

Кинетика и катализ, 2022, T. 63, № 6, стр. 749-759
Сравнительное исследование каталитической активности сульфокатионитов макропористой и гелевой структуры в процессе этерификации жирных кислот
К. В. Отопкова a, b, *, А. Л. Есипович a, b, Е. А. Канаков a, Т. А. Чарыкова a, В. Е. Байдаченко a, Т. А. Рябова a
a ФГБОУ ВО Нижегородский государственный технический университет им. Р.Е. Алексеева
603950 Нижний Новгород, ул. Минина, 24, Россия
b ФГАОУ ВО Национальный исследовательский Нижегородский государственный университет
им. Н.И. Лобачевского
603022 Нижний Новгород, пр. Гагарина, 23, Россия
* E-mail: k.otopkova@gmail.com
Поступила в редакцию 26.05.2022
После доработки 22.06.2022
Принята к публикации 26.06.2022
- EDN: UKULGD
- DOI: 10.31857/S0453881122060090
Аннотация
В работе исследованы активность и стабильность катионобменных смол в процессе этерификации жирных кислот с метиловым спиртом. Катиониты охарактеризованы с помощью методов ИК-спектроскопии, электронной микроскопии, низкотемпературной адсорбции–десорбции азота (метод БЭТ). Показано, что развитая поверхность и больший объем пор позволяют легче десорбироваться продуктам реакции, предотвращая блокирование каталитических центров и снижение активности катализатора. Среди рассмотренных катионообменных смол наилучшие результаты получены при использовании катионита с макропористой структурой Lewatit MonoPlus SP 112.
ВВЕДЕНИЕ
Метиловые эфиры жирных кислот (МЭЖК, биодизель) – широко известное альтернативное биотопливо со свойствами, близкими к свойствам минерального дизельного топлива. Биодизельное топливо не содержит соединений серы, нетоксично и разлагается в окружающей среде [1]. При использовании 100% биодизеля выбросы парниковых газов снижаются более чем на 70% по сравнению с нефтяным дизельным топливом [2].
МЭЖК, применяемые в настоящее время в качестве топлива, являются перспективным сырьем для химической промышленности [3]. МЭЖК предлагается использовать для производства ПАВ [4], ингибиторов коррозии [5], жирных спиртов [6], полимеров [7] и других ценных продуктов. Кроме того, МЭЖК служат пластификаторами для ПВХ изделий медицинского и пищевого назначения [8], а продукты их эпоксидирования предлагается применять в качестве эффективных стабилизаторов разложения [9].
Основной промышленный способ получения биодизеля – метанолиз растительных масел в присутствии гомогенных щелочных катализаторов [3, 10]. На сегодняшний день 95% производимого в мире биодизеля производится из пищевых масел, таких как рапсовое, соевое, подсолнечное и пальмовое масла.
Перспективным сырьевым источником для получения МЭЖК являются непищевые масла – льняное масло, масло Ятрофы, масло Karanja, животные жиры и рыбий жир. Еще один вид дешевого сырья для производства МЭЖК – отработанное растительное масло после жарки. Такое масло в 3–4 раза дешевле очищенного пищевого растительного масла, что способствует значительному уменьшению общей стоимости производства МЭЖК.
К недостаткам использования непищевых и отработанных масел относится высокое содержание свободных жирных кислот (СЖК). Для эффективного проведения процесса переэтерификации с обеспечением высокого выхода МЭЖК в присутствии гомогенных щелочных катализаторов необходимо соблюдать определенные требования к исходному маслу. Доля СЖК не должна превышать 0.5 мас. %, содержание воды – 0.3 мас. % [3, 11, 12]. СЖК дезактивируют катализаторы, в результате чего нужно использовать большее количество катализатора, а образующиеся соли жирных кислот (мыла) существенно усложняют сепарацию реакционной смеси. Таким образом, традиционный процесс получения МЭЖК в присутствии гомогенных оснóвных катализаторов требует применения высококачественного сырья, что предполагает высокие производственные затраты.
В настоящее время для переработки сырья с высоким содержанием СЖК предлагается осуществлять процесс в две стадии. На первой стадии протекает этерификация СЖК в присутствии кислотных катализаторов, а на второй стадии – переэтерификация в присутствии оснований.
Для проведения стадии этерификации СЖК используются гомогенные кислотные катализаторы – минеральные кислоты и органические сульфокислоты, обеспечивающие высокую скорость реакции и выход МЭЖК [13]. Однако применение минеральных кислот характеризуется существенными недостатками – высокая коррозионная активность требует наличия дорогостоящего оборудования, а необходимость стадии нейтрализации продуктов реакции приводит к снижению производительности процесса и образованию больших количеств трудно утилизируемых отходов.
