

Кинетика и катализ, 2022, T. 63, № 6, стр. 837-844
Исследование процессов с участием адсорбированного озона, стимулированных резонансным возбуждением колебательных состояний
О. С. Песцов a, Т. Р. Аминев a, А. А. Цыганенко a, *, **
a ФГБОУ ВО Санкт-Петербургский государственный университет
199034 Санкт-Петербург, Университетская набережная, д. 7–9, Россия
* E-mail: atsyg@yandex.ru
** E-mail: a.tsyganenko@spbu.ru
Поступила в редакцию 11.05.2022
После доработки 26.06.2022
Принята к публикации 26.06.2022
- EDN: OCHJZU
- DOI: 10.31857/S0453881122060107
Аннотация
Исследовано воздействие лазерного ИК-излучения на процессы разложения озона и озонолиза цис-дихлорэтилена, адсорбированного на диоксиде титана. Обнаружено, что резонансное возбуждение озона смешанного изотопного состава на частоте составного колебания ν1 + ν3 молекул, адсорбированных на оксиде титана, приводит к незначительному уменьшению относительной интенсивности полосы изотопной модификации, на частоте которой производится облучение. Облучение ускоряет протекание реакции озонолиза дихлорэтилена, однако наблюдение изотопной селективности этого процесса затруднено одновременным образованием нескольких продуктов, полосы поглощения которых перекрываются.
ВВЕДЕНИЕ
Идея использования резонансного лазерного возбуждения для селективного инициирования различных процессов в молекулярных системах привлекает внимание специалистов вот уже более полувека [1]. Большинство исследований резонансного взаимодействия лазерного излучения с веществом было выполнено для газов. Однако энергии активации гомогенных химических реакций обычно превышают энергию квантов инфракрасного лазера, а ангармонизм молекулярных колебаний препятствует многофотонному резонансному возбуждению молекул до высоких энергий [2].
Гетерогенные процессы характеризуются низкой энергией активации по сравнению с гомогенными, и отличаются высокой чувствительностью к небольшим изменениям энергии возбуждения участвующих в них молекул. Поглощение одного инфракрасного кванта молекулой может существенно изменить ее поведение, что позволяет использовать относительно низкие мощности лазерного излучения.
Карлов и др. [2] сообщили о первых результатах экспериментов, когда колебательное возбуждение определенных изотопсодержащих молекул СО2 приводило к различию сорбционных свойств на охлаждаемой поверхности по сравнению с невозбужденными молекулами. Совсем недавно [3] было показано, что инфракрасное лазерное излучение влияет на процесс кластеризации молекул CF3Br в их смеси с аргоном в зоне газодинамического расширения. Обнаружено, что резонансное колебательное возбуждение молекул непрерывным излучением CO2-лазера ведет к селективному по изотопам брома подавлению кластеризации.
Более перспективным представляется применение лазеров для инициирования поверхностных процессов, индуцированных прямым возбуждением колебательных состояний адсорбированных частиц. Были предприняты многочисленные попытки осуществить резонансную лазерную десорбцию, когда возбуждение инфракрасным лазерным излучением способствует десорбции тех адсорбированных молекул, частота колебаний которых совпадает с частотой колебаний возбуждающего излучения. Важнейшей прикладной задачей этих исследований была оценка возможности использования резонансного возбуждения колебательных состояний поверхностных соединений для разделения изотопов.
Резонансное возбуждение колебаний адсорбированных молекул осуществлялось в экспериментах по лазерной десорбции СО [4–6], CH3F [7] и аммиака [8, 9]. Однако было показано, что облучение смеси на частоте колебаний одного изотополога не приводит к его ожидаемой селективной десорбции, по-видимому, по этой причине интерес к подобным исследованиям в последующие годы несколько ослаб. Можно отметить лишь статьи по резонансной десорбции N2O с поверхности хлорида натрия [10] и CD3F с поверхности NaCl [11].
