

Кинетика и катализ, 2022, T. 63, № 6, стр. 789-797
Двухкомпонентная Ni–Mg–O/V–Mg–O каталитическая система. II. Особенности протекания процесса дегидрирования этана
Г. Б. Веселов a, *, Е. В. Ильина a, А. А. Ведягин a
a ФГБУН Институт катализа им. Г.К. Борескова СО РАН
630090 Новосибирск, просп. Акад. Лаврентьева, 5, Россия
* E-mail: g.veselov@catalysis.ru
Поступила в редакцию 27.05.2022
После доработки 28.07.2022
Принята к публикации 28.07.2022
- EDN: KSZYSM
- DOI: 10.31857/S045388112206017X
Аннотация
Детально изучены особенности протекания процесса дегидрирования этана на каталитической системе Ni–Mg–O/V–Mg–O, полученной золь-гель методом. Одновременное использование оксидной компоненты V–Mg–O, катализирующей целевую реакцию дегидрирования, и оксидной компоненты Ni–Mg–O, способной поглощать выделяемый водород, приводит к существенному повышению выхода этилена. Активность Ni–Mg–O-системы также исследована в предполагаемых побочных реакциях – гидрогенолиза этана и разложения этилена с отложением углерода. Максимальные значения конверсии этана в реакции гидрогенолиза наблюдаются в температурном интервале 440–560°C, а при температурах выше 625°C образец практически неактивен. С другой стороны, выход углерода при разложении этилена наибольший как раз при повышенных температурах (670°C). Методом ТПВ-H2 показано, что никель восстанавливается из твердого раствора NixMg1 – xO в системе Ni–Mg–O преимущественно в результате реакции с водородом, выделяющимся при дегидрировании этана на V–Mg–O-катализаторе. Полученные данные подтверждают предположение о том, что метан образуется при взаимодействии водорода с углеродными отложениями на металлическом никеле.
ВВЕДЕНИЕ
В последние годы особое внимание привлекают процессы, в которых применяются химические циклы. Данный технологический подход основан на использовании соединений-интермедиатов, способных запасать кислород и другие атомы [1]. Можно привести несколько примеров практического применения таких соединений, когда технология химических циклов позволяет существенно улучшить показатели целевого процесса. Прежде всего, следует упомянуть риформинг метана (I), (II) и жидких топлив (III), (IV). Решеточный кислород оксида переходного металла в прямом процессе расходуется на риформинг субстрата, а на стадии регенерации восполняется за счет кислорода воздуха.
(I)
${\text{M}}{{{\text{O}}}_{x}} + {\text{C}}{{{\text{H}}}_{4}} \to {\text{M}}{{{\text{O}}}_{x}}_{{\,--\,\,1}} + 2{{{\text{H}}}_{2}} + {\text{CO}},$(II)
${\text{M}}{{{\text{O}}}_{x}}_{{\,--\,1}} + 0.5{{{\text{O}}}_{2}}{\text{/}}{{{\text{H}}}_{{\text{2}}}}{\text{O/C}}{{{\text{O}}}_{2}} \to {\text{M}}{{{\text{O}}}_{x}} + {{{\text{H}}}_{2}}{\text{/CO}},$(III)
$\begin{gathered} {\text{M}}{{{\text{O}}}_{x}} + {{{\text{C}}}_{n}}{{{\text{H}}}_{m}}{{{\text{O}}}_{k}} + {{{\text{H}}}_{{\text{2}}}}{\text{O }} \to \\ \to {\text{M}}{{{\text{O}}}_{{x\,}}}_{{--\,\,1}} + {{{\text{H}}}_{2}} + {\text{CO }} + {\text{C}}{{{\text{O}}}_{2}}, \\ \end{gathered} $(IV)
${\text{M}}{{{\text{O}}}_{x}}_{{\,--\,1}} + 0.5{{{\text{O}}}_{2}} \to {\text{M}}{{{\text{O}}}_{x}}.$Другим примером является улавливание CO2 в химических циклах. Такой подход делает возможным получение чистого водорода при использовании в качестве его источника различных углеводородов [2, 3]. Для решения этой задачи особенно привлекательны системы на основе оксида магния, который способен улавливать CO2 в виде карбонатов (V). Обратимость процесса образования карбонатов магния позволяет сделать данный химический цикл замкнутым [3–5].