Более перспективны гетерогенные катализаторы, которые легко отделяются и могут быть повторно использованы. Их применение не требует стадии нейтрализации и промывки, в результате чего значительно снижается количество отходов.
В качестве гетерогенных кислотных катализаторов этерификации СЖК были испытаны цеолиты [14, 15], гетерополикислоты [16–18], сульфатированные оксиды [19] и другие каталитические системы [20, 21]. Однако все они характеризуются низкой стабильностью или активностью и требуют проведения процесса в более жестких условиях [22].
В последнее время значительное внимание исследователей привлекают катионообменные смолы в качестве катализаторов этерификации СЖК. Катиониты представляют собой пористые полимерные твердые катализаторы. За счет высокого содержания кислотных центров они дают возможность осуществлять процесс этерификации в более мягких условиях, при этом обладают достаточно высокой стабильностью в реакционной системе, легко отделяются, позволяют существенно сократить образование сточных вод и не вызывают коррозии оборудования [23].
В научной литературе представлено довольно много результатов изучения активности катионитов в процессе этерификации СЭК [24–26]. К сожалению, авторами не обосновывается выбор той или иной смолы, используемой для испытаний. Кроме того, недостаточно внимания уделено вопросу стабильности катионообменных смол, в том числе причинам их дезактивации. Поэтому в настоящей работе проведено исследование влияния текстурных и структурных характеристик катионитов на их активность и стабильность в процессе этерификации жирных кислот; изучен вопрос их дезактивации и возможности регенерации.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реагенты и материалы
Метанол квалификации ХЧ (ГОСТ 6995-77) с содержанием основного вещества не менее 99.5 мас. % использовали без дополнительной очистки.
Масло подсолнечное (ГОСТ 1129-2013), кислотное число (КЧ) – 0.098 мг КОН/г, применяли без дополнительной очистки. Жирнокислотный состав подсолнечного масла, определенный по ГОСТ 30418-96, представлен в табл. 1.
Таблица 1.
Масложировой состав подсолнечного масла
| № п/п | Наименование* | Массовая доля, % |
|---|---|---|
| 1 | Миристиновая кислота (14 : 0) | 0.19 |
| 2 | 9-Гексадеценовая кислота (16 : 1) | 0.10 |
| 3 | Пальмитиновая кислота (16 : 0) | 6.14 |
| 4 | Олеиновая кислота (18 : 1) | 28.91 |
| 5 | Линолевая кислота (18 : 2) | 59.61 |
| 6 | Стеариновая кислота (18 : 0) | 4.34 |
| 7 | 11-Эйкозеновая кислота (20 : 1) | 0.13 |
| 8 | Эйкозановая кислота (20 : 0) | 0.19 |
| 9 | Докозановая кислота (22 : 0) | 0.32 |
| 10 | Лигноцериновая кислота (24 : 0) | 0.07 |
МЭЖК получали переэтерификацией подсолнечного масла метанолом в присутствии едкого натра по методике, описанной в работе [27].
Жирные кислоты (кислотное число – 200 мг KOH/г) синтезировали омылением МЭЖК подсолнечного масла. В круглодонный реактор загружали 200 г МЭЖК и 400 мл 20%-ного спиртового раствора гидроксида калия. Смесь кипятили с обратным холодильником в течение 15 мин. Далее реактор охлаждали и смесь с помощью разбавленной серной кислоты (концентрация кислоты ~25%) доводили до pH 3.0. Жирные кислоты (ЖК) выделяли многократной экстракцией с гексаном. Полученный экстракт промывали большим количеством дистиллированной деионизированной воды до достижения нейтральной среды в промывных водах. Конечные следы растворителя удаляли вакуумной дистилляцией.
В работе использовали сильнокислотные сульфокатиониты КУ-2-8 (ГОСТ 20298-74, Россия), Amberlyst 36DRY (“DOW”, Франция) и Amberlite IR-120 (“DOW”, Франция), Lewatit MonoPlus SP 112 (“LANXESS”, Германия), Tulsion T-52 (“Thermax”, Индия), Tokem-103 W MTBE (“TOKEM”, Росиия), Purolite PPC 100H (“Purolite Ltd.”, Великобритания). Свойства катионитов представлены в табл. 2.
Таблица 2.
