

Кинетика и катализ, 2022, T. 63, № 6, стр. 774-788
Влияние добавки гадолиния на морфологию активной фазы, физико-химические и каталитические свойства катализатора MoVSbNbGdOx/SiO2 в реакции окислительного дегидрирования этана в этилен
Г. А. Зенковец a, *, А. А. Шутилов a, В. М. Бондарева a, В. И. Соболев a, И. П. Просвирин a, Е. А. Супрун a, А. В. Ищенко a, А. С. Марчук a, С. В. Цыбуля a, В. Ю. Гаврилов a
a ФГБУН Институт катализа им. Г.К. Борескова СО РАН
630090 Новосибирск, просп. Акад. Лаврентьева, 5, Россия
* E-mail: zenk@catalysis.ru
Поступила в редакцию 04.04.2022
После доработки 26.07.2022
Принята к публикации 27.07.2022
- EDN: QVJCFE
- DOI: 10.31857/S0453881122060181
Аннотация
В работе исследовано влияние добавок гадолиния на морфологию, фазовый состав и каталитические свойства катализаторов MoVSbNbGdOx/SiO2 в реакции окислительного дегидрирования этана в этилен (ОДЭ). Показано, что концентрация гадолиния оказывает значительное воздействие на его каталитические свойства. При оптимальном содержании гадолиния (Gd/Mo = 0.01–0.015) регистрируется увеличение каталитической активности и селективности по этилену: при температуре 400°C выход этилена достигает 72% при конверсия этана более 91% и селективности образования этилена выше 79%. По данным рентгенофазового анализа, сканирующей и просвечивающей электронной микроскопии основным компонентoм катализатора является фаза M1, стабилизированная как внутри, так и на поверхности частиц SiO2. Mодифицирование катализатора гадолинием существенно влияет на морфологию частиц фазы М1. При оптимальном содержании гадолиния при термообработке в основном формируются частицы фазы М1 игольчатой морфологии с выходящей на поверхность наиболее развитой плоскостью [001], проявляющие максимально высокую активность в ОДЭ. Частицы фазы М1 данной морфологии главным образом стабилизированы внутри пористых сферических частиц SiO2, что предотвращает их сильную агломерацию и спекание. Меньшая доля частиц фазы М1 тарельчатой морфологии, характеризующихся более низкой каталитической активностью, так же как и частицы фазы М2, стабилизируются на внешней поверхности частиц SiO2. В катализаторе, не содержащем добавки Gd, в основном формируются частицы фазы М1 тарельчатой морфологии, что приводит к снижению активности катализатора. Полученный катализатор длительное время устойчиво работает в реакционной среде без ухудшения свойств.
ВВЕДЕНИЕ
В настоящее время получение этилена прямым окислительным дегидрированием этана является перспективным направлением. Это один из наиболее многообещающих способов производства этилена с использованием природного газа, обеспечивающий значительное преимущество в энергоэффективности по сравнению с традиционными паровым пиролизом и каталитическим крекингом [1–6]. Согласно литературным данным наиболее эффективны для этого процесса многокомпонентные катализаторы MoVTe(Sb)NbOx [1–16]. Так, на лучших Te-содержащих катализаторах MoVTeNbOx при температуре 350–500°C достигается достаточно высокий выход этилена – 72–76% [2, 3, 9, 13–18]. Из литературы известно, что многочисленные Sb-катализаторы MoVSbNbOx менее активны и селективны в образовании этилена по сравнению с Te-содержащими. Так, на одном из лучших Sb-катализаторов MoVSbNbCaOx при температуре 400°С выход этилена составляет 52% [19–21].
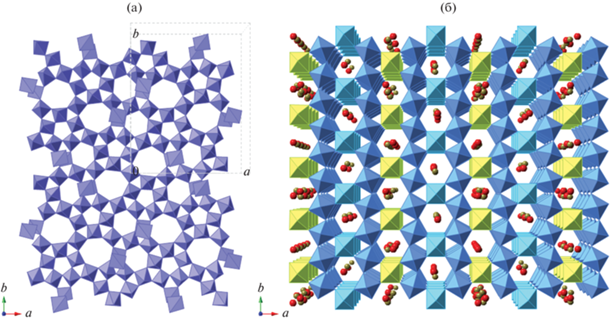
Однако в наших недавних работах [22, 23] были синтезированы Sb-содержащие катализаторы MoVSbNbCeOx/SiO2, модифицированные добавками оксида Ce, обеспечивающие при температуре 400–550°C выход этилена 74%. В одинаковых условиях реакционной среды они демонстрируют каталитические свойства, близкие к таковым одного из самых эффективных Te-катализаторов [2, 13, 14]. Катализатор содержит орторомбическую (M1) и гексагональную (M2) кристаллические фазы в количестве около 70 и 30% соответственно [22]. Как показано в [12, 15, 24–30], в MoVTe(Sb)NbOx фаза M1 (пространственная группа Pba2) имеет слоистую структуру, образованную сетями октаэдров MO6 (M = Mo, V), соединенных вершинами, которые, накладываясь трансляционно друг на друга в направлении [001], формируют пентагональные, гексагональные и гептагональные каналы, проходящие через всю структуру фазы М1. В структуре М1 сети октаэдров образуют в слое пентагональные, гексагональные и гептагональные кольца октаэдров (рис. 1а). Считается, что ниобий стабилизируется в пентагональных каналах, в то время как теллур или сурьма – в гексагональных каналах. Фаза M2 имеет структуру гексагональной бронзы (пространственная группа P6мм), в которой катионы в октаэдрическом кислородном окружении образуют каркас с гексагональными каналами, в которых расположены атомы Te или Sb (рис. 1б).
Рис. 1.
Структурные модели орторомбической М1 (а) и гексагональной М2 (б) фаз вдоль направления [001].
В литературе отмечается, что основным недостатком использования Te-содержащих каталитических систем MoVTeNbOx в ОДЭ является высокая летучесть теллура. При термообработке в инертной среде, а также в процессе каталитической реакции происходит частичное восстановление соединений теллура до металлического состояния, что приводит к его последующей сублимации из катализатора, разрушению кристаллической структуры катализаторов и ухудшению их каталитических свойств [1, 2, 13, 14, 31, 32].
