Zheng Y., Wang L., Zhong F., Cai G., Xiao Y., Jiang L. // Ind. Eng. Chem. Res. 2020. V. 59. P. 5636.
Pechenkin A., Potemkin D., Rubtsova M., Snytnikov P., Plyusnin P., Glotov A. // Catalysts. 2021. V. 11. № 10. P. 1151.
Bentabet A., Chaoui Z., Aydin A., Azbouche A. // Vacuum. 2010. V. 85. № 2. P. 156.
Bentabet A. // Mod. Phys. Lett. B. 2014. V. 28. № 1. P. 1450006.
Борщ В.Н., Жук С.Я., Вакин Н.А., Смирнов К.Л., Боровинская И.П., Мержанов А.Г. // Докл. АН. 2008. Т. 420. № 4. С. 496. (Borshch V.N., Zhuk S.Ya., Vakin N.A., Smirnov K.L., Borovinskaya I.P., and Merzhanov A.G. // Doklady Phys. Chem. 2008. V. 420. Part 2. P. 121.)
Борщ В.Н., Пугачева Е.В., Жук С.Я., Смирнова Е.М., Демихова Н.Р., Винокуров В.А. // Изв. РАН. Сер. Хим. 2020. № 9. С. 1697. (Borshch V.N., Pugacheva E.V., Zhuk S.Ya., Smirnova E.M., Demikhova N.R., Vinokurov V.A. // Russ. Chem. Bull. Int. Ed. 2020. V. 69. № 9. Р. 1697.)
Матус Е.В., Нефедова Д.В., Сухова О.Б., Исмагилов И.З., Ушаков В.А., Яшник С.А., Никитин А.П., Керженцев М.А., Исмагилов З.Р. // Кинетика и катализ. 2019. Т. 60. № 4. С. 532. (Matus E.V., Nefedova D.V., Sukhova O.B., Ismagilov I.Z., Ushakov V.A., Yashnik S.A., Nikitin A.P., Kerzhentsev M.A., Ismagilov Z.R. // Kinet. Catal. 2019. V. 60. P. 496.)
Zigla A.A., Kox T., Mevoa D., Assaouka H.T., Nsangou I.N., Daawe D.M., Kenmoe S., Kouotou P.M. // Catalysts. 2022. V. 12. № 4. P. 411.
Ma L., Seo C.Y., Chen X., Sun K., Schwank J.W. // Appl. Catal. B: Env. 2018. V. 222. P. 44.
Владимирова С.А. Дис. … к. х. н. М.: МГУ, 2019.
Liu Z., Li J., Wang R.Ph.D. // J. Coll. Interf. Sci. 2020. V. 560. P. 91.
Muroyama H., Tsuda Y., Asakoshi T., Masitah H., Okanishi T., Matsui T., Eguchi K. // J. Catal. 2016. V. 343. P. 178.
Aljishi A., Veilleux G., Lalinde J.A.H., Kopyscinski J. // Appl. Catal. A: Gen. 2018. V. 549. P. 263.
Li W., Liu Y., Mu M., Ding F., Liu Z., Guo X., Song C. // Appl. Catal. B: Env. 2019. V. 254. P. 531.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