Характеристики катионообменных смол
| Наименование | Катионообменные смолы | ||||||
|---|---|---|---|---|---|---|---|
| КУ-2-8 | Purolite PPC 100H |
Amberlite IR-120 |
Tulsion T-52 H |
Tokem-103 W MTBE | Amberlyst 36 DRY |
Lewatit MonoPlus SP 112 | |
| Структура* | Г | Г | Г | Г | МП | МП | МП |
| Матрица | Cополимер стирола и дивинил-бензола | Cополимер стирола и дивинил-бензола | Сополимер стирола и дивинил-бензола | Сополимер полистирола | Сополимер стирола и дивинил-бензола | Сополимер стирола и дивинил-бензола | Сополимер стирола и дивинил-бензола |
| Внешний вид | Сферические зерна от желтого до темно-коричневого цвета | Сферические зерна | Янтарные гранулы | Сферические гранулы от светло- желтого до темно-коричневого цвета | Сферические зерна от серого или коричневого до черного цвета | Сферические зерна непрозрачные | Сферические зерна бежево-серый, непрозрачный |
| Функциональная группа | –SO3H | –SO3H | –SO3H | –SO3H | –SO3H | –SO3H | –SO3H |
| Исходная (товарная) ионная форма | Na+ | H+ | Na+ | H+ | H+ | H+ | Na+ |
| Размер зерен, мкм | 315–1250 | 300–1200 | 620–830 | 300–1200 | 400–1250 | 600–850 | 650 |
| Насыпная плотность, г/л | 750–800 | 790–830 | 840 | 810–850 | 750–800 | 770 | 750 |
| Влагопоглощение, % | 48–58 | 51–55 | 45 | 43 | 50–60 | 55 | 52–57 |
| Максимальная рабочая температура, °С | 120 | 120 | 135 | 120 | 120 | 150 | 140 |
Методика подготовки катализатора
Перед исследованием свойств и испытанием в процессе этерификации для удаления нежелательных примесей катиониты были промыты несколько раз дистиллированной водой до изменения цвета промывных вод от светло-коричневого до бесцветного. Смолы, поставляемые в натриевой форме, после промывки переводили в кислотную форму по методике ГОСТ 10896-78. После промывки смолы сушили в вакуумном шкафу при 60°C в течение ночи и хранили в вакуумном эксикаторе в присутствии поглотителя влаги.
Исследование свойств катализатора
Измерение удельной поверхности свежих и отработанных катализаторов проводили методом БЭТ по физической адсорбции азота на приборе Sorbi MS (“META”, Россия). Подготовка проб включала обезвоживание образца в течение 40 мин при 105°С и его предварительное термическое выдерживание при 105°С.
ИК-спектры свежих и отработанных катализаторов записывали на воздухе при комнатной температуре на ИК-Фурье спектрометре IRAffinity-1 (“Shimadzu”, Япония) в таблетках KBr в области 400–4000 см–1 с шагом сканирования 1 см–1.
Кислотность (содержание протонных кислотных центров) свежих и отработанных катализаторов измеряли титриметрическим методом c использованием 0.1 н раствора едкого натра в качестве титранта и индикатора фенолфталеин.
Элементный состав и морфологию поверхности катализаторов определяли с помощью сканирующего электронного микроскопа JSM-7600F (“JEOL”, Япония) с энергодисперсионным анализатором INCA Energy TEM 250 Х-Мах.
Методика проведения процесса этерификации
Исследование активности катализаторов выполняли на лабораторной установке периодического действия, состоящей из реактора с рубашкой, обратного холодильника и магнитной мешалки.
Так как скорость набухания гелевых катионитов существенно ниже макропористых, все испытываемые катализаторы выдерживали в метаноле в течение суток непосредственно перед проведением процесса. Для этого в реактор загружали расчетное количество метанола и катализатора.
После выдерживания катализатора в метаноле в течение суток реактор нагревали до температуры 60°С при включенном перемешивающем устройстве. При достижении заданной температуры в реактор загружали расчетное количество ЖК, после чего отбирали начальную пробу и фиксировали время начала реакции. Для контроля за ходом протекания процесса через определенные промежутки времени брали пробы.
Методики проведения анализа продуктов реакции
Качественный и количественный состав МЭЖК в реакционной массе анализировали методом хромато-масс-спектрометрии на газовом хроматографе GC-2010 Plus (“Shimadzu”, Япония), оснащенного МАСС- и ПИД-детекторами, на капиллярной колонке VertiBond 1701 (длина – 30 м, внутренний диаметр – 0.25 мм, толщина пленки – 0.25 мкм). Температура термостата колонок – 200°С, испарителя – 250°С, детектора – 250°С. Газ-носитель – гелий. В качестве стандарта для определения концентраций МЭЖК в реакционных смесях использовали метилстеарат.