В настоящее время большое внимание уделяется улучшению каталитических характеристик систем, в состав которых входит Sb, в реакции ОДЭ. В связи с этим изучение влияния модифицирования Sb-содержащих катализаторов MoVSbNbOx/SiO2 добавками различной природы на их физико-химические и каталитические свойства представляется актуальным. В настоящей работе были синтезированы и исследованы новые многокомпонентные системы MoVSbNbGdOx/SiO2, характеризующиеся высокой каталитической активностью в реакции ОДЭ. Исследовано влияние концентрации модифицирующей добавки Gd на физико-химические и каталитические свойства образцов. Определен оптимальный химический состав, обеспечивающий высокую активность катализатора.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Катализаторы состава 50 мас. % Mo1V0.24Sb0.23Nb0.08Gd0–0.02Ox/50 мас. % SiO2 готовили смешением водных растворов парамолибдата аммония, метаванадата аммония, трехокиси сурьмы, нитрата церия, SiO2 и оксалата ниобия при температуре 50–80°C с последующей сушкой суспензии на лабораторной распылительной сушилке Mini Spray Dryer B-290 (“Buchi Labortechnik AG”, Швейцария) при температуре 105°C. Полученные порошки таблетировали и прокаливали в токе Ar при 350°C в течение 2 ч, а затем при 600°C в течение 2 ч. Для приготовления катализаторов использованы следующие исходные вещества: парамолибдат аммония (NH4)6Mo7O24 · 4H2O (“Вектон”, Россия), метаванадат аммония NH4VO3 (“Вектон”, Россия), Sb2O3 (“Реахим”, Россия), нитрат гадолиния Gd(NO3)3 · 6H2O (“Acros Organics”, США), оксалат ниобия, который синтезировали из пентахлорида ниобия NbCl5 (“Acros Organics”, США) по методике, приведенной в [33], золь SiO2 (“Nalco-2327”, США).
Химический состав прокаленного катализатора определяли методом атомно-эмиссионной спектрометрии с индуктивно-связанной плазмой на спектрометре OPTIMA 4300 DV (“Perkin Elmer”, США) [34].
Рентгенографические исследования образцов проводили на дифрактометре Bruker D8 (“Bruker”, Германия) с монохроматическим CuKα-излучением. Рентгенограммы снимали в режиме сканирования в области 2θ = 5°–70° с шагом 0.02° и накоплением в каждой точке 3 с. Для детектирования сигнала использовали многоканальный детектор LynxEye. Количественный фазовый анализ выполняли методом Ритвельда с помощью программного пакета TOPAS v.4.2 [35], применяя структурные параметры, приведенные в [37], и базы структурных данных ICSD [36].
Рентгеновские фотоэлектронные спектры (РФЭС) записывали на спектрометре SPECS (Германия) с анализатором PHOIBOS-150-MCD-9 и монохроматором FOCUS-500 (излучение AlK, h = 1486.74 эВ, 200 Вт). Шкала энергий связи была предварительно откалибрована с использованием уровней положений пиков Au4f7/2 (Есв = 84.0 эВ) и Cu2p3/2 (Есв = 932.67 эВ). Энергию связи калибровали по положению пика C1s (Есв = = 284.8 эВ), соответствующего углеводородным отложениям на поверхности образца [37]. Образец в виде порошка наносили на проводящий двухсторонний медный скотч. Обзорный спектр и индивидуальные спектры элементов регистрировали при энергии пропускания анализатора 20 эВ. Атомные отношения элементов рассчитывали из интегральных интенсивностей фотоэлектронных пиков, которые были скорректированы с помощью соответствующих факторов чувствительности на основе сечений фотоионизации Скофилда [38]. Обработку и анализ спектральных данных выполняли с помощью программного обеспечения XPS Peak 4.1 [39].
Пористую структуру катализаторов изучали методом низкотемпературной адсорбции азота при 77.4 K на приборе DigiSorb-2600 (“Micromeritics”, США) и методом ртутной порометрии на приборе AutoPore IV 9500 (“Micromeritics”, США). Катализаторы предварительно тренировали в вакууме при температуре 200°C и давлении 10–4 мм рт. ст. в течение 5 ч. Распределение мезопор по размерам и преобладающий размер пор рассчитывали классическим методом Баррета–Джойнера–Халенды (BJH). Величины суммарного объема пор (VΣ) и распределение макропор по размерам были получены из данных ртутной порометрии.
Морфологию образцов исследовали методом сканирующей электронной микроскопии (СЭМ) при помощи микроскопа Regulus SU8230 FESEM (“Hitachi”, Япония) при энергии электронов пучка 2 кэВ с использованием Lower (L), Upper (U) и Top (T) детекторов, регистрирующих комбинацию вторичных электронов (SE) и обратно рассеянных электронов (BSE). Комбинация режимов регистрации электронов позволила получить микроскопические изображения в фазовом, ориентационном и топографическом контрастах. Микроскоп укомплектован энергодисперсионным спектрометром ULTIM MAX 100 (“Oxford Instruments Analytical”, Великобритания) с системой микроанализа Aztec, позволяющей выполнять качественный и количественный анализы. Микроанализ образцов проводили при энергии электронов зонда 20 кэВ.
Микроструктуру образцов изучали методом просвечивающей электронной микроскопии высокого разрешения (ПЭМВР) с использованием электронного микроскопа Themis-Z 3.1 (“Thermo Fisher Scientific”, США) c ускоряющим напряжением 200 кВ и предельным разрешением по решетке 0.07 нм. Микроскоп оборудован X-FEG монохроматором и двойным CS/S корректором. Пошаговый локальный элементный энергодисперсионный рентгеновский (EDX) анализ и картирование по всем элементам были получены с помощью детектора Super-X EDS (энергетическое разрешение около 120 эВ). Образцы для исследования готовили путем ультразвукового диспергирования в этаноле с последующим нанесением суспензии на “дырчатую” углеродную пленку, закрепленную на медной сетке.
Каталитические свойства образцов в реакции окислительного дегидрирования этана (ОДЭ) изучали в проточной установке с online хроматографическим анализом компонентов реакционной смеси, который выполняли на хроматографе Цвет-500 (Россия) с использованием детектора по теплопроводности (газ носитель – гелий, поток газа носителя – 3.2 л/ч, температура детектора – 250°С) и 2-х колонок (длина – 2 м, внешний диаметр – 4 мм), заполненных Porоpak-Q и молекулярными ситами СаА. На первой колонке при комнатной температуре анализировали СО2, этан и этилен, на второй – кислород, азот и СО.
Эксперименты проводили в трубчатом реакторе с коаксиально расположенным термопарным карманом с неподвижным слоем катализатора (фракция 0.25–0.50 мм) при атмосферном давлении и температуре 400°C и составе исходной реакционной смеси (об. %) C2H6 : O2 : N2 = 10 : 10 : 80. Время контакта (от 1.5 до 35.0 с) изменяли за счет варьирования как скорости потока реакционной смеси (от 2 до 16 л/ч), так и объема загрузки катализатора (от 4 до 20 см3). Отсутствие гомогенного окисления в указанном температурном интервале подтверждено опытами с пустым реактором.
