

Кинетика и катализ, 2022, T. 63, № 6, стр. 760-771
Новые катализаторы асимметрического окисления прохиральных сульфидов на основе комплексов ванадия с производными левопимаровой кислоты
В. Н. Конев a, *, И. В. Ельцов b, З. П. Пай a, Т. Б. Хлебникова a
a ФГБУН ФИЦ Институт катализа им. Г.К. Борескова СО РАН
630090 Новосибирск, просп. Акад. Лаврентьева, 5, Россия
b ФГАОУ ВО Новосибирский национальный исследовательский государственный университет
630090 Новосибирск, ул. Пирогова, 2, Россия
* E-mail: konevv@catalysis.ru
Поступила в редакцию 15.04.2022
После доработки 07.06.2022
Принята к публикации 9.06.2022
- EDN: ZGJKMF
- DOI: 10.31857/S0453881122060077
Аннотация
С применением методов УФ-, видимой и 51V ЯМР-спектроскопии изучено образование ванадиевых комплексов с синтезированными энантио- и диастереомерно чистыми полидентатными тетрагидросалицилиденовыми лигандами дитерпенового ряда. Полученные каталитические комплексы протестированы в реакции окисления прохиральных сульфидов водным 35% раствором пероксида водорода. В результате исследований окисления прохиральных сульфидов было показано, что хиральные сульфоксиды образуются с выходами 28–90% и энантиомерным избытком (ЭИ) до 96%. Обнаружено, что природа используемого растворителя определяет конфигурацию сульфоксида. Установлено, что в этаноле образуются R-сульфоксиды, а в хлористом метилене – S-сульфоксиды.
ВВЕДЕНИЕ
Хиральные сульфоксиды находят широкое применение в синтетической органической химии и представляют интерес как синтоны для создания С–С- и С–Х-связей [1, 2]. Многие из них используются в качестве медицинских препаратов с различной биологической активностью [3]. Оптически чистые хиральные сульфоксиды применяются как лиганды металлокомплексных катализаторов [4, 5] или в качестве органических катализаторов [5] асимметрических реакций. В связи с этим разработка новых эффективных методов синтеза оптически активных сульфоксидов представляет собой актуальную задачу. Асимметрическое окисление прохиральных сульфидов, катализируемое хиральными металлокомплексами, – наиболее эффективный и простой способ синтеза энантиомерно обогащенных сульфоксидов [6]. В качестве окислителей для асимметрического превращения сульфидов в сульфоксиды используют трет-бутилгидропероксид, кумилгидропероксид, иодозобензол и пероксид водорода [6]. Самый доступный, дешевый и экологически безопасный из перечисленных реагентов для сульфоксидирования – пероксид водорода, поскольку продуктом его превращения является вода, а содержание активного кислорода составляет 47.1% по массе [7, 8]. Однако применение водного раствора пероксида водорода в качестве окислителя накладывает ограничения на используемые каталитические системы, а именно катализатор не должен разрушаться в водном растворе.
За последние десятилетия разработано и изучено значительное количество катализаторов окисления прохиральных сульфидов пероксидом водорода, представленных в основном комплексами титана, ванадия, железа, марганца и молибдена [7, 9]. Наиболее изучены катализаторы асимметрического окисления, основанные на металлокомплексах с лигандами салицилиденового (саленового) типа [7]. Одним из недостатков этих лигандов, содержащих азометиновые группы, является гидролиз в содержащих воду растворителях [10, 11]. Восстановление двойных связей саленов приводит к образованию более стабильных и стерически лабильных тетрагидросалицилиденов, называемых салановыми лигандами.
Хиральные салицилиденовые лиганды в комплексах с титаном и ванадием активно применяются как катализаторы окисления прохиральных органических сульфидов различного строения, приводящего к сульфоксидам с высокой хемо- и энантиоселективностью [6, 7]. Катализаторы асимметрического сульфоксидирования с тетрагидросалицилиденовыми лигандами в основном представлены комплексами титана и в значительно меньшей степени – комплексами ванадия [6]. В нескольких работах описан синтез и исследование комплексов ванадия(IV) и ванадия(V) с тетрагидросалицилиденовыми лигандами, однако их энантиоселективность в реакции окисления сульфидов не превышает 50% [12]. В целом каталитические свойства комплексов ванадия с салановыми лигандами в различных стереоселективных реакциях также изучены в меньшей степени, чем ванадий-саленовые комплексы. Известны примеры использования салановых комплексов ванадия как катализаторов асимметрических вариантов пинаконового сочетания, сульфоксидирования, эпоксидирования [13, 14].
Существенным недостатком катализаторов с тетрагидросалицилиденовыми лигандами является их менее жесткая структура по сравнению с салицилиденовыми металлокомплексами [11, 14, 15]. Это приводит к появлению нескольких диастереомерных активных интермедиатов, что снижает энантиоселективность превращения. Более объемные лиганды со стерически затрудненными донорными атомами могли бы способствовать ограничению количества образующихся диастереомеров каталитически активных комплексов ванадия и, таким образом, увеличить энантиоселективность процесса получения сульфоксидов. Удобной матрицей для создания вышеупомянутых объемных лигандов могут служить дитерпеновые кислоты и их производные благодаря большому количеству хиральных центров, жесткой и объемной структуре. В более ранних наших работах [16, 17] были синтезированы бисаминофенолы тетрагидросалицилиденового типа на основе трансформаций природного дитерпена – левопимаровой кислоты (схема 1 ). Каталитическая активность комплексов меди(II) с аминофенолами 3a–3f была изучена в нитроальдольной реакции при взаимодействии ароматических альдегидов и нитрометана с образованием нитроальдолей, где ЭИ достигал 82% [17]. Результаты исследований асимметрической индукции металлокомплексов меди с производными левопимаровой кислоты показывают, что предложенный подход создания лигандов с использованием дитерпенов в качестве хиральной матрицы имеет большой потенциал.