Содержание моно-, ди- и триглицеридов находили методом гельпроникающей хроматографии (ГПХ) на жидкостном хроматографе ЖХ-301 (“Хромос”, Россия), оборудованном ВЭЖХ-насосом, системой колонок для ГПХ, состоящей из двух последовательно соединенных колонок Phenogel 00H-0441-K0 (“Phenomenex”, США, (300 × × 7.8 мм)), заполненных сшитым полистиролом (размер частиц – 5 мкм, диаметр пор – 50 Å). Для защиты колонок от загрязнений использовали предколонку Guards 03B-2088-K0 (“Phenomenex”, США, размер частиц – 5 мкм). Анализ проводили при температуре 25°С. В качестве элюента применяли тетрагидрофуран со скоростью подачи 1.0 мл/мин. Расчет концентраций МЭЖК выполняли методом абсолютного градуировочного графика с метилстеаратом в качестве стандарта. Расчет концентраций глицеридов ЖК производили методом абсолютного градуировочного графика с использованием моноолеата, диолеата и тристеарата глицерина как стандарта.
Кислотное число реакционной массы определяли по ГОСТ Р 52110-2003 титриметрическим методом в среде изопропилового спирта, титрантом служил 0.1 н раствор гидроксида калия, индикатором – фенолфталеин.
Конверсию ЖК вычисляли по формуле (1):
(1)
${\text{Конверсия}}\,\,{\text{ЖК}} = \frac{{{\text{КЧ}} \times {\text{279}}{\text{.3}}}}{{{\text{56}} \times {\text{1000}}}} \times {\text{100\% ,}}$Для исследования стабильности сульфокатионитов после проведения процесса этерификации катализатор отделяли от реакционной массы, высушивали на фильтровальной бумаге на воздухе в течении ночи, после чего навеску катализатора использовали на повторном цикле.
Для регенерации сульфокатионита после завершения процесса этерификации катализатор отделяли от реакционной массы и промывали метанолом при комнатной температуре до обесцвечивания промывного раствора. После промывки катализатор сушили при температуре 60°С в течении 3 ч.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Результаты каталитических испытаний катионообменных смол в процессе этерификации ЖК метанолом представлены на рис. 1.
Рис. 1.
Сравнение каталитической активности сульфокатионитов с гелевой (а) и макропористой (б) структурой в процессе этерификации. Условия реакции: температура – 60°С, мольное соотношение метанол : ЖК = 10 : 1, загрузка катионита – 10 мас. % от массы ЖК (0.01 ммоль –SO3H/г ЖК).
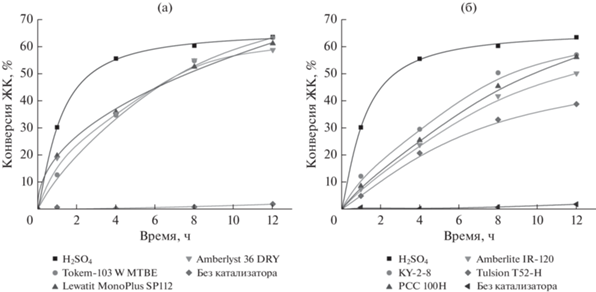
Без использования катализатора даже за 12 ч конверсия ЖК была не более 2.0%. В то же время осуществление процесса этерификации с в присутствии серной кислоты (0.01 ммоль/г ЖК) обеспечивает высокую скорость процесса. Уже за 4 ч с момента начала реакции конверсия достигает 55.6%, а за 12 ч – 63.6%.
При применении катионита с макропористой структурой Lewatit MonoPlus SP 112 и с гелевой структурой КУ-2-8 (0.01 ммоль –SO3H/г ЖК) за 12 ч проведения процесса конверсия ЖК составила 61.5 и 57.1% соответственно. Наименее активным среди испытанных оказался катионит с гелевой структурой Tulsion T-52 H, в его присутствии конверсия ЖК была 38.9%.
Следует отметить, что максимальная конверсия ЖК (63.4%) достигнута при использовании катионита Tokem-103 W MTBE. Однако после каталитических испытаний зерна катионита были полностью разрушены в результате механического воздействия.
Активность катионитов существенно зависит от содержания каталитически активных сульфогрупп. В табл. 3 представлены значения кислотности использованных в работе смол.
Таблица 3.