Время контакта рассчитывали по формуле:
где Vкат – объем катализатора (мл), U – скорость потока исходной реакционной смеси (л/ч).Конверсию этана (X) и селективности (Si) образования продуктов реакции (этилен и оксиды углерода) определяли по [2]:
Выход этилена (В) рассчитывали по уравнению:
Значения конверсии этана, селективности образования этилена и его выхода, полученные в параллельных экспериментах, воспроизводились с высокой точностью: ±1.1, ±0.3 и ±1 отн. % соответственно (см. рис. 4). Поскольку в настоящем исследовании используется реакционная смесь, содержащая 10 об. % этана, 10 об. % кислорода и 80 об. % N2, то коэффициент изменения объема близок к 1, и им можно пренебречь при расчетах конверсии, селективностей образования продуктов и выхода этилена. Баланс по углероду во всех экспериментах составляет 98 ± 2%. В приведенных выше условиях на всех исследуемых катализаторах реакция протекает в кинетической области (модуль Тиле и степень использования поверхности катализаторов равны 0.015 ± 0.01 и 0.9999 соответственно).
Для сравнения активности катализаторов удельную скорость расходования этана (W1) и удельную скорость образования этилена (W2), отнесенные к м2 удельной поверхности, при конверсии этана 10% находили по уравнению:
где U – скорость потока реакционной смеси (л/с); SBET– величина удельной поверхности (м2/г); m – вес катализатора (г).
Также были рассчитаны удельные скорости (W1 и W2), отнесенные к 1 г катализатора и 1 г активной массы катализатора.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
По данным химического анализа составы приготовленных катализаторов MoVSbNbGdOx/SiO2, MoVSbNbOx/SiO2 соответствуют расчетному составу: 50 вес. % Mo1V0.24Sb0.23Nb0.08Gd0.01÷0.02Ox/50 вес. % SiO2 и 50 вес. % Mo1V0.24Sb0.23Nb0.08Ox/50 вес. % SiO2.
На рис. 2 приведены зависимости селективностей образования этилена и оксидов углерода от конверсии этана для катализатора Mo1V0.24Sb0.23Nb0.08Gd0–0.02Ox/SiO2 с разным соотношением Gd/Mo. Из рисунка видно, что во всех случаях с увеличением конверсии этана селективность по этилену постепенно снижается, а селективность по продуктам полного окисления возрастает. Согласно [3, 18, 40] такой вид зависимости позволяет описать процесс окислительного дегидрирования этана на данных катализаторах последовательно-параллельной схемой (схема 1 ):

Схема 1 . Последовательно-параллельная схема описания процесса окислительного дегидрирования этана на исследуемых катализаторах.
Рис. 2.
Зависимости селективностей образования этилена и продуктов полного окисления (CO + CO2) от конверсии этана для катализаторов Mo1V0.24Sb0.23Nb0.08Gd0–0.02Ox/SiO2 с различным соотношением Gd/Mo.

При прямом окислительном дегидрировании этана образуется этилен, а при глубоком окислении этана и доокислении этилена – оксиды углерода. При малой конверсии этана селективность по этилену составляет 96%, при достаточно высокой конверсии этана (>65%) селективности образования продуктов реакции зависят от содержания гадолиния в катализаторе. Следовательно, добавление гадолиния в катализатор MoVSbNbOx/SiO2 не оказывает действия на параллельное направление протекания реакции, но значительно влияет на доокисление этилена. Это также можно видеть из рис. 3, на котором показан выход этилена в зависимости от конверсии этана.
Рис. 3.
Зависимости выхода этилена от конверсии этана для катализаторов MoVSbNbGdOx/SiO2 с различным соотношением Gd/Mo.
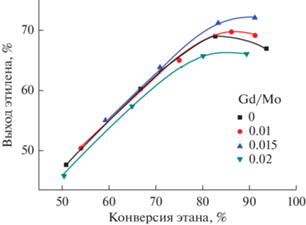
Рис. 4.
Удельные скорости расходования этана (W1) и образования этилена (W2) при конверсии этана 10 %, рассчитанные на 1 м2 (а), на 1 г катализатора (б) и на 1 г активного компонента в присутствии катализаторов MoVSbNbGdOx/SiO2 с разным соотношением Gd/Mo: 0 (1), 0.01 (2), 0.015 (3) и 0.02 (4).

Из рис. 3 следует, что при введении гадолиния в катализатор MoVSbNbOx/SiO2 до соотношения Gd/Mo = 0.015 при высокой конверсии этана (~90%) выход этилена увеличивается с 68 до 72%, но дальнейшее увеличение содержания Gd до Gd/Mo = 0.02 приводит к снижению этого показателя до 66%. Полученные результаты устойчиво воспроизводимы при многократном синтезе и испытании катализаторов в данной реакции.
На рис. 4 для всех исследуемых катализаторов приведены удельные скорости превращения этана (W1) и образования этилена (W2). Можно видеть, что W1 и W2, рассчитанные на 1 м2 поверхности, на 1 г катализатора и на 1 г активной массы катализатора, проходят через небольшой максимум в области соотношений Gd/Mo = 0.01–0.015. Следовательно, наибольшие активность и выход этилена регистрируются на катализаторе MoVSbNbGdOx/SiO2 с соотношением Gd/Mo = 0.015. Как видно из рис. 5, полученный катализатор устойчиво работает в реакционной смеси при проведении длительных испытаний в течение 36 ч, сохраняя высокую активность и селективность по этилену.
Рис. 5.
Зависимость конверсии этана (1) и селективностей образования этилена (2) и оксидов углерода (3) от длительности испытания для катализатора MoVSbNbGdOx/SiO2 с соотношением Gd/Mo = 0.015.

На рис. 6 представлены рентгенограммы свежеприготовленных образцов MoVSbNbGdOx/SiO2 с соотношением Gd/Mo = 0–0.02, прокаленных при 600°С в токе Ar. На этом же рисунке также показаны рентгенограммы фаз M1 и M2, рассчитанные методом Ритвельда из структурных данных, приведенных в [24, 27]. На рентгенограммах исследуемых катализаторов четко виден интенсивный пик в области 2θ = 22.20°, соответствующий фазе M1, и пик в области 2θ = 36.1°, относящийся к фазе M2, а также другие пики, характерные для этих фаз. Пики, принадлежащие к иным кристаллическим фазам, на рентгенограммах не регистрируются. Наряду с пиками присутствующих в образцах фаз M1 и M2 в области 2θ = 18°–25° на рентгенограммах всех образцов наблюдается гало SiO2. Таким образом, по своему фазовому составу полученные катализаторы MoVSbNbGd0–0.02Ox/SiO2 представляет собой смесь кристаллических фаз M1 и М2.