Следует также упомянуть другие преимущества MgO. Указанный оксид представляет собой тугоплавкий материал. Он может быть получен в виде дисперсного порошка с развитой удельной поверхностью, что дает возможность рассматривать его в качестве перспективного инертного разбавителя для оксидов переходных металлов [6]. Эксплуатация массивных оксидов без разбавителя в химических циклах при повышенных температурах приводит к их быстрому спеканию и существенному снижению эффективности процесса.
Отдельным классом процессов, в которых селективное удаление водорода из реакционного объема смещает химическое равновесие и увеличивает выход целевого продукта, является дегидрирование алканов. Среди промышленных способов получения практически важных мономеров (этилен, пропилен и др.) наиболее распространены термический крекинг и каталитическое неокислительное дегидрирование. Термодинамика реакции дегидрирования неблагоприятна – для достижения значимых значений конверсии алканов требуются достаточно высокие температуры [7]. В ряде работ было показано, что селективное удаление водорода, например, с помощью водородпроницаемых мембран, позволяет повысить выход непредельных углеводородов на порядок [8–10]. Процесс дегидрирования алканов также может быть реализован на основе технологии химических циклов. Использование интерметаллидов, таких как Zr2Ni и Nd2Co7, дает возможность связывать водород за счет образования стабильных гидридов (VI) [11, 12]. Твердофазные окислители, также называемые переносчиками кислорода, связывают водород в воду (VII) [13–16]. Например, Ванг (Wang C.) и соавт. изучали катализаторы типа Ni/цеолит, в которых часть никеля формирует центры Ni2+, активные в дегидрировании, а другая часть образует наночастицы NiO, ответственные за поглощение водорода [17]. Восстановление исходного состояния оксида происходит по реакции (IV).
(VI)
${{{\text{M}}}_{{\text{1}}}}{{{\text{M}}}_{2}} + x{{{\text{H}}}_{2}} \rightleftarrows {{{\text{M}}}_{{\text{1}}}}{{{\text{M}}}_{{\text{2}}}}{{{\text{H}}}_{{2x}}},$(VII)
${\text{M}}{{{\text{O}}}_{x}} + {{{\text{C}}}_{n}}{{{\text{H}}}_{{2n\,\, + \,\,2}}} \to {\text{M}}{{{\text{O}}}_{x}}_{{\,--\,\,1}} + {{{\text{C}}}_{n}}{{{\text{H}}}_{{2n}}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}}.$В наших предыдущих работах были изучены оксидные системы Cu–Mg–O, Ni–Mg–O, Fe–Mg–O и Co–Mg–O, приготовленные с использованием золь-гель технологии. Методом ТПВ-H2 была продемонстрирована воспроизводимость окислительно-восстановительного поведения таких систем [18–22]. Показано, что каждый оксид способен воспроизводимо поглощать водород в определенном температурном интервале. В первой части представленной работы была рассмотрена эффективность смешанной оксидной системы Ni–Mg–O/V–Mg–O в процессе окислительного дегидрирования этана [23]. В настоящей статье более подробно изучены особенности протекания процесса дегидрирования этана в присутствии данной системы и исследовано изменение ее каталитической активности во времени. Испытана активность системы Ni–Mg–O в возможных побочных реакциях, способных приводить к снижению селективности, таких как гидрогенолиз этана и разложение этилена с образованием углерода. Методом ТПВ-H2 охарактеризовано состояние образцов после проведения реакции дегидрирования.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приготовление образцов
Оксид магния синтезировали с помощью золь-гель метода. Магний (1 г) растворяли в 43 мл метанола. Для полного растворения смесь нагревали до 50°C в течение 1 ч при постоянном перемешивании. После окончания реакции образования метоксида магния в смесь добавляли толуол (гель-стабилизатор) в объемном соотношении 1 : 1. Далее, проводили медленный гидролиз метоксида магния добавлением дистиллированной воды по каплям. Полученный гель перемешивали в течение 1 ч, после чего сушили при комнатной температуре в течение 2 ч. Образовавшийся ксерогель помещали в муфельную печь, нагревали до 500°C со скоростью 1°C/мин и выдерживали 1 ч. Такой режим термической обработки обеспечивал разложение гидроксида магния и формирование мезопористого оксида MgO.