Содержание протонных кислотных центров свежеприготовленных сульфокатионитов
| Наименование смолы | Кислотность, ммоль/г |
|---|---|
| КУ-2-8 | 4.584 |
| PPC 100H | 4.314 |
| Amberlite IR-120 | 4.058 |
| Tulsion T-52 H | 3.987 |
| Tokem-103 W MTBE | 5.370 |
| Amberlyst 36 DRY | 4.225 |
| Lewatit MonoPlus SP 112 | 4.377 |
На рис. 2 показаны зависимости конверсии ЖК от кислотности применяемых смол. С целью предотвращения влияния скорости набухания смол на их активность смолы перед проведением процесса выдерживали в метаноле в течение суток.
Рис. 2.
Зависимость конверсии ЖК от кислотности макропористых и гелевых сульфокатионитов: 1 – Amberlyst 36DRY; 2 – Lewatit MonoPlus SP 112; 3 – Tokem-103 W MTBE; 4 – Tulsion T-52 H; 5 – Amberlite IR-120; 6 – PPC 100H; 7 – КУ-2-8. Время проведения процесса: 1 ч (а, б), 8 ч (в). Условия реакции: температура – 60°С, мольное соотношение метанол : ЖК = 10 : 1, загрузка катионита – 10 мас. % от массы ЖК. ◼ – макропористая структура (МП); ◆ – гелевая структура (Г).
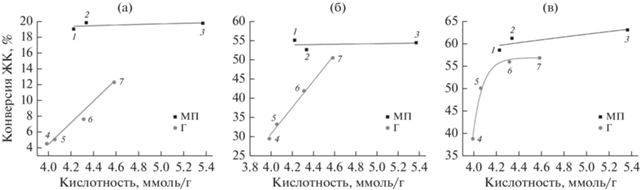
Как видно из рис. 2, для смол гелевой структуры характерна линейная зависимость конверсии ЖК от кислотности. Несмотря на то, что кислотность макропористых катионитов Amberlyst 36DRY и Lewatit MonoPlus SP 112 ниже таковой гелевого катионита КУ-2-8, их активность существенно выше.
В табл. 4 представлены текстурные характеристики свежих сульфокатионитов.
Таблица 4.
Текстурные характеристики свежеприготовленных сульфокатионитов
| Катионит | Удельная площадь поверхности (БЭТ), м2/г | Общий объем пор, см3/г | Средний размер пор (D50), нм |
|---|---|---|---|
| КУ-2-8 | 0.2 | – | – |
| PPC 100H | 0.4 | – | – |
| Amberlite IR-120 | 0.2 | – | – |
| Tulsion T-52 H | 1.2 | – | – |
| Tokem-103 W MTBE | 7.4 | 0.027 | 66.4 |
| Amberlyst 36DRY | 18.1 | 0.089 | 27.6 |
| Lewatit MonoPlus SP 112 | 20.2 | 0.085 | 25.2 |
Удельные площади поверхности гелевых сульфокатионитов в сухом состоянии составляют всего 0.2–1.2 м2/г. В сухих гелевых смолах отсутствует пористая структура. Для образования пор в полимерной сетке необходимо набухание полимерной матрицы в результате проникновения растворителя в пространство между полимерными звеньями. При этом размер пор равен порядка 1 нм.
Макропористые смолы пронизаны сквозными и тупиковыми порами, за счет чего могут иметь гораздо большую площадь поверхности в сухом состоянии, чем смолы гелевого типа. Значения удельной поверхности для смол Lewatit MonoPlus SP 112 и Amberlyst 36DRY составляют 20.2 и 18.1 м2/г соответственно. Менее развитая поверхность характерна для катионита Tokem-103 W MTBE – 7.4 м2/г. Средний размер пор для макропористых катионитов равен 25–66 нм.
По-видимому, макропористая структура катионитов обеспечивает лучший доступ к каталитическим центрам объемных молекул ЖК. Кроме того, развитая поверхность и больший объем пор позволяют легче десорбироваться продуктам реакции, предотвращая блокирование каталитических центров и снижение активности катализатора. Это подтверждается результатами ИК-спектрометрии образцов катализатора до и после проведения процесса (рис. 3).
Рис. 3.
ИК-спектры катионообменных смол – свежих (а) и после проведения одного цикла каталитических испытаний (б): 1 – Tulsion T-52 H; 2 – Amberlite IR-120; 3 – PPC 100H; 4 – КУ-2-8; 5 – Amberlyst 36 DRY; 6 – Lewatit MonoPlus SP 112; 7 – Tokem-103 W MTBE.