Рис. 6.
Рентгенограммы свежеприготовленных MoVSbNbGdOx/SiO2 катализаторов с соотношением Gd/Mo, равным 0.015 (1), 0.010 (2), 0 (3), 0.02 (4), и расчетные рентгенограммы фаз M1 (5), M2 (6).

Из приведенных в табл. 1 данных видно, что содержание фаз M1 и M2 в активном компоненте катализатора, не содержащего гадолиний, составляет 77 и 23% соответственно. При введении в катализатор Gd до соотношения Gd/Mo = 0.015–0.02 наблюдается некоторое увеличение содержания фазы M1 в активном компоненте до 80–82% при содержании фазы M2 20–18%. Следует отметить, что после испытаний катализаторов в реакционной смеси их фазовый состав не изменяется.
Таблица 1.
Фазовый состав активного компонента катализаторов MoVSbNbGdOx/SiO2 с различным соотношением Gd/Mo
| Катализатор | Фазовый состав | Содержание фаз, вес. % | Параметры решетки, Å | ||
|---|---|---|---|---|---|
| а | b | c | |||
| Mo1V0.24Sb0.23Nb0.08Ox/50 вес. % SiO2 | M1 | 77 | 21.091 | 26.592 | 4.000 |
| M2 | 23 | 12.592 | 7.302 | 4.021 | |
| Mo1V0.24Sb0.23Nb0.08 Gd0.010Ox/50 вес. % SiO2 | M1 | 73 | 21.091 | 26.595 | 4.011 |
| M2 | 27 | 12.620 | 7.296 | 3.987 | |
| Mo1V0.24Sb0.23Nb0.08 Gd0.015Ox/50 вес. % SiO2 | M1 | 80 | 21.095 | 26.597 | 3.999 |
| M2 | 20 | 12.59 | 7.299 | 4.019 | |
| Mo1V0.24Sb0.23Nb0.08 Gd0.02Ox/50 вес. % SiO2 | M1 | 82 | 21.120 | 26.617 | 4.024 |
| M2 | 18 | 12.649 | 7.302 | 4.000 | |
Величина параметров кристаллической решетки a и b фазы M1 в катализаторах MoVSbNbGdOx/SiO2 несколько возрастает с увеличением соотношения Gd/Mo. Так повышение соотношения Gd/Mo в катализаторе от 0 до 0.02 приводит в фазе M1 к росту параметрa a от 21.091 до 21.120 нм и параметра b от 26.592 до 26.617 нм. Это может быть обусловлено внедрением атомов Gd в решетку соединения MoVNbOx. Известно, что радиус иона Gd3+ больше радиусов ионов Mo, V, Nb, что должно вести к увеличению параметров решетки в фазе M1, если в ней дополнительно стабилизируются ионы гадолиния.
Состояние элементов в катализаторах MoVSbNbGdOx/SiO2 с соотношением Gd/Mo = = 0, 0.015, 0.02, исходных и после испытаний в реакционной смеси, определено методом РФЭС. На рис. 7 приведен обзорный РФЭ-спектр свежеприготовленного катализатора MoVSbNbGdOx/SiO2 с соотношением Gd/Mo = 0.015, который характеризуется наиболее высокой активностью в реакции ОДЭ. Видно, что элементный состав поверхности образца соответствует заданному, и никаких дополнительных элементов не обнаружено. Аналогичные обзорные спектры регистрируются и для всех остальных исследованных катализаторов. Для определения состояния элементов и их поверхностных концентраций были проанализированы узкие спектральные регионы отдельных элементов. На рис. 8а–8е показаны спектральные регионы элементов Mo, V, Nb, Sb, Gd, Si свежеприготовленного катализатора MoVSbNbGdOx/SiO2 с соотношением Gd/Mo = 0.015.
Рис. 7.
Обзорный РФЭ-спектр свежеприготовленного катализатора MoVSbNbGdOx/SiO2 с соотношением Gd/Mo = 0.015.

Рис. 8.
РФЭ-спектр регионов индивидуальных элементов свежеприготовленного катализатора MoVSbNbGdOx/SiO2 с соотношением Gd/Mo = 0.015: Mo3d (а), V2p3/2 (б), Nb3d (в), Sb3d + O1s (г), Gd3d5/2 (д), Si2p (е).
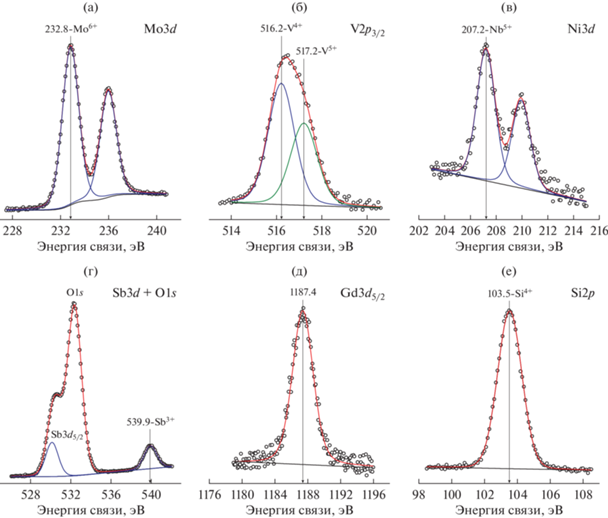
В спектре Mo3d значение энергии связи Mo3d5/2 составляет 232.8 ± 0.1 эВ, что соответствует состоянию Mo6+ [41, 42]. В спектре V2p пик V2p3/2 описывается двумя компонентами с энергиями связи 516.2 ± 0.1 и 517.2 ± 0.1 эВ, которые относятся к состояниям V4+ и V5+ [44, 45]. Проведенные оценки показывают, что соотношение V5+/V4+ на поверхности этого катализатора равно ~0.70. В спектре Nb3d энергия связи пика Nb3d5/2 составляет 207.1 ± 0.1 эВ и характеризует состояние Nb5+ [46, 47]. В связи с тем, что интенсивный пик Sb3d5/2 перекрывается с линией O1s, идентификацию химического состояния сурьмы проводили по пику Sb3d3/2. Значение энергии связи этого пика равно 539.9 ± 0.1 эВ, что соответствует состоянию Sb3+ [48, 49]. В спектре Gd3d энергия связи пика Gd3d5/2 составляет 1187.4 ± 0.1 эВ и относится к состоянию Gd3+ [50]. Энергия связи пика Si2p (103.5 эВ) характерна для SiO2 [37].