Redlich и др. [12] изучали индуцированную ИК-лазером десорбцию из многослойной структуры различных изотопологов молекул метана, конденсированных на монокристаллической поверхности NaCl (100) с использованием времяпролетной масс-спектрометрии. Десорбция из слоев чистого CH4, CD4 или CD3H, а также из слоя смеси двух разных изотопных соединений исследовалась в зависимости от длины волны воздействующего инфракрасного излучения перестраиваемого лазера на свободных электронах. Десорбция метана происходила только тогда, когда возбуждающий свет находился в резонансе с внутренними колебательными модами молекул. Для слоев смешанных изотопных соединений десорбция наблюдалась только на резонансных частотах, и не зависела от плотности энергии возбуждающего лазера. Было показано, что возбуждение колебаний одного изотополога вызывает десорбцию другого.
Очевидно, что в слое адсорбированных молекул происходит достаточно быстрый обмен энергией колебательного возбуждения. Чтобы понять причины отсутствия изотопной селективности, необходимо проанализировать данные о механизме и эффективности этого обмена, о времени жизни и скорости диссипации колебательного возбуждения и, насколько возможно, о вероятности стимулирования десорбции или реакции колебательно возбужденных поверхностных соединений. Такой анализ был нами произведен [13], и в результате него были сформулированы требования к системам, где можно было бы ожидать изотопной селективности.
Время диссипации колебательной энергии поглощающей молекулы должно быть достаточно большим, т.е. это должны быть не металлы, в которых возбуждение электронно-дырочной пары приводит к быстрому затуханию колебательного возбуждения, а скорее изоляторы или широкозонные полупроводники. Чтобы минимизировать генерацию фононов, молекулы должны быть слабо связаны с поверхностью, а частота возбужденных молекулярных колебаний должна быть как можно дальше от частот фононных мод. Учитывая, что для большинства оксидных адсорбентов колебания решетки лежат вблизи 1000 см–1 или ниже, частоту возбуждающего излучения предпочтительно выбирать не ниже 2000 см–1. Энергия кванта при такой частоте (>25 кДж/моль) достаточно велика для десорбции слабосвязанных молекул, для инициирования изомерных переходов в системах с изомерией связывания или для стимулирования реакций промежуточных поверхностных соединений, стабилизированных при ‒196°С. Она существенно выше средней энергии теплового движения, что исключает спонтанное возбуждение подобных колебательных состояний даже при комнатной температуре.
Обмен колебательной энергией в слое адсорбированных молекул происходит по механизму резонансного диполь-дипольного взаимодействия [14]. Его эффективность зависит от колебательной поляризуемости молекулы, определяющей коэффициент экстинкции соответствующего колебания, и от расстояния между взаимодействующими молекулами. Минимизировать обмен энергией можно путем выбора молекул с не слишком высокой интенсивностью колебательных полос и использования сравнительно малых покрытий поверхности.
Кроме того, для исключения обратной реакции процесс должен быть по возможности необратимым. Совместить все перечисленные требования в одной системе не представляется возможным. Придавая первоочередное значение необратимости, в качестве объекта исследования в нашей работе мы выбрали озон. Его диссоциацию в газовой фазе при интенсивном многофотонном возбуждении CO2-лазером наблюдали Proch и Schröder [15]. Озон, адсорбированный на некоторых оксидных катализаторах, легко разлагается при комнатной температуре, а на активном CeO2 уже при –196°С эта реакция может иметь взрывной характер [16]. ИК-исследования показали, что на поверхности оксидов титана и церия молекулы озона деформируются, теряя свою симметрию [16, 17], а их полосы поглощения заметно смещены по отношению к частотам газообразного O3. Предполагалось, что эта форма адсорбции является промежуточной в реакции разложения озона. Полоса составного колебания ν1 + ν3 в области 2100 см–1 спектра озона смешанного изотопного состава, хемосорбированного на TiO2, расщеплена на восемь отдельных пиков, каждый из которых удалось однозначно интерпретировать [18], что позволяет избирательно возбуждать молекулы конкретного изотополога озона, определенным образом связанного с поверхностным атомом титана. Попыток инициировать селективное разложение адсорбированного озона с помощью резонансного возбуждения ИК-лазером, насколько нам известно, ранее не предпринималось.