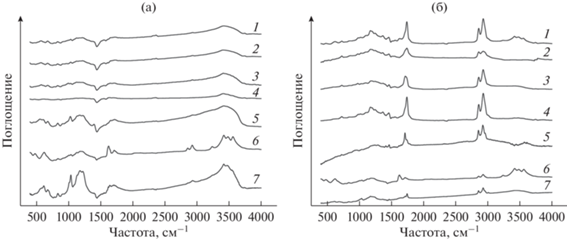
На рис. 3а представлены ИК-спектры свежих сухих катионообменных смол. Все ИК-спектры показали наличие общих характерных полос поглощения [28]. Полосы в интервалах 600–700, 1010–1080 и 1150–1260 см–1 соответствуют колебания ‒SO3H-групп. Широкая полоса в интервале 3400–3700 см–1 связана с колебаниями ОН-связи. Пик при 1645 см–1 к характерен для колебаний связей C=C, а полосы в области 1000–1250 см–1 – различных деформаций связей C–H, относящихся к ароматическим ядрам полимерной матрицы. В тоже время на ИК-спектрах образцов после проведения процесса (рис. 3б) наблюдается появление интенсивных пиков, соответствующих жирным кислотам, эфирам и спиртам [28]. Полоса в диапазоне 1735–1750 см–1 связана с колебаниями C=O насыщенных алифатических сложных эфиров. Полоса, наблюдаемая около 1076 cм–1, характеризует колебания C–O-связей в спиртах, полосы в области 2800–3000 cм–1 – колебания CH3-групп. Полоса поглощения 1469 cм–1 может быть отнесена к колебаниям C–H-связей.
В результате блокирования поверхности исходными веществами и продуктами реакции происходит снижение содержания протонных кислотных центров катализаторов. При этом падение кислотности макропористых катионитов после одного цикла каталитических испытаний (<1%) значительно меньше, чем у катионитов гелевой структуры (табл. 5).
Таблица 5.
Содержание протонных кислотных центров сульфокатионитов после 1 цикла каталитических испытаний
| Катионит | Кислотность, ммоль/г | Относительное снижение содержания протонных кислотных центров по сравнению с исходным образцом, % |
|---|---|---|
| КУ-2-8 | 4.328 | 5.6 |
| PPC 100H | 3.914 | 9.3 |
| Amberlite IR-120 | 3.814 | 6.0 |
| Tulsion T-52 H | 3.908 | 2.0 |
| Tokem-103 W MTBE | 5.030 | 6.3 |
| Amberlyst 36 DRY | 4.195 | 0.7 |
| Lewatit MonoPlus SP 112 | 4.350 | 0.6 |
На рис. 4 представлены результаты каталитических испытаний макропористого катионита Lewatit MonoPlus SP 112 и гелевого катионита КУ‑2-8 в течение 5 циклов. Образцы после проведения каждого цикла отделяли от реакционной массы центрифугированием и использовали далее без дополнительной обработки.
Рис. 4.
Стабильность сульфокатионитов после 5 циклов (без промывки катализатора перед испытаниями). Условия реакции: температура – 60°С, мольное соотношение метанол : ЖК = 10 : 1, загрузка катионита – 10 мас. % от массы ЖК (0.01 ммоль –SO3H/г ЖК).
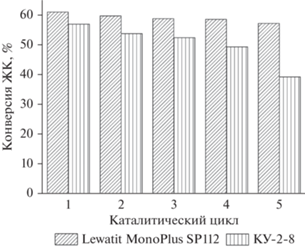
Как видно из рис. 4, только на 5 цикле наблюдается некоторое снижение активности макропористого катионита Lewatit MonoPlus SP 112, при этом активность гелевого катионита КУ-2-8 постоянно падает от цикла к циклу.
При промывке катионита КУ-2-8 метанолом перед повторным применением его стабильность повышается (рис. 5).
Рис. 5.
Стабильность сульфокатионитов после 5 циклов (с промывкой катализатора перед испытаниями). Условия реакции: температура – 60°С, мольное соотношение метанол : ЖК = 10 : 1, загрузка катионита – 10 мас. % от массы ЖК (0.01 ммоль –SO3H/г ЖК).
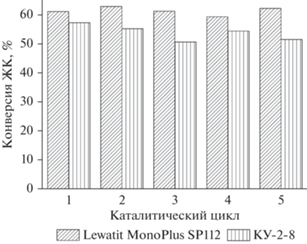
Катионит Lewatit MonoPlus SP 112 сохраняет свою активность даже после 20 каталитических циклов, а небольшое ее снижение (конверсия ЖК составляет 58.8%) обусловлено механическим истиранием зерен и потерей катализатора при сепарировании.
В табл. 6 представлены значения кислотности сульфакатионитов после каждого цикла испытаний.
Таблица 6.