Исследование состояния окисления индивидуальных элементов в образце MoVSbNbOx/SiO2, не содержащем добавки гадолиния, и в образце MoVSbNbGdOx/SiO2 с соотношением Gd/Mo = = 0.02 показывает, что в этих катализаторах молибден присутствует в состоянии Mo6+, ванадий – в состояниях V4+ и V5+, ниобий – в состоянии Nb5+, гадолиний – в состоянии Gd3+, сурьма – в состоянии Sb3+ и кремний – в состоянии Si4+. Видно, что состояния элементов в этих катализаторах соответствуют таковым в вышеуказанном промотированном гадолинием образце MoVSbNbGdOx/SiO2 с Gd/Mo = 0.015. Изменяется лишь соотношение V5+/V4+ с изменением в них содержания гадолиния. В исходном катализаторе без добавки Gd поверхностное соотношение V5+/V4+ составляет 0.64, при введении гадолиния до Gd/Mo = 0.015 оно увеличивается до 0.70, а при дальнейшем повышении содержания гадолиния до Gd/Mo = 0.02 уменьшается до 0.57 (табл. 2).
Таблица 2.
Соотношение V5+/V4+ в катализаторах MoVSbNbGdOx/SiO2 с различным содержанием гадолиния, исходных и после работы в реакционной смеси
| Соотношение Gd/Mo |
Соотношение V5+/V4+ |
|
|---|---|---|
| исходный | после реакции | |
| 0 | 0.63 | 0.68 |
| 0.015 | 0.70 | 0.79 |
| 0.02 | 0.57 | 0.65 |
После проведения каталитической реакции состояния всех элементов в катализаторах не изменяются, а соотношение V5+/V4+ возрастает по сравнению со свежеприготовленными образцами (табл. 2). Поверхностный состав активного компонента в катализаторах MoVSbNbGdOx/SiO2 (Gd/Mo = 0.015) и MoVSbNb/SiO2 по данным РФЭС соответствует Mo1V0.18Sb0.45Nb0.06Gd0.01Ox и Mo1V0.17Sb0.49Nb0.07Ox. Видно, что на поверхности присутствуют все элементы катализатора, при этом она значительно обогащена сурьмой.
В табл. 3 приведены величины удельной поверхности и текстурные параметры катализаторов с различным соотношением гадолиния. Можно видеть, что введение гадолиния приводит к некоторому увеличению удельной поверхности, пористая структура образцов практически не изменяется. При этом во всех катализаторах регистрируются мезопоры, объем которых составляет Vs = 0.11–0.12 см3/г, и макропоры объемом Vmac = 0.10–0.16 см3/г.
Таблица 3.
Параметры пористой структуры катализаторов MoVSbNbGdOx/SiO2 с различным соотношением Ga/Mo
| MoVSbNbGdOx/SiO2, Gd/Mo |
SBET, м2/г | VΣ, см3/г | Vs, см3/г | Dmes, нм | Vmac, см3/г | Dmac, мкм |
|---|---|---|---|---|---|---|
| 0 | 13.6 | 0.20 | 0.114 | 18.1 | 0.09 | 0.5–1.5 |
| 0.01 | 14.9 | 0.25 | 0.117 | 20.3 | 0.13 | 0.5–1.5 |
| 0.015 | 15.7 | 0.28 | 0.120 | 21.8 | 0.16 | 0.5–1.5 |
| 0.020 | 20.2 | 0.26 | 0.118 | 21.5 | 0.14 | 0.5–1.5 |
Распределение объемов мезопор по размерам для катализаторов MoVSbNbGdOx/SiO2 с различным соотношением Gd/Mo показано на рис. 9. Видно, что во всех катализаторах регистрируются мезопоры с диаметром 10–50 нм с максимумом на кривой распределения в области 20–40 нм. Размер макропор во всех катализаторах одинаковый и составляет 0.5–1.5 мкм.
Рис. 9.
Распределение объемов мезопор по размерам для катализаторов MoVSbNbGdOx/SiO2 с различным соотношением Gd/Mo: 0 (1), 0.015 (2), 0.020 (3).

На рис. 10а и 10б приведен электронно-микроскопический снимок катализатора MoVSbNbGdOx/SiO2 (Gd/Mo = 0.015), прокаленного при 600°C, и распределение его частиц по размерам. Частицы катализатора имеют сферическую форму с преимущественным размером 1–5 мкм, хотя имеется и небольшая доля частиц большего размера вплоть до 9 мкм. Данный тип морфологии частиц и их размер в нашем случае обусловлены методом приготовления, а именно использованием распылительной сушки катализаторной суспензии на сушилке с диаметром сопла 1 мм. Следует отметить, что получаемые частицы достаточно прочные, они не разрушаются при сушке и сохраняют свою первоначальную морфологию даже после термообработки при 600°C в токе Ar. Упаковка частиц катализатора указанных размера и морфологии обусловливает формирование макропористой части пористого пространства с объемом пор Vmac = 0.1–0.16 см3 /г и размером 0.5–1.5 мкм (табл. 3).
Рис. 10.
Микрофотография СЭМ (a) и распределение размеров частиц по размерам (б) для катализатора MoVSbNbGdOx/SiO2 (Gd/Mo = 0.015), прокаленного при 600°C.

При большем увеличении заметны два типа частиц (рис. 11а–11г). Это сферические пористые частицы аморфного SiO2, наполненные частицами катализатора, которые наблюдаются как контрастные маленькие белые черточки на их поверхности. Кроме того, на поверхности большинства сферических частиц регистрируются ограненные частицы тарельчатой формы с размером 1–1.5 мкм (рис. 11а). Как можно видеть из рис. 11б, на котором представлен электронно-микроскопический снимок сферических частиц, они состоят из смеси аморфного SiO2 с частицами катализатора игольчатой морфологии размером до 500 нм. Согласно литературным данным, для катализаторов MoVTeNbOx такая морфология характерна для частиц фазы М1 [13, 18, 29, 51]. По аналогии с Te-содержащими катализаторами можно полагать, что и в нашем случае наблюдаемые частицы игольчатой морфологии соответствуют фазе M1. Из снимков СЭМ также следует, что в объеме сферических частиц не отмечается сильной агломерации и спекания частиц фазы М1, чему, видимо, препятствует присутствие SiO2.