В адсорбированном состоянии озон легко реагирует с некоторыми молекулами. Так, озонолиз этилена, адсорбированного на оксиде кремния, активно идет даже при –196°С [19]. С хлорпроизводными этилена реакция происходит медленнее, но поверхность оксида титана катализирует протекание озонолиза дихлорэтилена [20], скорость которого можно замедлить, гидратируя поверхность TiO2. Попытка стимулировать озонолиз резонансным возбуждением колебательных состояний адсорбированного озона представляется весьма привлекательной, поскольку мы имеем возможность детектировать продукты реакции, отсутствовавшие в исходном спектре, тогда как наблюдать изменения интенсивности полос поглощения самого озона, который при –196°С, имея достаточное давление паров (около 10–2 Торр), продолжает адсорбироваться и десорбироваться, представляется более сложной задачей.
В связи со сказанным выше задачей настоящей работы было исследование возможности инициирования резонансным возбуждением колебательных состояний озона, адсорбированного на диоксиде титана, процессов его разложения и реакции озонолиза совместно адсорбированного цис-дихлорэтилена. Применение изотопного замещения должно дать сведения о селективности процессов, инициируемых возбуждением определенного изотополога озона.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Образцы TiO2 были спрессованы из порошка коммерческого производства Hombifine N Anatase с удельной поверхностью 340 м2/г. Образец подвергали термовакуумной обработке при 450°С в течение 30 мин, затем следовали прогрев при той же температуре в кислороде (10–20 Торр) в течение 15 мин и еще 15 мин вакуумирования. Образец охлаждали до комнатной температуры в присутствии около 1 Торр кислорода, чтобы избежать восстановления его поверхности. После, для понижения его реакционной способности, при комнатной температуре в объем с образцом напускали несколько Торр паров H2O и снова подвергали термовакуумной обработке при 90°С в течение 20 мин, после чего кювету охлаждали и изучали эффект резонансного воздействия ИК-излучения на частоте составного колебания ν1 + ν3 адсорбированного озона. Адсорбцию озона и облучение, как правило, осуществляли при температуре кипения жидкого азота (–196°С). Для исследования озонолиза напуск цис-дихлорэтилена производили при –50°С, после чего следовало охлаждение до температуры кипения жидкого азота и адсорбция озона.
Для обработки образца и синтеза озона применяли кислород, образующийся при нагревании перманганата калия. Для приготовления изотопозамещенного озона использовали 18O2 c обогащением 76%. Озон получали в высокочастотном разряде, создаваемом с помощью трансформатора Тесла при вымораживании жидким азотом.
Обработку образца и регистрацию спектров проводили с помощью низкотемпературной вакуумной кюветы из нержавеющей стали, устройство которой подробно описано в работе [21]. Давление измеряли с помощью абсолютных мембранно-емкостных датчиков Barocel (“Edwards”, Великобритания) с пределами измерения 0.1–1000 и 10–3–10 Торр. Держатель с образцом для термообработки без нарушения герметизации переносили магнитом в кварцевую трубку, присоединенную к верхней части кюветы, а для адсорбции и регистрации спектров помещали в нижнюю охлаждаемую часть, снабженную окнами из селенида цинка. Температуру измеряли с помощью термопарного датчика, помещаемого в охлаждаемую часть кюветы. Для достижения теплового контакта образца с холодными стенками кюветы в объем с образцом добавляли 0.2–1 Торр гелия.
Спектры регистрировали с помощью ИК-Фурье спектрометра IR-Prestige-21 (“Shimadzu”, Япония) с разрешением 4 см–1. Для уменьшения влияния поглощения атмосферной воды и углекислого газа внутренний объем спектрометра продували сухим азотом. Регистрацию спектров и последующую их обработку осуществляли с помощью программ IR-Solution и MagicPlot.