Содержание протонных кислотных центров сульфокатионитов Lewatit MonoPlus SP 112 и КУ-2-8 в ходе каталитических испытаний
| Наименование смолы | Кислотность, ммоль/г | ||||
|---|---|---|---|---|---|
| до синтеза | после 1 цикла | после 2 цикла | после 3 цикла | после 5 цикла | |
| Lewatit MonoPlus SP 112 (без промывки) |
4.377 | 4.350 | 4.255 | 4.348 | 3.849 |
| Lewatit MonoPlus SP 112 (с промывкой метанолом) |
4.377 | 4.405 | 4.380 | 4.396 | 4.425 |
| КУ-2-8 (без промывки) | 4.584 | 4.328 | 4.287 | 4.162 | 3.722 |
| КУ-2-8 (с промывкой метанолом) | 4.584 | 4.359 | 4.361 | 4.349 | 4.335 |
Как уже было показано выше, кислотность катионитов за счет блокирования продуктами реакции понижается. Однако промывка метанолом позволяет очистить поверхность катализатора и деблокировать каталитические центры, в результате чего кислотность восстанавливается. Это также подтверждается данными элементного анализа поверхностей катализаторов (табл. 7). После проведения каталитических испытаний содержание серы на поверхности исследуемых образцах падает, а после промывки метанолом восстанавливается.
Таблица 7.
Элементный состав поверхности свежих катионитов и после каталитических испытаний
| Наименование смолы | Соотношение углерод/кислород/сера, ат. % | ||
|---|---|---|---|
| углерод | кислород | сера | |
| Lewatit MonoPlus SP, 112 свежий | 73.3 | 19.5 | 7.2 |
| Lewatit MonoPlus SP 112, после 5 циклов, без промывки | 77.9 | 16.4 | 5.7 |
| Lewatit MonoPlus SP 112, после 5 циклов, промытый метанолом | 74.3 | 19.0 | 6.7 |
| КУ-2-8, свежий | 69.2 | 23.1 | 7.7 |
| КУ-2-8, после 5 циклов, без промывки | 82.9 | 16.4 | 0.7 |
| КУ-2-8, после 5 циклов, промытый метанолом | 78.8 | 15.8 | 5.4 |
Результаты исследования текстурных характеристик катионита Lewatit MonoPlus SP 112 также свидетельствуют о восстановлении поверхности катализатора после его промывки метанолом (рис. 6, табл. 8).
Рис. 6.
Микрофотографии катионита Lewatit MonoPlus SP 112: свежего (а), после 5 каталитических циклов (б), после промывки метанолом (в).

Таблица 8.
Удельная поверхность катионита Lewatit MonoPlus SP 112 свежего и после каталитических испытаний
| Катионит | Удельная площадь поверхности по методу БЭТ, м2/г |
|---|---|
| Свежий | 20.2 |
| Без промывки | |
| После 1 цикл | 3.0 |
| После 3 цикл | 2.2 |
| После 5 цикл | 2.0 |
| С промывкой метанолом | |
| После 3 цикла | 20.4 |
| После 5 цикла | 20.4 |
Таким образом, использование макропористого катионита с периодической промывкой метанолом в мягких условиях позволяет достигать выхода ЖК 60% с высокой стабильностью.
Для моделирования процесса получения МЭЖК из масел с высокой кислотностью была приготовлена смесь жирных кислот и растительного масла в соотношении (мас. %) 25 : 75 (КЧ = = 67 мг KOH/г). При такой кислотности проведение переэтерификации в присутствии оснóвных катализаторов невозможно из-за образования солей жирных кислот.
Для снижения содержания жирных кислот их этерифицировали при температуре 60°С, мольном соотношении метанол : ЖК = 10 : 1 и загрузке катализатора 10 мас. % (0.01 ммоль –SO3H/г ЖК). За 12 ч процесса конверсия ЖК составила 59.9%. Следует отметить, что в данных условиях превращений глицеридов жирных кислот за счет переэтерификации не наблюдается, что подтверждается результатами анализа реакционной массы методом гельпроникающей хроматографии.
Полученное после этерификации масло использовали для процесса переэтерификации метанолом. Процесс осуществляли при температуре 60°С и мольном соотношении метанол : масло = = 4 : 1 в течение 3 ч в присутствии 3 мас. % едкого натра. Суммарный выход МЭЖК был 88%.