Рис. 11.
Снимки СЭМ катализатора MoVSbNbGdOx/SiO2 (Gd/Mo = 0.015), прокаленного при 600°C, при различном увеличении.

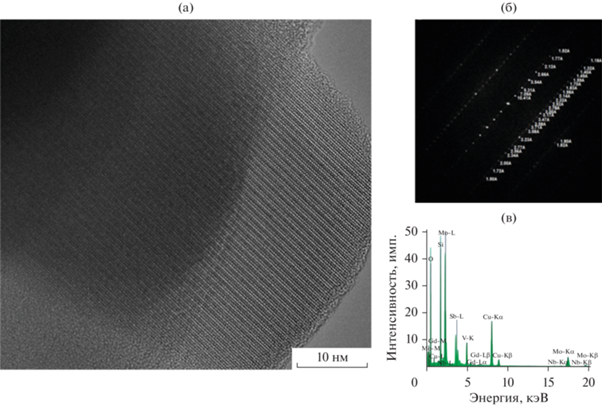
Для уточнения кристаллической структуры частиц игольчатой морфологии (рис. 11б), они были дополнительно исследованы методом ПЭМВР. На рис. 12 представлен электронно-микроскопический снимок высокого разрешения, соответствующая ему Фурье-дифрактограмма и EDX-спектр, полученные с частицы игольчатой морфологии. Анализ показывает, что межплоскостные расстояния в структуре этой частицы соответствуют фазе М1 с орторомбической структурой с наиболее развитой плоскостью [001]. По данным EDX-анализа соотношение компонентов в поверхностном слое катализатора соответствует Mo1V0.32Sb0.35Nb0.045Gd0.001.
Рис. 12.
Снимок ПЭМВР (а), соответствующая ему Фурье-дифрактограмма (б) и EDX-спектр (в) фазы M1 игольчатой морфологии в катализаторе MoVSbNbGdOx/SiO2 (Gd/Mo = 0.015), прокаленном при 600°C.
Следует отметить, что частицы тарельчатой морфологии явно не видны внутри частиц SiO2, они в основном стабилизированы на внешней поверхности. Поскольку по результатам рентгенофазового анализа в катализаторе не зарегистрированы другие кристаллические фазы, кроме М1 и М2, можно полагать, что вышеуказанные частицы также представляют собой данные фазы. Ранее в работах [51, 52] авторами наблюдалось формирование фаз M1 и M2 тарельчатой морфологии в Te-содержащих катализаторах MoVTeNbOx. На рис. 13 приведен электронно-микроскопический снимок ПЭМВР, полученный с края одной из частиц тарельчатой морфологии, и Фурье-дифрактограмма. Проведенный анализ показывает, что кристаллическая структура этой частицы соответствует фазе М1 с наибольшим вкладом плоскости [010], что согласуется с данными [51]. Следовательно, можно заключить, что в данной системе MoVSbNbGdOx/SiO2 формируются частицы фазы М1 игольчатой и тарельчатой морфологии.
Рис. 13.
Снимок ПЭМВР (а) и соответствующая ему Фурье-дифрактограмма (б) частицы тарельчатой морфологии в катализаторе MoVSbNbGdOx/SiO2 (Gd/Mo = 0.015), прокаленном при 600°C.

Для того чтобы понять, как происходит формирование частиц тарельчатой морфологии, мы просмотрели достаточно большое количество микрофотографий СЭМ, исследуемого катализатора. В результате удалось найти снимок, приведенный на рис. 11г, на котором видно, что в объеме частицы тарельчатого типа толщиной 0.05 мкм наблюдаются неупорядоченные более мелкие частицы с размером 0.05–0.1 мкм, которые, по-видимому, при термообработке упорядочиваются с образованием кристаллических фаз М1 и M2. Из сравнения снимков, представленных на рис. 10в и 10г, можно предположить, что фазы М1 и/или М2 тарельчатой морфологии, стабилизированные на внешней поверхности частиц SiO2, формируются во время спекания более мелких частиц, выделившихся из объема частиц SiO2. Интересно отметить, что частицы тарельчатой морфологии, в основном, стабилизируются на поверхности частиц SiO2, и не существуют в виде многочисленных отдельных агломератов. Вероятно, это может свидетельствовать об их структурной связи с частицами, стабилизированными в объеме. На поверхности сформированных частиц тарельчатой морфологии также можно видеть дефекты в виде террас и ступеней. Однако вопрос о формировании частиц тарельчатой морфологии в исследуемых катализаторах требует дальнейшего изучения.
Исследование морфологии частиц катализатора, не содержащего добавку гадолиния, методом СЭМ свидетельствует, что концентрация в нем частиц тарельчатой морфологии, расположенных на поверхности сферических частиц SiO2, значительно возрастает по сравнению с образцом, в состав которого входит гадолиний (рис. 11а, 13 и 14). На поверхности между частицами тарельчатой морфологии наблюдаются частицы игольчатой морфологии.

ЗАКЛЮЧЕНИЕ
Таким образом, на основании проведенных исследований можно заключить, что введение добавки гадолиния в катализатор MoVSbNbOx/SiO2 в оптимальных количествах (Gd/Mo = 0.01–0.015) приводит к возрастанию его активности и селективности образования этилена в ОДЭ, а также повышению выхода этилена до 72%, что сопоставимо с показателями лучших Te-содержащих катализаторов, известных из литературы. Однако дальнейшее увеличение содержания гадолиния до соотношения Gd/Mo = 0.02 снижает его активность и уменьшает выход этилена до 66%. Это обусловлено более интенсивным протеканием реакции доокисления этилена в продукты полного окисления. Для объяснения полученных результатов нами проведено сопоставление физико-химических и каталитических свойств приготовленных катализаторов с разным содержанием гадолиния. Как следует из табл. 1, при изменении соотношения Gd/Mo от 0 до 0.02 содержание фазы M1 в активном компоненте возрастает от 77 до 82%, а содержание фазы М2 снижается от 23% до 18%. Однако из простого сопоставления данных РФА с каталитическими свойствами прямой корреляции между повышением активности катализатора и увеличением содержания фазы М1 не обнаружено. По-видимому, морфология частиц фазы M1 также оказывает существенное влияние на каталитические свойства. Как было показано в работах [51, 52], в многокомпонентных Te-содержащих катализаторах MoVSbNbOx наблюдались частицы фазы М1 игольчатой и тарельчатой морфологии, активность которых в ОДЭ была разная. Это связано с тем, что в частицах фазы М1 игольчатой морфологи наиболее развитой плоскостью, выходящей на поверхность, является плоскость [001], что обусловливает их повышенную активность в реакции ОДЭ по сравнению с частицами фазы М1 тарельчатой морфологии, в которых самой развитой плоскостью является менее активная плоскость [010].