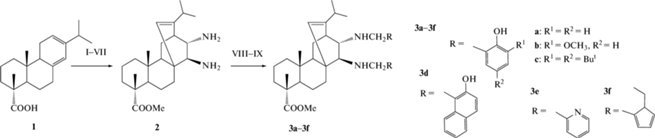
Схема 1 . Схема синтеза аминофенолов 3a–3f [16, 17]. Условия реакций: I – CH3I, K2CO3, ацетон, 6 ч; II – фумаровая кислота, 180°C, Ar, 6 ч; III – SOCl2, ДМФА, толуол, 110°C, 3 ч; IV – NaN3, PhCH3, 5°C, 2 ч; V – 110°C, 2 ч; VI – HCl (35%), 110°C, 2 ч; VII – H2O, NaHCO3; VIII – RCHO; IX – NaBH4, EtOH.
В связи с этим целью настоящей работы являлось изучение комплексов ванадия(V) с азотсодержащими производными левопимаровой кислоты 3a–3f, используемых в качестве катализаторов асимметрического варианта реакции окисления прохиральных сульфидов в сульфоксиды.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Прохиральные сульфиды 4a, 4f, 4g (содержание 98–99%, “Alfa Aesar”) использовали без дополнительной очистки. Сульфиды 4b [18], 4c [19], 4d [20], 4e [21] синтезировали по описанным в литературе методикам. Аминофенолы 3a–3f получали по методике [17]. Физико-химические данные соединений 3a–3f аналогичны описанным в литературе [16]. Растворители квалификации “х. ч.” и “ос. ч.” хранили над слоем молекулярных сит (3 Å) и применяли без дополнительной очистки. Для колоночной хроматографии использовали силикагель фирмы “Merck” (40–63 мкм).
Физико-химические методы исследования
Спектры ЯМР 1H и 51V регистрировали на спектрометрах AV-300, AM-400 и Avance III 500 (“Bruker”, Германия). Химические сдвиги для 1H ЯМР были записаны в м. д. (δ) относительно тетраметилсилана (0 м. д.), сигналы растворителей служили в качестве внутреннего стандарта (CDCl3, δ 7.25 м. д.). Химические сдвиги для ЯМР 51V были записаны в м. д. с использованием сигнала VOCl3 (0 м. д.) в качестве внутреннего стандарта. Углы оптического вращения измеряли на поляриметре polAAr 3005 (“Index Instruments Ltd.”, Великобритания). Анализ методом ВЭЖХ осуществляли на хроматографе ProStar с диодно-матричным детектором ProStar 335 (“Varian”, США) с применением хиральных колонок Chiralcel OD-H или AS-H. Запись спектров в УФ- и видимой областях (190–1000 нм) выполняли на спектрометре UV-1800 (“Shimadzu”, Япония). Анализ методом ГЖХ проводили на хроматографе ГХ-1000 (“Хромос”, Россия), оснащенном пламенно-ионизационным детектором (капиллярная колонка BPX-5, изотерма 180°С, газ-носитель – гелий). Тонкослойную хроматографию осуществляли с использованием пластин ПТСХ-АФ-A (“Сорбфил”) (проявитель – 10% раствор фосфорномолибденовой кислоты в этаноле).
Приготовление раствора катализатора
К навеске аминофенола 3a (17.6 или 35.2 мг) добавили раствор ацетилацетоната ванадила VO(acac)2 (7.6 мг, 0.03 ммоль) или изопропоксида ванадила VO(OiPr)3 (7.3 мг, 0.03 ммоль) в хлористом метилене (10 мл). Смесь перемешивали в закрытой виале на магнитной мешалке при 23 или 35°C в течение нескольких часов. Условия приготовления приведены в табл. 1.
Таблица 1.
Условия приготовления растворов катализаторов
| № | 3a | VO(acac)2 | VO(OiPr)3 | Время, ч | T, °C | |||
|---|---|---|---|---|---|---|---|---|
| n, ммоль | m, мг | n, ммоль | m, мг | n, ммоль | m, мг | |||
| 1 | 0.03 | 17.6 | 0.03 | 7.6 | – | – | 4; 240; 504 | 23 |
| 2 | 0.06 | 35.2 | 0.03 | 7.6 | – | – | 4; 240; 504 | 23 |
| 3 | 0.03 | 17.6 | – | – | 0.03 | 7.3 | 3; 72 | 23; 35 |
Для получения раствора катализатора в этаноле (или других растворителях, табл. 2–5) отбирали 1 мл раствора катализатора в хлористом метилене, приготовленного по вышеописанной методике, затем растворитель испаряли медленным потоком воздуха и к остатку добавляли 1 мл этанола (или другого растворителя, табл. 2–5). Растворы катализаторов из аминов 3b–3f в хлористом метилене получали аналогично способу для аминофенола 3a (табл. 1, № 3), затем по описанной выше методике CH2Cl2 замещали на этанол.
Таблица 2.
Исследование влияния растворителей в реакции окисления бензилфенилсульфида 4a пероксидом водорода в присутствии комплекса ванадия(V) с лигандом 3a
| № | Растворитель | Выход 5a, % | Выход 6a, % | ЭИ 5a, % (R или S) |
|---|---|---|---|---|
| 1 | CH2Cl2 | 60 | 32 | 40(S) |
| 2 | CHCl3 | 68 | 29 | 27(S) |
| 3 | PhCH3 | 47 | 36 | 9(S) |
| 4 | (CH2)4O | 60 | 25 | 12(R) |
| 5 | EtOAc | 50 | 34 | 5(R) |
| 6 | CH3CN | 70 | 28 | 8(R) |
| 7 | (CH3)2CO | 66 | 29 | 24(R) |
| 8 | i-PrOH | 61 | 38 | 49(R) |
| 9 | CH3OH | 58 | 40 | 55(R) |
| 10 | C2H5OH | 60 | 40 | 76(R) |
| 11* | CH2Cl2 | 58 | 34 | 45(S) |
Условия: [4a ] 0 = 0.3 М, [4] : [H2O2] : [VO(OiPr)3] : [3a ] = 100 : 145 : 1 : 1, Т = 0°С, 60 ч. *[4a ] : [H2O2] : [VO(OiPr)3] : [3a ] = = 100 : 145 : 0.5 : 0.5.