ЗАКЛЮЧЕНИЕ
Проведенные в настоящей работе исследования показали, что на активность и стабильность сульфокатионитов в первую очередь оказывает влияние их текстурные свойства. Макропористая структура катионитов способствует более легкой десорбции продуктов реакции, предотвращая блокирование каталитических центров и снижение активности катализатора. Среди рассмотренных катионообменных смол наибольшую активность и стабильность проявил катионит с макропористой структурой Lewatit MonoPlus SP 112. Даже после 20 каталитических циклов без регенерации активность Lewatit MonoPlus SP 112 снижается незначительно. Катиониты с гелевой структурой менее стабильны, однако промывка метанолом позволяет регенерировать их поверхность, в результате чего активность восстанавливается.
Список литературы
Dhar A., Agarwal A.K. // Fuel. 2014. V. 119. P. 70.
Biodiesel Vehicle Emissions. – URL: https://www.afdc.energy.gov/vehicles/diesels_emissions.html (oбpaщeния 20.04.2022).
Belousov A.S., Esipovich A.L., Kanakov E.A., Otopkova K.V. // Sust. Energ. Fuels. 2021. V. 5. P. 4512.
Maag H., Am J. // Oil Chem. Soc. 1984. V. 61. P. 259.
Biermann U., Friedt W., Lang S., Luehs W., Machmueller G., Metzger J.O., Klaas M.R., Schaefer H.J., Schneider M.P. // Angew. Chem. Int. Ed. 2000. V. 39. P. 2206.
Farris R.D., Am J. // Oil Chem. Soc. 1979. V. 56. P. 770.
Dedov A.V. // Fibre Chem. 2013. V. 45. P. 104.
Pleissner D., Lau K. Y., Zhang C., Lin C. S. K. // ChemSusChem. 2015. V. 8. P. 1686.
Danov S.M., Kazantsev O.A., Esipovich A.L., Belo usov A.S., Rogozhin A.E., Kanakov E.A. // Catal. Sci. Technol. 2017. V. 7. № 17. P. 3659.
Shahid E.M., Jamal Y. // Renew. Sust. Energ. Rev. 2011. V. 15. P. 4732.
Alegría A., Cuellar J. //Appl. Catal. B: Env. 2015. V. 179. P. 530.
Esipovich A.L., Rogozhin A.E., Danov S.M., Belousov A.S., Kanakov E.A. // Chem. Eng. J. 2018. V. 339. P. 303.
Dias J.M., Alvim-Ferraz M.C.M., Almeida M.F. // Fuel. 2008. V. 87. P. 3572.
Doyle A.M., Albayati T.M., Abbas A.S., Alismaeel Z.T. // Renew. Energy. 2016. V. 97. P. 19.
Chung K.H., Park B.G. // J. Ind. Eng. Chem. 2009. V. 15. P. 388.
Li L., Zou C., Zhou L. // Renew. Energy. 2017. V. 107. P. 14.
Fernandes S.A., Cardoso A.L., Silva M.J.A. // Fuel Proc. Technol. 2012. V. 96. P. 98.
Caldas B.S., Nunes C.S., Souza P.R., Rosa F.A., Visentainer J.V., Júnior O.D.S., Muniz E.C. // Appl. Catal. B: Env. 2016. V. 181. P. 289.
Gardy J., Hassanpour A., Lai X., Ahmed M.H., Rehan M. // Appl. Catal. B: Env. 2017. V. 207. P. 297.
Zhang Q.Y., Wei F.F., Ma P.H., Zhang Y.T., Wei F.H., Chen H.L. // Waste Biomass Valorization. 2018. V. 9. P. 911.
Soltani S., Rashid U., Al-Resayes S.I., Nehdi I.A. // Energy Convers. Manag. 2017. V. 141. P. 183.
Soroush S., Umer R., Al-Resayes, Ibrahim S. Nehdi, Arbi I. // Energy Convers. Manag. 2016. https://doi.org/10.1016/j.enconman.2016.07.042
Rodrigues Keila L.T.; Pasa Vânya M.D.; Cren Érika C. // J. Env. Chem. Eng. 2018. V. 6. № 4. P. 4531.
Lim R., Kim D.-K., Lee J.-S. // Energies. 2020. V. 13. P. 1513.
Yunus N.M., Zainal Abidin S., Sim Yee C. // Energy Sources. Part A. 2018. V. 40. P. 2518.
Zhang Q., Zhang Y., Deng T., Wei F., Jin J., Ma P. // Biomass, Biofuels, Biochemicals. 2020. P. 407.
Esipovich A.L., Rogozhin A.E., Belousov A.S., Kanakov E.A., Danov S.M. // Fuel Proc. Technol. 2018. V. 173. P. 153.
Abidin S.Z., Yunus N.M., Ghani S.A.A., Roslan N.A., Yee C.S. // Biofuels. 2017. V. 11. № 5. P. 561. https://doi.org/10.1080/17597269.2017.1378990
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