Видимо, возрастание активности в ОДЭ катализаторов MoVSbNbGdOx/SiO2 с увеличением соотношения Gd/Mo от 0 до 0.015 может быть связано с повышением количества фазы М1 игольчатой морфологии. По данным сканирующей электронной микроскопии прокаленный катализатор MoVSbNbGdOx/SiO2 (Gd/Mo = 0.015) представляют собой пористые сферические частицы SiO2 размером 0.5–5 мкм, внутри которых стабилизируются частицы фазы М1 игольчатой морфологии. Внутри объемных частиц SiO2 формируется мезопористая структура с диаметром пор 10–50 нм. Благодаря достаточно высокой пористости частицы фазы М1 доступны для реакционной смеси, что обеспечивает протекание реакции ОДЭ в кинетический области. Как видно из рис. 13а, в катализаторе, не содержащем добавки гадолиния, наряду с частицами фазы М1 игольчатой морфологии регистрируется достаточно большое количество частиц фаз М1 и М2 тарельчатой морфологии, стабилизированных на внешней поверхности частиц SiO2. Следовательно, при добавлении в катализатор гадолиния концентрация частиц фаз М1 и М2 тарельчатой морфологии заметно снижается, и в основном регистрируются частицы фазы M1 игольчатой морфологии. Фаза M1 игольчатой морфологии обладает орторомбической структурой, образованной из пентагональных колец, связанных вместе октаэдрами MO6 (M = Mo, V), формирующих гексагональные и гептагональные каналы в плоскости [001], в которых стабилизируются атомы Sb или Te. Как следует из результатов рентгенофазового анализа, параметры решетки фазы М1 несколько увеличиваются при добавлении гадолиния в катализатор. По всей вероятности, гадолиний также стабилизируется в гексагональных каналах фазы M1 орторомбической структуры.
По данным РФЭС введение гадолиния в катализатор MoVSbNbOx/SiO2 до соотношения Gd/Mo = 0.015 приводит к увеличению поверхностного соотношения V5+/V4+, при дальнейшем повышении содержания гадолиния в катализаторе соотношение V5+/V4+ снижается. После работы катализатора в реакционной смеси поверхностное соотношение V5+/V4+ возрастает во всех катализаторах (табл. 2). Согласно литературным данным [13, 18, 26, 53] рост поверхностной концентрации ионов V5+ как в Те-содержащих катализаторах MoVТеNbOx, так и в Sb-содержащих MoVSbNbСeOx/SiO2 способствует увеличению активности и селективности катализатора в образовании этилена. В нашем случае также наблюдается корреляция между повышением активности и селективности катализатора и ростом поверхностной концентрации ионов V5+. По данным [26, 53, 54] возрастание поверхностной концентрации V5+ приводит к усилению акцепторных свойств активных центров, способных активировать молекулу этана. Следовательно, можно полагать, что промотирующая роль гадолиния в катализаторе при оптимальном его содержании заключается в увеличении количества активной фазы М1 игольчатой морфологии и в повышении поверхностной концентрации ионов V5+. Bведение в структуру катализатора Gd в количестве, превышающем оптимальное, приводит к уменьшению поверхностной концентрации V5+, что способствует более интенсивному протеканию процессов доокисления этилена и снижению активности катализатора. Таким образом, добавление гадолиния в систему MoVSbNbСeOx/SiO2, по-видимому, изменяет тарельчатую морфологию частиц фазы M1 на игольчатую и увеличивает соотношение V5+/V4+, что приводит к повышению активности катализатора.
Список литературы
Chu B., Truter L., Nijhuis T., Cheng Y. // Appl. Catal. A: Gen. 2015. V. 498. P. 99.
Бондарева В.М., Кардаш Т.Ю., Ищенко Е.В., Соболев В.И. // Катализ в промышленности. 2014. № 6. С. 38. (Bondareva V.M., Kardash T.Y., Ishchenko E.V., Sobolev V.I. // Catalysis in Industry. 2015. V. 7. № 2. P. 104.)
Botella P., Garcia-Gonzalez E., Dejoz A., Lopez Nieto J.M., Vazquez M.I., Ganzalez-Calbet J. // J. Catal. 2004. V. 225. P. 428.
Botella P., Dejoz A., Lopez Nieto J.M., Concepcion P., Vazquez M.I. // Appl. Catal. A: Gen. 2006. V. 298. P. 16.
Botella P., Dejoz A., Abello M.C., Vazquez M.I., Arria L., Lopez Nieto J.M. // Catal. Today. 2009. V. 142. P. 272.
Cavani F., Trifirò F. // Catal. Today. 1995. V. 24. P. 307.
Valente J.S., Armendariz-Herrera H., Quintana-Solorzano R., Angel P., Nava N., Masso A., Lopez Nieto J.M. // ACS Catal. 2014. V. 4. P. 1292.
Valente J.S., Quintana–Solorzano R., Armendariz–Herrera H., Barragan–Rodríguez G., Lopez Nieto J.M. // Ind. Eng. Chem. Res. 2014. V. 53. P. 1775.
Nguyen T.T., Burel L., Nguyen D.L., Pham-Huu C., Millet J.M. // Appl. Catal. A: Gen. 2012. V. 433. P. 41.
Nguyen T.T., Aouine M., Millet J.M.M. // Catal. Commun. 2012. V. 21. P. 22.
Nguyen T.T., Deniau B., Baca M., Millet J.M.M. // Top. Catal. 2016. V. 59. P. 1496.
Deniau B., Nguyen T.T., Delichere P., Safonova O., Millet J.M.M. // Top. Catal. 2013. V. 56. P. 1952.
Ishchenko E.V., Kardash T.Yu., Gulyaev R.V., Ishchenko A.V., Sobolev V.I., Bondareva V.M. // Appl. Catal. A: Gen. 2016. V. 514. P. 1.
Ishchenko E.V., Gulyaev R.V., Kardash T.Y., Ishchenko A.V., Gerasimov E.Yu., Sobolev V.I., Bondareva V.M. // Appl. Catal. A: Gen. 2017. V. 534. P. 58.
Xie Q., Chen L., Wenig W., Wan H. // J. Mol. Catal. A: Chem. 2005. V. 240. P. 191.
Lopez Nieto J.M., Botella P., Vzquez M.I., Dejoz A. U.S. Patent 7,319,179 B2, 2008; assigned to CSIC-UPV.