Таблица 3.
Окисление бензилфенилсульфида 4a пероксидом водорода в присутствии комплекса ванадия(V) с аминофенолами 3a–3f
| № | Аминофенол | Выход 5a, % | Выход 6a, % | ЭИ 5a, % (R или S) |
|---|---|---|---|---|
| 1 | 3a | 60 | 40 | 76(R) |
| 2 | 3b | 68 | 32 | 32(R) |
| 3 | 3c | 61 | 38 | 13(R) |
| 4 | 3d | 61 | 38 | 8(R) |
| 5 | 3e | 73 | 19 | 2(S) |
| 6 | 3f | 72 | 27 | 2(R) |
Таблица 4.
Оптимизация условий окисления бензилфенилсульфида 4a пероксидом водорода в присутствии комплекса ванадия(V) с лигандом 3a
| № | [4a ] : [H2O2] : [VO(OiPr)3] : [3a ] | Выход 5a, % | Выход 6a, % | ЭИ 5a, % (R или S) |
|---|---|---|---|---|
| 1 | 100 : 145 : 0 : 0 | <5 | 0 | – |
| 2 | 100 : 145 : 1 : 0 | 58 | 42 | – |
| 3 | 100 : 145 : 0 : 1 | <5 | 0 | 0 |
| 4 | 100 : 125 : 0.5 : 0.5 | 83 | 17 | 67(R) |
| 5 | 100 : 145 : 0.5 : 0.5 | 78 | 22 | 68(R) |
| 6 | 100 : 160 : 0.5 : 0.5 | 65 | 34 | 67(R) |
| 7 | 100 : 125 : 1 : 1 | 80 | 20 | 68(R) |
| 8 | 100 : 155 : 1 : 1 | 50 | 50 | 72(R) |
| 9* | 100 : 145 : 1 : 1 | 58 | 42 | 68(R) |
| 10** | 100 : 145 : 1 : 1 | 84 | 15 | 52(R) |
| 11 | 100 : 145 : 1.5 : 1.5 | 60 | 40 | 76(R) |
| 12 | 100 : 145 : 2 : 2 | 56 | 44 | 74(R) |
| 13*** | 100 : 145 : 0.5 : 0.5 | 76 | 1 | 0 |
| 14**** | 100 : 145 : 0.5 : 0.5 | 69 | 31 | 0 |
Таблица 5.
Окисление сульфидов различного строения пероксидом водорода в присутствии комплекса ванадия(V) с лигандом 3a
| № | Субстрат | Растворитель | Выход продуктов, % | ЭИ сульфоксида, % (R или S) | |
|---|---|---|---|---|---|
| Сульфоксид | Сульфон | ||||
| 1 |  |
EtOH |  71 |
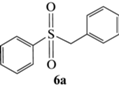 26 |
96(R) |
| 2 | CH2Cl2 | 48 | 44 | 58(S) | |
| 3 |  |
EtOH |  57 |
 41 |
73(R) |
| 4 | CH2Cl2 | 50 | 41 | 49(S) | |
| 5 |  |
EtOH | 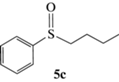 40 |
 45 |
86(R) |
| 6 | CH2Cl2 | 55 | 42 | 41(S) | |
| 7 | 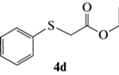 |
EtOH |  49 |
 42 |
94(R) |
| 8 | CH2Cl2 | 28 | 0 | 34(R) | |
| 9 | 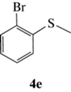 |
EtOH |  90 |
 0 |
67(R) |
| 10 | CH2Cl2 | 86 | 0 | 59(S) | |
| 11 | 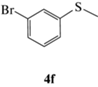 |
EtOH |  60 |
 36 |
51(R) |
| 12 | CH2Cl2 | 58 | 30 | 39(S) | |
| 13 |  |
EtOH |  59 |
 41 |
51(R) |
| 14 | CH2Cl2 | 48 | 43 | 57(S) | |
Методика проведения реакции каталитического сульфоксидирования
Асимметрическое окисление сульфидов проводили на установке для параллельного синтеза RCT basic (“IKA”, Германия), оборудованной платформой H 135.10 совместно с секциями для реакционных сосудов IKA H. 135.102, в реакторах G075X-17Kit10-H объемом 2.5 мл.
В стеклянный реактор объемом 2.5 мл, снабженный крышкой с тефлоновой прокладкой, помещали 1 мл раствора катализатора (30 мкмоль) (табл. 1) в соответствующем растворителе (табл. 2–5), добавляли сульфид 4a–4g (0.3 ммоль) и перемешивали в течение 30 мин при заданной температуре (табл. 2–5). Затем к реакционной смеси приливали необходимое количество 35% водного раствора пероксида водорода (табл. 2–5). Смесь выдерживали при перемешивании и заданной температуре необходимое время (табл. 2–5). Затем к реакционной смеси добавляли воду (5 мл). Из водного раствора продукты экстрагировали хлороформом (3 × 5 мл). Объединенные органические экстракты сушили безводным сульфатом натрия, хлороформ упаривали. Для расчета содержания сульфоксида 5a и сульфона 6a (табл. 2–4) в реакционной смеси остаток после упаривания хлороформа анализировали методом ГЖХ с использованием внутреннего стандарта (генэйкозан). Сульфоксиды 5a–5g и сульфоны 6a–6g (табл. 5) выделяли колоночной хроматографией на силикагеле (элюент – гексан : этилацетат = 1 : 1).
Физико-химические характеристики полученных энантиомерно обогащенных сульфоксидов 5a–5g.
(R)-Бензилсульфинилбензол (5a): выход 71%, ЭИ 96% (табл. 5). ЯМР 1H (500 МГц, CDCl3): δ = 7.49–7.34 (м, 5H), 7.32–7.19 (м, 3H), 7.02–6.93 (м, 2H), 4.09 (д, 12.5, 1H),
3.99 (д, 12.6, 1H). ВЭЖХ (Chiracel OD-H, н-гексан/изопропанол, 50 : 50 v/v, 1 мл/мин, 25°С, 224 нм): tr(R) = 17.1 мин, tr(S) = = 20.3 мин.  +225.0 (c 0.43, CH3COCH3), лит. [22]
+225.0 (c 0.43, CH3COCH3), лит. [22]  –164.2 (c 0.9, ЭИ 82%, CH3COCH3).