Для облучения образца применяли лазер MIRcat Tunable Mid-IR External Cavity Laser System (“Daylight Solution”, США). Луч лазера вводили в оптическую схему спектрометра после интерферометра прибора с помощью моторизированного поворотного зеркала и фокусировали на центральной части образца. Имелась возможность перестраивать частоту излучения лазера в пределах от 1989 до 2330 см–1. Лазер использовали в импульсном режиме с частотой импульсов 1 МГц при длительности импульса 100 нс. Средняя мощность лазера для частот 2100 и 2200 см–1 составляла около 45 мВт и отличалась от этой величины не более чем на 30%, а на частоте 2022 см–1 была равна примерно 10% от мощности на частоте 2200 см–1. Для некоторых экспериментов использовали режим “sweep mode”, при котором частота перестаивается в указанных пределах на протяжении всего времени облучения со скоростью 5 см–1/с.
Все эксперименты с применением лазерного излучения проводили на базе ресурсного центра научного парка СПбГУ “Оптические и лазерные методы исследования вещества” (https://researchpark.spbu.ru/laser-rus).
РЕЗУЛЬТАТЫ ЭКСПЕРИМЕНТОВ
Адсорбция озона на TiO2 при –196°С приводит к появлению и постепенному повышению интенсивности полос поглощения (п. п.) адсорбированных молекул. На рис. 1 показан спектр озона смешанного изотопного состава (76% 18O3) в области составного колебания ν1 + ν3, где все кривые нормированы по низкочастотной полосе 1996 см–1, принадлежащей молекулам 18О3. Спектр 1 записан через 70 мин после начала адсорбции, после чего была произведена серия последовательных сеансов облучения лазером на частоте 2030–2020 см–1 в общей сложности в течение 115 мин. Как видно из рис. 1, облучение вызывает некоторое уменьшение интенсивности п. п. 2022 см–1 относительно п. п. 1996 см–1. При этом в промежутках между сеансами с течением времени наблюдается восстановление исходного соотношения между этими полосами. Последующее облучение на частоте 2200 см–1 в области, где отсутствуют п. п. озона, приводит к ускорению возврата к исходному соотношению интенсивностей. Общая интегральная интенсивность полос озона, которая по мере облучения на частоте 2030–2020 см–1 возрастала, при облучении на частоте 2200 см–1 несколько уменьшается.
Рис. 1.
ИК-спектр озона (18O3 76%), адсорбированного на TiO2, спустя 70 мин после напуска (1), после облучения на частоте 2030–2020 см–1 в течение 115 мин (2) и последующего облучения на частоте 2200 см–1 в течение 45 мин (3). Спектры нормированы по полосе 1996 см–1.
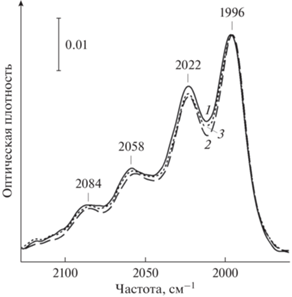
Влияние облучения наглядно видно на рис. 2, где показана зависимость отношения интегральных интенсивностей полос 2022 и 1996 см–1, определенных при разложении общего контура полосы на составляющие (гауссианы), от времени облучения. Уже после первого сеанса облучения в 25 мин на частоте 2030–2020 см–1 соотношение интенсивностей заметно изменяется в пользу низкочастотной полосы. Дальнейшее облучение на этой частоте увеличивает это соотношение, первоначально составляющее 1.32, до 1.49. Облучение на частоте 2200 см–1 показывает обратную динамику изменения соотношения интенсивностей полос поглощения.
Рис. 2.
Динамика изменения отношения интегральных интенсивностей полос поглощения при 1996 и 2022 см–1 в ИК-спектре озона при облучении на частоте 2030–2020 см–1 в течение 0 (1), 25 (2), 55 (3) и 115 мин (4) и последующем облучении на частоте 2200 см–1 в течение 20 (5) и 45 мин (6).

При адсорбции цис-дихлорэтилена на TiO2 в спектре появляются п. п. при 3165, 3080, 1590 и 1294 см–1, принадлежащие соответственно валентным C–H и C=C и деформационному CCH колебаниям адсорбированных молекул [22], п. п. поверхностных ОН-групп при 3720–3660 см–1 исчезают, и вырастает широкая полоса возмущенных гидроксилов при 3583 см–1. При охлаждении до температуры кипения жидкого азота (–196°С) существенных изменений в спектре адсорбированного цис-C2H2Cl2 не наблюдается.