Lopez Nieto J.M., Botella P, Vazquez M.I., Dejoz A. WO 03/064035 A1, 2003; assigned to CSIC-UPV.
Najari S., Saeidi S., Concepcion P., Dionysiou D.D., Bhargava S.K, Lee A.F., Wilson K. // Chem. Soc. Rev. 2021. V. 50. P. 4564.
McCain J.H. US Patent 5,524,236, 1985.
Lopez Nieto J.M., Botella P., Concepcion P., Dejoz A., Vazquez M.I. // Catal. Today. 2004. V. 91–92. P. 241.
Nieto J.M.L., Botella P., Vazquez M.I., Dejoz A. // Chem. Commun. 2002. P. 1906.
Zenkovets G.A., Shutilov A.A., Bondareva B.M., Sobolev V.I., Marchuk A.S., Tsybulya S.V., Prosvirin I.P., Ishchenko A.V., Gavrilov V.Yu. // ChemCatChem. 2020. V. 12. P. 4149.
Зенковец Г.А., Шутилов А.А., Бондарева В.М., Довлитова Л.С., Соболев В.И., Марчук А.С., Цыбуля С.В., Просвирин И.П. // Кинетика и катализ. 2021. Т. 62. С. 263. (Zenkovets G.A., Shutilov A.A., Bondareva V.M., Dovlitova L.S., Sobolev V.I., Marchuk A.S., Tsybulya S.V., Prosvirin I.P. // Kinet. Catal. 2021. V. 62. P. 315.)
DeSanto P., Butterey D., Grasseli R., Lugmair C., Volpe A., Toby B., Vogt T. // Krystallogr. Cryst. Mater. 2004. V. 219. P. 152.
Tu X., Niwa M., Arano A., Kimata Y., Okazaki E., Nomura S. // Appl. Catal. A: Gen. 2018. V. 549. P. 152.
Guliants V.V., Bhandari R., Brongersma H.H., Knoester A., Gaffney A.M., Han S. // J. Phys. Chem. B. 2005. V. 109. P. 10234.
Solsona B., Vazquez M.I., Ivars F., Dejoz A., Concepcion P., Lopez Nieto J.M. // J. Catal. 2007. V. 252. P. 271.
Murayama H., Vitry D., Ueda W., Fuchs G., Anne M., Dubois J.L. // Appl. Catal. A: Gen. 2007. V. 318. P. 137.
Woo J., Borisevich A., Koch C., Guliants V.V., Vogt T., Buttrey D.J. // ChemCatChem. 2015. V. 7. P. 3731.
Pyrz W.D., Blom D.A., Shiju N.R., Guliants V.V., Vogt T., Burtrey D.J. // J. Phys. Chem. C. 2008. V. 112. P. 10 043.
Ivars F., Solsona B., Hernandez S., Lopez-Nieto J.M. // Catal. Today. 2010. V.149. P. 260.
Svintsitskiy D.A., Kardash T.Yu., Lazareva E.V., Sara ev A.A., Derevyannikova E.A., Vorokhta M., Šmíd B., Bondareva V.M. // Appl.Catal. A: Gen. 2019. V. 579. P. 141.
Andrushkevich N.V., Popova G.Y., Chesalov Y.A., Ischenko E.V., Kramov M.I., Kaichev V.V. // Appl. Catal. A: Gen. 2015. V. 506. P. 109.
Price W.J. Analytical Atomic absorption Spectrometry. London–N.Y.–Rheine: Heyden & Son Ltd., 1972.
Topas 4.3. Bruker AXS. Germany. 2015.
Inorganic Crystal Structure Database, ICSD. In Release 2008. Fashinformationszentrum Karsruhe D #8211; 1754 Eggenstein #8211; Leopoldshafen, Germany, 2008.
Moudler J., Stickle W., Sobol P., Bomben K. Handbook of X-ray Photoelectron Spectroscopy, Perkin-Elmer Corp.: Eden. Prairie, MN, 1992.
Scofield J.H. // J. Electron Spectrosc. Relat. Phenom. 1976. V. 8. P. 129.
http://xpspeak.software.informer.com/4.1/
Gartner C.A., Veen A.C., Lercher J.A. // ChemCatChem. 2013. V. 5. P. 3196.
Sanders A.F.H., Jong A.M., Beer V.H.J., Veen A.C., Niemantsverdriet J.W. // Appl. Surf. Sci. 1999. V. 144–145. P. 380.
Saih Y., Segawa K. // Appl. Catal. A: Gen. 2009. V. 353. P. 258.
Demeter M., Neumann M., Reichelt W. // Surf. Sci. 2000.V. 454–456. P. 41.
Silversmit G, Depla D., Poelman H., Marin G.B., Gryse R. // J. Electron Spectrosc. Relat. Phenom. 2004. V. 135. P. 167.
Sawatzky G.A., Post D. // Phys. Rev. B. 1979. V. 20. P. 1546.
Farahani M.D., Dasireddy V.D.B.C., Friedrich H.B. // ChemCatChem. 2018. V. 10. P. 2059.
Xue J., Wang R, Zhang Z., Qiu S. // Dalton Trans. 2016. V. 45. P. 16519.
He G.-H., Liang C.-J., Ou Y.-D., Liu D.-N., Fang Y.-P., Xu Y.-H. // Mater. Res. Bull. 2013. V. 48. P. 2244.
Li L., Zhang Y.X., Fang X.S., Zhai T.Y., Liao M.Y., Wang H.Q., Li G.H., Koide Y, Bando Y., Golberg D. // Nanotechnology. 2011. V. 22. № 165704.
Zhang H, Malik V., Mallapragada S., Akinc M. // J. Magn. Magn. Mater. 2017. V. 423. P. 386.
Melzer D., Xu P., Harimann D., Zhu Y., Browning N.D., Sanchez-Sanchez M., Lercher J.A. // Angew. Chem. Int. Ed. 2016. V. 55. P. 8873.
Melzer D., Mestl G., Wanninger K., Zhu Y., Browning D., Sanchez-Sanchez M., Lerche J.A. // Nature Commun. 2019. V. 10. P. 4012.
Xin C., Wang F., Xu Q. // Appl. Catal. A: Gen. 2021.V. 610. P. 117946.
Yun Y.S., Lee M., Sung J., Yun D., Kim T.Y., Park H., Lee K.P., Song C.K., Kim Y., Lee J., Seo Y.-J., Song I.K., Yi J. // Appl. Catal. B: Env.2018. V. 237. P. 554.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