–164.2 (c 0.9, ЭИ 82%, CH3COCH3).
(S)-Бензилсульфинилбензол (5a): выход 48%, ЭИ 58% (табл. 5),  –110.7 (c 0.34, CH3COCH3), лит. [22]
–110.7 (c 0.34, CH3COCH3), лит. [22]  –164.2 (c 0.9, ЭИ 82%, CH3COCH3).
–164.2 (c 0.9, ЭИ 82%, CH3COCH3).
(R)-Метилсульфинилбензол (5b): выход 57%, ЭИ 73% (табл. 5). ЯМР 1H (500 МГц, CDCl3): δ = = 7.63–7.65 (м, 2H), 7.47–7.54 (м, 3H), 2.71 (с, 3H). ВЭЖХ (Chiracel AS-H,
н-гексан/изопропанол, 50 : 50 v/v, 1 мл/мин, 25°С, 254 нм): tr(R) = = 9.4 мин, tr(S) = 11.1 мин.  +109.2 (c 0.13, CH3COCH3), лит. [22]
+109.2 (c 0.13, CH3COCH3), лит. [22]  –85.4 (c 0.8, ЭИ 78%, CH3COCH3).
–85.4 (c 0.8, ЭИ 78%, CH3COCH3).
(S)-Метилсульфинилбензол (5b): выход 50%, ЭИ 49% (табл. 5),  –71.8 (c 0.11, CH3COCH3), лит. [22]
–71.8 (c 0.11, CH3COCH3), лит. [22]  –85.4 (c 0.8, ЭИ 78%, CH3COCH3).
–85.4 (c 0.8, ЭИ 78%, CH3COCH3).
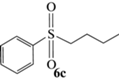
(R)-Бутилсульфинилбензол (5c): выход 40%, ЭИ 86% (табл. 5), ЯМР 1H (500 МГц, CDCl3): δ = = 7.61–7.63 (м, 2H), 7.47–7.54 (м, 3H), 2.77–2.80 (м, 2H), 1.69–1.78 (м, 1H),
1.56–1.64 (м, 1H), 1.36–1.52 (м, 2H), 0.92 (т, 3H). ВЭЖХ (Chiracel OD-H, н-гексан/изопропанол, 90 : 10 v/v, 1 мл/мин, 25°С, 254 нм): tr(R) = 10.1 мин, tr(S) = 12.1 мин.  +167.0 (c 0.26, CH3CH2OH), лит. [23]
+167.0 (c 0.26, CH3CH2OH), лит. [23]  –107.2 (c 0.86, ЭИ 46%, CH3CH2OH).
–107.2 (c 0.86, ЭИ 46%, CH3CH2OH).
(S)-Бутилсульфинилбензол (5c): выход 55%, ЭИ 41% (табл. 5),  –84.0 (c 0.31, CH3CH2OH), лит. [23]
–84.0 (c 0.31, CH3CH2OH), лит. [23]  –107.2 (c 0.86, ЭИ 46%, CH3CH2OH).
–107.2 (c 0.86, ЭИ 46%, CH3CH2OH).
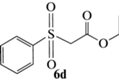
Этиловый эфир (R)-2-(фенилсульфинил)уксусной кислоты (5d): выход 49%, ЭИ 94% (табл. 5), ЯМР 1H (500 МГц, CDCl3): δ = 7.68–7.71 (м, 2H), 7.52–7.55 (м, 3H), 4.14 (м, 2H), 3.84 (д, 13.6 Гц, 1H),
3.65 (д, 13.6 Гц, 1H), 1,21 (т, 3H). ВЭЖХ (Chiracel OD-H, н-гексан/изопропанол, 90 : 10 v/v, 1 мл/мин, 25°С, 254 нм): tr(R) = 19.7 мин, tr(S) = = 23.0 мин.  +192.0 (c 0.29, CH3COCH3) (лит. [24]
+192.0 (c 0.29, CH3COCH3) (лит. [24]  –131 (c 1, ЭИ 90%, CH3COOEt)).
–131 (c 1, ЭИ 90%, CH3COOEt)).
(R)-1-Бром-2-(метилсульфинил)бензол (5e): выход 90%, ЭИ 67% (табл. 5), ЯМР 1H (500 МГц, CDCl3): δ = 7.94–7.96 (м, 1H), 7.56–7.60 (м, 2H), 7.36–7.39 (м, 1H), 2.82 (с, 3H). ВЭЖХ
(Chiracel AS-H, н-гексан/изопропанол, 50 : 50 v/v, 1 мл/мин, 25°С, 254 нм): tr(R) = 12.6 мин, tr(S) = = 14.3 мин.  179.6 (c 0.48, CHCl3), лит. [22]
179.6 (c 0.48, CHCl3), лит. [22] 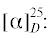 –91.7 (c 0.56, ЭИ 74%, тетрагидрофуран).
–91.7 (c 0.56, ЭИ 74%, тетрагидрофуран).
(S)-1-Бром-2-(метилсульфинил)бензол (5e): выход 86%, ЭИ 59% (табл. 5),  –143.4 (c 0.47, CHCl3), лит. [22]
–143.4 (c 0.47, CHCl3), лит. [22]  –91.7 (c 0.56, ЭИ 74%, тетрагидрофуран).
–91.7 (c 0.56, ЭИ 74%, тетрагидрофуран).
(R)-1-Бром-3-(метилсульфинил)бензол (5f): выход 60%, ЭИ 36% (табл. 5), ЯМР 1H (500 МГц, CDCl3): δ = 7.81 (м, 1H), 7.62–7.64 (м, 1H), 7.54–7.56 (м, 1H), 7.38–7.42 (м, 1H), 2.74
(с, 3H). ВЭЖХ (Chiracel AS-H, н-гексан/изопропанол, 50 : 50 v/v, 1 мл/мин, 25°С, 254 нм): tr(R) = = 12.3 мин, tr(S) = = 16.2 мин.  +50.0 (c 0.26, CH3COCH3), лит. [25]
+50.0 (c 0.26, CH3COCH3), лит. [25]  +105.4 (c 1.0, ЭИ 85%, CH3COCH3).