Влияние адсорбции озона и облучения проиллюстрировано на рис. 3. Напуск озона 16О3 при –196°С на образец с предварительно адсорбированным цис-дихлорэтиленом приводит к появлению и постепенному увеличению интенсивности п. п. адсорбированного озона при 2106 и 1034 см–1. Одновременно наблюдается рост полосы при 2342 см–1, очевидно, принадлежащей колебанию ν3 СО2, а также полосы 1694 см–1 с более или менее выраженным плечом при 1732 см–1 и сопутствующей ей более слабой полосы при 1338 см–1. Полосы цис-дихлорэтилена при этом несколько ослабевают, чему в разностных спектрах на рис. 3 соответствует полоса отрицательного поглощения на частоте 1590 см–1. В некоторых экспериментах отмечается также появление полос при 2186 и 2158 см–1. Облучение на частоте 2115–2100 см–1 в течение 10 мин (рис. 3, спектр 2) ведет к почти полному исчезновению п. п. 1694 см–1, росту п. п. при 1801, 1650 и 1570 см–1 и усилению поглощения в области 3550–3000 см–1. При этом продолжает увеличиваться интенсивность полосы СО2 при 2342 см–1 и уменьшается интенсивность полос колебаний озона. Дальнейшее облучение (рис. 3, спектр 3) в течение 20 мин не вызывает такого резкого изменения спектра, как после первого раза – интенсивность п. п. озона, СО2 и 1694 и 1650 см–1 несколько увеличивается. Еще меньший эффект производит 20-минутное облучение на частоте 2210–2195 см–1.
Рис. 3.
ИК-спектр продуктов озонолиза цис-C2H2Cl2, адсорбированного на TiO2: после напуска озона 16O3 спустя 67 мин (1); после облучения лазером в течение 10 мин на частоте 2115–2100 см–1 (2); после дополнительных 20 мин облучения лазером на той же частоте (3); после нагревания до –100°C (4). Спектр образца до напуска озона вычтен.

Следует отметить, что в ряде экспериментов включение облучения приводило к разрушению образца. По-видимому, подобно тому, как озонолиз этилена, адсорбированного на оксиде кремния, может происходить со взрывом [19], в нашем случае схожий процесс может инициироваться ИК-облучением, и, если образец не разрушается, в начальный момент быстрая реакция происходит в той части образца, где концентрация реагентов для этого достаточна, после чего продолжается диффузия озона вглубь таблетки, сопровождающаяся более медленными изменениями в спектре.
Повышение температуры кюветы приводит к увеличению интенсивности п. п. озона и СО2, очевидно, вследствие их испарения со стенок кюветы. Полоса 1694 см–1 при этом практически исчезает, и на месте плеча при 1732 см–1 вырастает интенсивная полоса поглощения.
Реакция адсорбированного цис-дихлорэтилена с озоном смешанного изотопного состава сопровождается изменениями спектра, проиллюстрированными на рис. 4. После напуска озона наблюдается рост полосы поглощения при 1697 см–1 с плечами при 1732 и 1665 см–1, который продолжается при облучении на частоте 2025–2015 см–1, когда относительная интенсивность плеча при 1732 см–1 несколько повышается, а плеча при 1665 см–1, напротив, понижается. Такие же изменения происходят при облучении на частоте 2060–2045 см–1 и продолжаются при повышении температуры, когда интенсивность полосы 1697 см–1 с плечом при 1732 см–1 резко возрастает.
Рис. 4.
ИК-спектры продуктов озонолиза цис-C2H2Cl2 адсорбированного на TiO2, в высокочастотной (а) и низкочастотной (б) областях: спустя 40 мин после напуска озона 18O3 (76%) (1); после облучения в течение 100 мин на частоте 2025–2015 см–1 (2); после дополнительного облучения в течение 70 мин на частоте 2060–2045 см–1 (3); после нагревания до –100°C (4). Спектр образца до напуска озона вычтен. В высокочастотной области спектры разнесены по вертикальной оси.