+105.4 (c 1.0, ЭИ 85%, CH3COCH3).
(S)-1-Бром-3-(метилсульфинил)бензол (5f): выход 58%, ЭИ 39% (табл. 5),  –35.2 (c 0.33, CH3COCH3), лит. [25]
–35.2 (c 0.33, CH3COCH3), лит. [25]  +105.4 (c 1.0, ЭИ 85%, CH3COCH3).
+105.4 (c 1.0, ЭИ 85%, CH3COCH3).
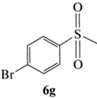
(R)-1-Бром-4-(метилсульфинил)бензол (5g): выход 59%, ЭИ 51% (табл. 5), ЯМР 1H (500 МГц, CDCl3): δ = 7.66–7.69 (д, 8.5 Гц, 2H), 7.51–7.54 (д, 8.5 Гц, 2H), 2.71 (с, 3H). ВЭЖХ (Chiracel
AS-H, н‑гексан/изопропанол, 50 : 50 v/v, 1 мл/мин, 25°С, 254 нм): tr(R) = 11.4 мин, tr(S) = 13.4 мин. 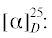 +60.6 (c 0.41, CH3COCH3), (лит. [22]
+60.6 (c 0.41, CH3COCH3), (лит. [22] 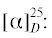 –64.0 (c 1.1, ЭИ 74%, CH3COCH3)).
–64.0 (c 1.1, ЭИ 74%, CH3COCH3)).
(S)-1-Бром-4-(метилсульфинил)бензол (5g): выход 48%, ЭИ 57% (табл. 5),  –50.0 (c 0.31, CH3COCH3), лит. [22]
–50.0 (c 0.31, CH3COCH3), лит. [22] 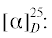 –64.0 (c 1.1, ЭИ 74%, CH3COCH3).
–64.0 (c 1.1, ЭИ 74%, CH3COCH3).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Синтез и исследование каталитических систем
В качестве катализаторов реакции сульфоксидирования применяют как комплексы ванадия(IV), так и комплексы ванадия(V) [7, 11, 26]. Известно, что для синтеза катализаторов без выделения индивидуальных комплексов ванадия с тридентатными иминами используется от 1 до 2 экв. лиганда на 1 экв. ацетилацетоната ванадила [27–31]. По-видимому, избыток лиганда необходим для смещения равновесия в сторону образования ванадий-иминовых комплексов. Для получения комплекса [V+4 (3a)] смесь VO(acac)2 и 3a в хлористом метилене перемешивали в инертной атмосфере (аргон) в течение нескольких часов, однако цвет раствора не изменился. Это может свидетельствовать о том, что формирования комплекса [V+4(3a)] не произошло, поэтому были проведены эксперименты для синтеза соединений ванадия(V). Известно, что салановые комплексы ванадия(V) образуются из комплексов ванадия(IV) при окислении кислородом воздуха или при использовании исходного соединения V(V) [12, 32]. Комплексы ванадия(V) синтезировали из VO(acac)2 и 3a (табл. 1) при перемешивании в хлористом метилене на воздухе. Через 4 ч растворы поменяли цвет с сине-зеленого на красно-фиолетовый, что говорит об образовании комплекса V(V).
В электронных спектрах полученных растворов комплексов в хлористом метилене в видимой области присутствуют полосы поглощения при 375 и 494 нм (рис. 1), интенсивность которых растет при увеличении времени выдержки до 21 дня. Известно, что данные сигналы в спектрах характерны для салановых комплексов ванадия в степени окисления +5, тогда как для ванадия(IV) характерными являются полосы 550–600 нм [12]. Как видно из рис. 1, для комплекса VO(acac)2/3a интенсивность сигналов (рис. 1, спектры 2–7) увеличивается на протяжении десятков дней, что свидетельствует о низкой скорости образования целевого комплекса ванадия(V), по-видимому, связанной с медленной стадией окисления V+4 в V+5 кислородом воздуха [12].
Рис. 1.
Электронные спектры поглощения растворов полученных комплексов ванадия с лигандом 3a в хлористом метилене (табл. 1): ацетилацетонат ванадила (1); [3a ] : [VO(acac)2] = 1 (табл. 1, № 1), время выдержки 4 (2), 240 (4) и 504 ч (6); [3a ] : [VO(acac)2] = 2 (табл. 1, № 2), время выдержки 4 (3), 240 (5) и 504 ч (7); [3a ] : [VO(OiPr)3] = 1 (табл. 1, № 3), время выдержки 72 ч (8).
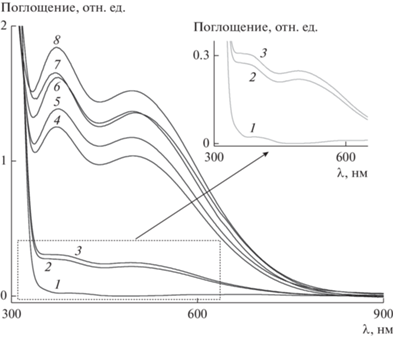
Для увеличения скорости реакции образования комплекса ацетилацетонат ванадила заменили на изопропоксид ванадила, в котором ванадий уже находится в степени окисления +5. После смешивания аминофенола 3а с изопропоксидом ванадила ([3a ] : [VO(OiPr)3] = 1, табл. 1, № 3) цвет раствора сразу меняется на красно-коричневый. Через 3 ч после добавления изопропоксида ванадила к раствору аминофенола 3a в хлористом метилене в спектре ЯМР 51V полученной реакционной смеси наблюдается сложный набор сигналов комплексов ванадия (–429, –470, –495, –510, –528, –613, –620 м. д.), в том числе исходного изопропоксида ванадила при –629 м. д. [33] (рис. 2, спектр 1). После выдержки реакционной смеси при 35°С в течение 3 дней в спектре ЯМР 51V присутствуют выраженный сигнал при –528 м. д., соответствующий основному продукту комплексообразования, и сигналы с низкой интенсивностью при –441, –471, –496, –563 и –612 м. д., соответствующие минорным продуктам (рис. 2, спектр 2). В электронных спектрах поглощение полученного через 3 дня раствора комплекса VO(OiPr)3/3a (табл. 1, № 3) (коэффициенты экстинкции – 5567 М–1 см–1 (375 нм) и 4583 М–1 см–1 (495 нм)) больше, чем в спектрах комплексов, синтезированных из ацетилацетоната ванадила (рис. 1). Таким образом, на примере лиганда 3a показано, что использование изопропоксида ванадила позволяет за 3 дня приготовить раствор катализатора, содержащий преимущественно один металлокомплекс в качестве основного продукта. В связи с этим в дальнейших исследованиях катализаторы сульфоксидирования синтезировали с применением VO(OiPr)3 по вышеописанной методике.

Исследование активности и энантиоселективности полученных катализаторов
Для изучения асимметрической индукции металлокомплексных катализаторов ванадия с лигандами дитерпенового ряда в качестве модельного субстрата был выбран бензилфенилсульфид 4a (схема 2 ), а в качестве окислителя – 35% (по массе) водный раствор пероксида водорода.

Схема 2 . Асимметрическое окисление бензилфенилсульфида 4a.
Исследование влияния растворителя. Известно, что высокие значения энантиомерного избытка сульфоксида в асимметрическом окислении сульфидов достигаются при использовании хлорсодержащих растворителей (дихлорэтана, хлористого метилена и хлороформа) [28, 29], а также толуола [29, 31, 34]. Отметим, что в некоторых случаях энантиомерный избыток был выше в кислородсодержащих растворителях, например, в ацетоне или ТГФ [35]. В работе [36] было установлено, что добавление к хлористому метилену сорастворителя метанола приводит к увеличению ЭИ сульфоксида. Тем не менее, в большинстве случаев применение протонных растворителей, в частности спиртов, для проведения реакции окисления приводило к рацемической смеси или низким значениям ЭИ сульфоксидов [37]. Несмотря на то, что в целом энантиомерный избыток сульфоксидов выше в хлоруглеводородах, предпочтительнее использовать экологически безопасные растворители. В связи с этим было изучено влияние растворителей различной природы на выходы асимметрического окисления бензилфенилсульфида 4a пероксидом водорода при 0°С в присутствии комплекса ванадия с лигандом 3a (схема 2 ).
Данные (табл. 2) указывают на то, что растворитель, в котором проводят реакцию окисления, влияет на конфигурацию образующегося бензилфенилсульфоксида. Так, в слабополярных растворителях (толуол и галогенированные углеводороды) получен S-энантиомер (табл. 2, № 1–3), а в растворителях, содержащих донорные атомы (кислород или азот), – R-бензилфенилсульфоксид (табл. 2, №№ 4–10). Примеры таких изменений конфигурации сульфоксида при смене типа растворителя в реакции каталитического окисления прохиральных сульфидов в литературе не найдены. Как правило, для синтеза того или другого энантиомера необходимо использовать металлокомплексы с другими лигандами. Например, R- или S-сульфоксиды образуются в результате V(V)-катализируемого окисления этил-2-нафтилсульфида с применением полученных из 3-экзо-аминоизоборнеола и 3-эндо-аминоборнеола диастереомерных салицилиденовых лигандов с противоположной конфигурацией асимметрических центров [34]. Также в нашей ранней работе [38] на примере реакции Анри было показано, что изменение стерического окружения асимметрического центра в лиганде металлокомплекса при такой же конфигурации хиральных центров приводит к обращению конфигурации преобладающего энантиомера в продукте реакции.
В нашем случае при окислении бензилфенилсульфида в присутствии комплекса ванадия с лигандом 3a растворитель влияет не только на конфигурацию преобладающего сульфоксида, но и на энантиоселективность. Более высокие значения ЭИ получены для реакций в спиртах (табл. 2, №№ 8–10), причем лучший результат среди них отмечен при использовании этанола (табл. 2, № 10).
Обнаруженный эффект влияния растворителя на конфигурацию преобладающего продукта реакции, по-видимому, обусловлен его способностью координироваться к атому ванадия в каталитическом комплексе. В условиях катализа комплексом ванадия с дитерпеновым лигандом 3a в толуоле и хлорированных углеводородах, которые не участвуют в формировании каталитически активного интермедиата, преимущественно образуется S-бензилфенилсульфоксид. R-Бензилфенилсульфоксид является преобладающим в кислород- и азотсодержащих растворителях, предположительно играющих роль дополнительного лиганда в каталитически активном металлокомплексе.
Исследование активности и энантиоселективности полученных катализаторов с разными лигандами. Исследование асимметрической индукции лигандов 3a–3f с различными типами ароматических заместителей при атомах азота в составе комплексов ванадия(V) показало, что наилучшей стереоселективностью обладает катализатор с лигандом 3a с наименее объемными заместителями в ароматическом кольце (табл. 3, № 1). Увеличение объемной структуры функциональных групп в лиганде снижает энантиоселективность катализатора, а использование соединений 3e и 3f (схема 1 ), не содержащих гидроксиароматические группы, приводит к появлению почти рацемических сульфоксидов (табл. 3, №№ 5–6). Объяснением отсутствия стереоселективности в данных случаях может являться то, что амины 3e и 3f не образуют комплексы с ионом ванадия(V).