Соотношение интенсивностей полос различных изотопологов СО2 и озона, показанных на рис. 4, позволяет оценить степень обогащения обеих молекул по 18О. Такая оценка для озона дает величину близкую к 76%, тогда как для СО2 получается около 70%.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Реакция разложения озона на поверхности оксида титана спонтанно происходит уже при ‒196°С, однако адсорбция воды с последующим удалением ее избытка откачкой при 90°С значительно понижают ее скорость, и становится возможным наблюдать за влиянием резонансного возбуждения колебаний озона ИК-излучением на частоте колебательного перехода ν1 + ν3 на ход реакции. Спектры адсорбированного СО показывают, что на поверхности такого гидратированного образца присутствуют координационно ненасыщенные ионы титана [23], на которых согласно квантово-химическому расчету молекулы озона хемосорбируются, связываясь терминальным атомом кислорода [24]. Положение полос озона, адсорбированного на гидратированном TiO2, немного отличается от данных предшествующих работ [17, 18]
К сожалению, интенсивность п. п. адсорбированного озона при –196°С в ходе эксперимента непостоянна. Его количество вначале пополняется за счет продолжающейся адсорбции из газовой фазы, а после того, как возрастающая с увеличением покрытия скорость спонтанного разложения озона сравнивается со скоростью его прибыли, начинается постепенное уменьшение покрытия из-за расходования общего количества озона в кювете. В результате наблюдать непосредственно убыль интенсивности п. п. озона под действием облучения не удается.
Трудность наблюдения за изменением относительной интенсивности п. п. адсорбированного озона усугубляется тем, что в условиях эксперимента в спектре присутствует поглощение как физически адсорбированного, так и хемосорбированного озона, причем интенсивность полос последнего крайне мала, и отслеживать, как они меняются, не представляется возможным. Так, в полосу при 2022 см–1 вносят вклад колебания как хемосорбированного 18O3, так и физически сорбированного 161818O3 [18]. Кто из них более подвержен разложению под действием ИК-облучения, не ясно, а область генерации используемого лазера не позволяет светить в полосу 1990 см–1 физически сорбированного 18O3.
Тем не менее, в опытах с озоном смешанного изотопного состава облучение на частоте 2030–2020 см–1 приводит к незначительному, но отчетливо наблюдаемому изменению соотношения между интенсивностями п. п. разных изотопологов. При этом относительная интенсивность полосы, на частоте которой производится облучение, падает. Со временем в отсутствие облучения соотношение интенсивностей постепенно возвращается к первоначальному, очевидно, в результате продолжающейся адсорбции озона и обмена адсорбированных молекул с присутствующими в газовой фазе. Этот процесс несколько ускоряется при облучении вне полосы поглощения озона.
Стимулирование озонолиза адсорбированных молекул путем резонансного возбуждения колебаний адсорбированного озона весьма привлекательно, поскольку чувствительность метода при детектировании появления новых полос поглощения гораздо выше, чем при измерении интенсивности. Проведенные опыты по озонолизу цис-дихлорэтилена показали, что даже на гидратированном диоксиде титана процесс спонтанно идет, приводя к образованию продукта с характерными п.п. при 1694 см–1 и более слабой п. п. при 1338 см–1. При повышении температуры происходит некоторое его преобразование, в процессе которого в спектре вместо п. п. 1694 см–1 вырастает другая при 1732 см–1. Эти полосы по положению близки к частотам формилхлорида HCOCl, п. п. которого в газовой фазе наблюдаются при 1784 и 1307 см–1 [25] и который, как можно предположить, является основным продуктом неполного окисления цис-дихлорэтилена в процессе озонолиза.
Полное окисление приводит к появлению СО2, слабые полосы которого начинают расти после напуска озона и усиливаются при облучении. Учитывая его исчезающе малое давление паров при –196°С, можно заключить, что диоксид углерода образуется при озонолизе непосредственно в образце, а не попадает на него как примесь в озоне, полностью избавиться от которой не удается. Из соотношения интенсивностей компонентов расщепленной полосы СО2, наблюдаемой при использовании 18О3 (76%), была определена степень обогащения кислорода в составе диоксида. Такая оценка дает величину около 70%. Это означает, что почти весь кислород происходит из озона, а небольшое понижение содержания изотопа 18О может быть следствием обмена с кислородом поверхности.
Гораздо большее количество СО2 образуется при лавинообразном озонолизе цис-дихлорэтилена, инициированном лазерным облучением. Исчезновение полосы 1694 см–1 объясняется тем, что происходит доокисление образовавшегося формилхлорида, однако полоса 1338 см–1 при этом остается. Видимо, значительная часть молекул HCOCl сохраняется, но изменившееся окружение влияет на колебание С–О так, что оно дает вклад в поглощение при иных частотах. В то же время другая часть молекул из-за резкого скачка температуры претерпевает характерное для формилхлорида разложение на СО и HCl [26]. При этом СО остается в адсорбированном состоянии, давая п. п. при 2186 и 2158 см–1, тогда как HCl адсорбируется диссоциативно, обнаруживая себя по поглощению в области водородно-связанных гидроксильных групп при 3550–3000 см–1.
Попытки облучать систему озон–цис-дихлорэтилен излучением разной частоты не привели к сколь-нибудь заметному различию в спектрах. Более того, облучение вне области поглощения озона или повышение температуры вызывают примерно такие же изменения. Если же и допустить, что эффект есть, и в результате облучения на частоте определенной изотопной модификации озона образуется формилхлорид с преобладанием какого-либо изотопа, засвидетельствовать это практически невозможно. Мало того, что полосы продукта достаточно широки, даже при использовании обычного озона 16О3 мы наблюдаем появление нескольких перекрывающихся полос продукта. Кроме того, в условиях, когда должна преобладать высокочастотная полоса карбонильной группы при 1732 см–1, при применении 18О3 полоса поглощения ее изотопного аналога попадает на 1697 см–1, почти совпадая с полосой 1694 см–1 обычного изотопа, а полоса изотопозамещенного аналога последней при 1665 см–1 накладывается на другую полосу при 1650 см–1 (рис. 3).
ЗАКЛЮЧЕНИЕ
Воздействие лазерного излучения на частоте составного колебания ν1 + ν3 определенных изотопологов озона, адсорбированных на оксиде титана, приводит к незначительному, но надежно фиксируемому уменьшению относительной интенсивности полосы, на частоте которой производится облучение. Эффект со временем ослабевает, что объясняется продолжающейся адсорбцией озона при температуре жидкого азота.
Резонансное возбуждение озона ускоряет процесс озонолиза адсорбированного цис-дихлорэтилена, который может иметь взрывообразный характер, когда образующийся при озонолизе формилхлорид разлагается с образованием СО, СО2 и продуктов адсорбции HCl. Наблюдение селективного озонолиза дихлорэтилена затруднено одновременным появлением нескольких продуктов, полосы поглощения которых перекрываются.
Подводя итог, можно сделать вывод, что озон, видимо, является не лучшим объектом для осуществления изотопно-селективных процессов, инициируемых резонансным возбуждением колебательных состояний, хотя полученные результаты указывают на реальность осуществления лазерно-стимулированного разложения озона и на возможность оптимизации условий реализации этой реакции. Так, можно было бы пытаться создать условия, когда на поверхности присутствовал бы только хемосорбированный озон или выбрать другую молекулу для осуществления озонолиза, стимулированного ИК-облучением.
Достоинством озона для нашей цели является необратимость происходящих реакций, однако, как видно, такой выбор имеет и ряд недостатков. Во-первых, используется возбуждение составного колебания, что означает возможность обмена колебательной энергией между фундаментальными колебаниями, в том числе разных изотопологов озона. Этому способствует высокая интенсивность полос поглощения, например, колебания ν3. Из-за обмена колебательной энергией изотопная селективность желаемых процессов понижается. Во-вторых, частоты фундаментальных колебаний озона близки к частотам колебаний оксида, поэтому скорость диссипации колебательного возбуждения должна быть велика, и квантовый выход искомых реакций не может быть высок. Кроме того, интенсивность полосы составного колебания весьма низкая, и крайне малая часть излучения может быть использована на интересующие нас процессы.