Оптимизация условий каталитического окисления сульфидов. Из данных, приведенных в табл. 2, видно, что природа растворителя не оказывает существенного воздействия на выход сульфоксида 5a. Результаты исследования влияния катализаторов, полученных in situ на основе комплексов ванадия(V), на протекание реакции, проводимой в этаноле, приведены в табл. 3. В случае использования только изопропоксида ванадия (без хирального лиганда) сульфоксид и сульфон получены с выходами 58 и 42% (табл. 4, № 2), а в присутствии комплекса ванадия с аминофенолом 3a выходы составили 60 и 40% соответственно (табл. 4, № 10). При этом в отсутствие соединений ванадия (табл. 4, №№ 1 и 3) реакция практически не идет, а органокаталитический вариант реакции не реализуется (табл. 4, № 3). Невысокие значения выходов сульфоксида 5a (табл. 4) в изучаемых условиях вызваны его дальнейшим окислением в сульфон, сопровождающимся кинетическим разделением. Это подтверждается данными, полученными при варьировании количества пероксида водорода (при неизменных загрузках катализатора 1.0 и 0.5%). Повышение соотношения [H2O2] : [4а] с 1.25 до 1.6 (табл. 4, №№ 4, 6–8) приводит к сокращению выхода сульфоксида с 80–83 до 50–65%. Увеличение загрузки катализатора до 2% (табл. 4, № 12) также незначительно снижает выход сульфоксида за счет образования сульфона. На энантиоселективность окисления количество пероксида водорода оказывает меньшее действие. Так, при загрузке катализатора в количестве 0.5% варьирование соотношения [H2O2] : [4a ] от 1.25 до 1.6 практически не влияет на величину ЭИ сульфоксида 5a (табл. 4, №№ 4–6). В экспериментах с 1.0% катализатора максимальное значение ЭИ получено при соотношении [H2O2] : : [4a ] = 1.45 (табл. 2, № 10). При неизменном соотношении [H2O2] : [4a ] = 1.45 также наблюдается уменьшение энантиомерного избытка сульфоксида 5a при самой низкой (0.5%, табл. 4, № 5) и самой высокой (2.0%, табл. 4, № 12) загрузках катализатора, а наибольшее значение достигается при 1.0–1.5 мол. % (ЭИ 76%, табл. 2, № 10, табл. 4, № 11).
Увеличение концентрации субстрата с 0.3 до 0.6 М приводит к неожиданному результату – селективность образования сульфоксида возрастает с 60 до 84%, но ЭИ падает с 76 до 52% (табл. 4, № 10). Напротив, уменьшение концентрации субстрата с 0.30 до 0.15 М практически не влияет на выход сульфоксида, но немного снижается энантиоселективность образования продукта с 76 до 68% (табл. 4, № 9). Применение других окислителей (трет-бутилгидропероксид (табл. 4, № 13) и кумилгидропероксид (табл. 4, № 14) приводит к рацемическим сульфоксидам, что, по-видимому, связано с разрушением хирального катализатора и образованием неорганического дипероксокомплекса ванадила.
Скрининг субстратов в реакции сульфоксидирования в присутсвии катализатора ванадия(V) с лигандом 3a
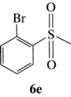
Изучение субстратоселективности каталитической системы VO(OiPr)3/3a в реакции энантиоселективного сульфоксидирования водным раствором пероксида водорода проводили с использованием сульфидов с ароматическими и алифатическими заместителями в двух типах растворителей (этаноле и хлористом метилене). В присутствии комплекса ванадия(V) с лигандом 3a реакция окисления сульфидов различных структурных типов в этаноле протекает с образованием R-сульфоксидов, тогда как в хлористом метилене приводит к S-энантиомерам. Исключение составляет этиловой эфир фенилтиогликолевой кислоты, продуктом окисления которого как в этаноле, так и хлористом метилене является R-сульфоксид (табл. 5, №№ 7–8). По-видимому, данный субстрат или его сульфоксид координируется к атому ванадия как бидентатный лиганд, что обеспечивает одинаковую конфигурацию активного каталитического комплекса вне зависимости от растворителя, в котором проводится окисление.
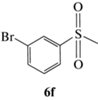
Как следует из данных, представленных в табл. 5, в обоих растворителях ЭИ сульфоксидов 5a–5g в значительной степени зависит от объема заместителей при атоме серы субстрата. Почти во всех случаях в этаноле энантиоселективность катализатора выше, чем в хлористом метилене, из чего можно сделать вывод, что этиловый спирт координируется к иону ванадия, создавая дополнительную стереодифференциацию. Основной вклад в энантиоселективность сульфоксидирования в этаноле, по-видимому, вносят объемы как алифатического (табл. 5, №№ 1, 3, 5, 7), так и ароматического (табл. 5, №№ 3, 9, 11, 13) заместителей при атоме серы в субстрате. В хлористом метилене наряду со стерическим фактором на энантиоселективность реакции, возможно, оказывает влияние и электронный эффект заместителя в ароматическом кольце субстрата, как следует из результатов окисления тиоанизолов (табл. 5, №№ 4, 10, 12, 14).
Стерическое окружение атома серы в субстрате также влияет и на способность комплекса ванадия с лигандом 3a катализировать окисление сульфоксида в сульфон, что можно видеть из величин выходов продуктов окисления бромзамещенных тиоанизолов (табл. 5, №№ 9–14). При этом окисление наиболее стерически затрудненного сульфида, о-бромтиоанизола, приводит к появлению только сульфоксида (табл. 5, №№ 9–10). Данный факт объясняется тем, что для образования сульфона сульфоксид должен координироваться к атому ванадия в переходном состоянии, но объемный заместитель при атоме серы препятствует этому, и каталитического окисления сульфида не происходит. Во всех остальных экспериментах (табл. 5, №№ 1–8, 11–14) в отсутствие стерических затруднений в непосредственной близости к атому серы субстрата VO(OiPr)3/3a катализирует дальнейшее окисление сульфидов в сульфоны, сопровождающееся кинетическим разделением.
ЗАКЛЮЧЕНИЕ
Таким образом, разработана каталитическая система на основе комплексов ванадия с лигандом, полученным из левопимаровой кислоты, для энантиоселективного окисления прохиральных сульфидов водным раствором пероксида водорода. Сульфоксидирование с использованием этого катализатора в различных растворителях (спиртах или галогенированных углеводородах) приводит к продуктам с противоположной конфигурацией асимметрического атома, что позволяет синтезировать целевой энантиомер сульфоксида только путем замены растворителя и не прибегать к дорогостоящей процедуре производства диастереомера катализатора. Наибольшая энантиодифференцирующая способность полученных катализаторов наблюдается в этаноле при окислении субстратов с объемными заместителями у атома серы. Значительный вклад в ЭИ образующегося сульфоксида вносит кинетическое разделение: высокие значения энантиомерного избытка (до 96%) наблюдаются при средних значениях выхода сульфоксида (40–70%).
Список литературы
Pellissier H. // Tetrahedron. 2006. V. 62. № 24. P. 5559.
Salom-Roig X., Bauder C. // Synthesis. 2020. V. 52. № 7. P. 964.
Fernández I., Khiar N. // Chem. Rev. 2003. V. 103. № 9. P. 3651.
Pellissier H. // Tetrahedron. 2007. V. 63. № 6. P. 1297.
Otocka S., Kwiatkowska M., Madalińska L., Kiełbasiński P. // Chem. Rev. 2017. V. 117. № 5. P. 4147.
Han J., Soloshonok V.A., Klika K.D. Drabowicz J., Wzorek A. // Chem. Soc. Rev. 2018. V. 47. P. 1307.
Bryliakov K.P. // Chem. Rev. 2017. V. 117. № 17. P. 11 406.
Landaeta V.R., Rodríguez-Lugo R.E. // Inorganica Chim. Acta. 2015. V. 431. P. 21.
Srour H., Le Maux P., Chevance S., Simonneaux G. // Coord. Chem. Rev. 2013. V. 257. № 21. P. 3030.
Correia I., Pessoa J.C., Duarte M.T., Henriques R.T., Piedade M.F.M., Veiros L.F., Jakusch T., Kiss T., Dörnyei Á., Castro M.M.C.A., Geraldes C.F.G.C., Avecilla F. // Chem. A Eur. J. 2004. V. 10. № 9. P. 2301.
Pessoa J.C., Correia I. // Coord. Chem. Rev. 2019. V. 388. P. 227.
Adão P., Costa Pessoa J., Henriques R.T., Kuznetsov M.L., Avecilla F., Maurya M.R., Kumar U., Correia I. // Inorg. Chem. 2009. V. 48. № 8. P. 3542.
Sun J., Dai Z., Li C., Pan X., Zhu C. // J. Organomet. Chem. 2009. V. 694. № 20. P. 3219.
Pellissier H. // Coord. Chem. Rev. 2020. V. 418. P. 213 395.
Ding Z., Yang Y. // Kinet. Catal. 2017. V. 58. № 3. P. 290.
Khlebnikova T.B., Konev V.N., Pai Z.P. // Data Br. 2018. V. 18. P. 1642.
Khlebnikova T.B., Konev V.N., Pai Z.P. // Tetrahedron. 2018. V. 74. № 2. P. 260.
Degennaro L., Tota A., De Angelis S., Andresini M., Cardellicchio C., Capozzi M.A., Romanazzi G., Luisi R. // Eur. J. Org. Chem. 2017. V. 2017. № 44. P. 6486.
Zhang Y., Tan R., Gao M., Hao P., Yin D. // Green Chem. 2017. V. 19. № 4. P. 1182.
Reddy A.C.S., Anbarasan P. // Org. Lett. 2019. V. 21. № 24. P. 9965.
Liu J., Chen G., Xing J., Liao J. // Tetrahedron Asymmetry. 2011. V. 22. № 5. P. 575.
Liu Z.-M., Zhao H., Li M.-Q., Lan Y.-B., Yao Q.-B., Tao J.-C., Wang X.-W. // Adv. Synth. Catal. 2012. V. 354. № 6. P. 1012.
Ruppenthal S., Brückner R. // J. Org. Chem. 2015. V. 80. № 2. P. 897.
Guan X.Y., Liu Z.Q., Huang H.X., Yang L.P., Jian L., Hu W.H. // Synlett. 2009. № 13. P. 2183.
Tang J., Huang F., Wei Y., Bian H., Zhang W., Liang H. // Dalton Trans. 2016. V. 45. № 19. P. 8061.
Марков А.А., Долин С.П., Моисеева Н.И., Гехман А.Е., Моисеев И.И. // Кинетика и катализ. 2009. V. 50. № 5. С. 656. (Markov A.A., Dolin S.P., Moiseeva N.I., Gekhman A.E., Moiseev I.I. // Kinet. Catal. 2009. V. 50. № 5. P. 656.)
Wu Y., Liu J., Li X., Chan A.S.C. // Eur. J. Org. Chem. 2009. V. 2. № 16. P. 2607.
Liu H., Wang M., Wang Y., Yin R., Tian W., Sun L. // Appl. Organomet. Chem. 2008. V. 22. № 5. P. 253.
Wang Y., Wang M., Wang Y., Wang X., Wang L., Sun L. // J. Catal. 2010. V. 273. № 2. P. 177.
Drago C., Caggiano L., Jackson R.F.W. // Angew. Chemie Int. Ed. 2005. V. 44. № 44. P. 7221.
Cogan D.A., Liu G., Kim K., Backes B.J., Ellman J.A. // J. Am. Chem. Soc. 1998. V. 120. № 32. P. 8011.
Bryliakov K.P., Talsi E.P. // Kinet. Catal. 2003. V. 44. № 3. P. 334.
Hillerns F., Rehder D. // Chem. Ber. 1991. V. 124. № 10. P. 2249.
Chuo T.H., Boobalan R., Chen C. // ChemistrySelect. 2016. V. 1. № 10. P. 2174.
Liu H., Wang M., Wang Y., Wang Y., Sun H., Sun L. // Catal. Commun. 2009. V. 11. № 4. P. 294.
Ohta C., Shimizu H., Kondo A., Katsuki T. // Synlett. 2002. № 1. P. 161.
Sutradhar M., Martins L.M.D.R.S., Guedes da Silva M.F.C., Pombeiro A.J.L. // Coord. Chem. Rev. 2015. V. 301–302. P. 200.
Konev V.N., Pai Z.P., Khlebnikova T.B. // Russ. J. Org. Chem. 2020. V. 56. № 4. P. 604.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