Список литературы
Велихов Е.П., Баранов В.Ю., Летохов В.С., Рябов Е.А., Старостин А.Н. Импульсные СО2-лазеры и их применение для разделения изотопов. Москва: Наука, 1983. 304 с.
Карлов Н.В., Прохоров А.М. // Успехи физ. наук. 1976. Т. 123. С. 57.
Apatin V.M., Makarov G.N., Ogurok N.D., Petin A.N., Ryabov E.A. // J. Exp. Theor. Phys. Lett. 2018. V. 127. P. 244.
Heidberg J., Stahmer K., Stein H., Weiss H. // J. Electron. Spectrosc. Relat. Phenom. 1987. V. 45. P. 87.
Heidberg J., Brase B., Stahmer K., Suhren M. // Appl. Surf. Sci. 1990. V. 46. P. 44.
Heidberg J., Kampshoff E., Stein H., Weiss H., Warskulat M. // Mikrochimica Acta. 1988. V. 95. № 1–6. P. 105.
Heidberg J., Stein H., Riehl E. // Surf. Sci. 1983. V. 126. P. 183.
Hussla I., Seki H., Chuang T.J., Gortel Z.W., Kreuzer H.J., Piercy P. // Phys. Rev. B. 1985. V. 32. P. 3489.
Chuang T.J., Seki H., Hussla I. // Surf. Sci. 1985. V. 158. P. 525.
Redlich B., van der Meer L., Zacharias H., Meijer G., von Helden G. // Nucl. Instrum. Methods. Phys. Res. Sect. A. 2003. V. 507. P. 556.
Redlich B., Zacharias H., Meijer G., von Helden G. // Phys. Chem. Chem. Phys. 2002. V. 4. P. 3448.
Redlich B., Zacharias H., Meijer G., von Helden G. // J. Chem. Phys. 2006. V. 124. P. 044704.
Tsyganenko A.A., Kompaniets T.N., Novikov R.G., Pestsov O.S. // Curr. Opin. Chem. Eng. 2019. V. 24. P. 69.
Dobrotvorskaia A.N., Pestsov O.S., Tsyganenko A.A. // Top. Catal. 2017. V. 60. № 19–20. P. 1506.
Proch D., Schröder H. // Chem. Phys. Lett. 1979. V. 61. P. 426.
Bulanin K.M., Lavalley J.C., Lamotte J., Mariey L., Tsyganenko N.M., Tsyganenko A.A. // J. Phys. Chem. B. 1998. V.102. P. 6809.
Bulanin K.M., Lavalley J.C., Tsyganenko A.A. // J. Phys. Chem. 1995. V. 99. № 25. P. 10294.
Tsyganenko A., Aminev T., Baranov D., Pestsov O. // Chem. Phys. Let. 2020. V. 761. P. 138071.
Manoilova O.V., Lavalley J.C., Tsyganenko N.M., Tsyganenko A.A. // Langmuir. 1998. V. 14. № 20. P. 5813.
Tsyganenko A.A., Zakharov N.V., Tsyganenko N.M. // Symposium “Molecular Photonics” Dedicated to Academician A.N. Terenin. St. Petersburg. Russia, 21–28 June, 2009. Abstracts. P. 214.
Tsyganenko A.A. // Top. Catal. 2013. V. 56. № 11. P. 905.
Свердлов Л.М., Ковнер М.А., Крайнов Е.П. Колебательные спектры многоатомных молекул. М.: Наука, 1970. 559 с.
Родионова Т.А., Цыганенко А.А., Филимонов В.Н. / В сб. Адсорбция и адсорбенты. Киев: Наукова Думка, 1982. Т. 10. С. 33.
Aminev T., Krauklis I., Pestsov O., Tsyganenko A. // Appl. Sci. 2021. V. 11. P. 7683.
Barnes I., Becker K.H., Starcke J. // Chem. Phys. Lett. 1995. V. 246. P. 594.
Dowideit P., Mertens R., von Sonntag C. // J. Am. Chem. Soc. 1996. V. 118. № 45. P. 11288.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