Оксидную систему Ni–Mg–O синтезировали по схожей методике. В этом случае гидролиз метоксида магния был осуществлен не дистиллированной водой, а предварительно приготовленным водным раствором нитрата никеля. Использование неорганического предшественника позволило удешевить и упростить процедуру синтеза без серьезной потери в текстурных характеристиках и равномерности распределения оксида никеля [24]. Сушку и прокаливание образцов проводили в соответствии с процедурами, описанными выше. После разложения гидроксида магния и нитрата никеля была получена оксидная система Ni–Mg–O с массовым содержанием NiO 15%.
Синтез катализатора V–Mg–O выполняли следующим способом. В свежеприготовленный гель гидроксида магния добавляли раствор предшественника (ацетилацетонат ванадила) в смеси 20 мл метанола, 10 мл толуола и 1.8 мл H2O и оставляли на ночь при непрерывном перемешивании. Далее совместный гель помещали в автоклав и нагревали со скоростью 1.3°C/мин. Удаление растворителя осуществляли при сверхкритических условиях (конечное давление в автоклаве – 80 атм, температура – 265°C). Полученный в результате этих операций ванадий-магниевый аэрогель нагревали в муфельной печи со скоростью 1°C/мин до 500°C и выдерживали при этой температуре в течение 1 ч. Массовое содержание ванадия в образце составило 5%.
Тестирование образцов
Исследование образцов методом температурно-программированного восстановления в атмосфере водорода (ТПВ-H2) проводили следующим образом. 200 мг образца загружали в кварцевый реактор, подавали газовую смесь (10% H2, 90% N2, объемная скорость подачи – 57.8 мл/мин) и производили нагрев до 700°C со скоростью 10°C/мин. Концентрацию водорода (мол. %) определяли с помощью газоанализатора ГАММА-100 (ФГУП “СПО “Аналитприбор”, Россия), снабженного детектором по теплопроводности. После этого систему выдерживали 15 мин при 700°C и охлаждали до комнатной температуры. До испытания в следующем цикле ТПВ-H2 образец регенерировали посредством термической обработки в потоке воздуха (нагрев до 500°C со скоростью 20°C/мин, выдержка 30 мин при 500°C). Циклы восстановления/окисления повторяли требуемое количество раз.
Эксперименты по гидрогенолизу этана проводили в интервале температур 200–700°C согласно следующей методике. 100 мг образца (фракция 0.25–0.5 мм) загружали в кварцевый реактор и подавали смесь водорода и гелия (140 и 350 мл/мин соответственно). Реактор нагревали до 200°C и выдерживали до достижения стационарного состояния. В газовый поток добавляли этан (10 мл/мин) и пропускали смесь через реактор в течение 3 мин, после чего отбирали пробу на хроматографический анализ (газовый хроматограф Кристалл-2000М, ЗАО СКБ “Хроматэк”, Россия), а подачу этана в реактор прекращали. В течение следующих 10 мин образец в реакторе продували только смесью водорода и гелия. Процедуру повторяли 5 раз в каждой температурной точке.
Каталитический пиролиз этилена на образце Ni–Mg–O изучали в проточном реакторе с весами Мак-Бена [25]. Кинетику разложения этилена исследовали в температурном интервале 550–700°C. 100 мг образца Ni–Mg–O помещали в корзинку из вспененного кварца, подвешивали на калиброванную пружину и помещали в кварцевый реактор. Реактор нагревали до заданной температуры со скоростью 10°C/мин в потоке инертного газа (аргон). В нагретый реактор подавали реакционную смесь, содержащую 18 об. % этилена, 59 об. % водорода, остальное – аргон. Скорость реакционного потока составляла 66 л/ч. Изменение массы образца регистрировали с помощью катетометра. Съемку кинетической кривой проводили до того момента, пока масса образца не переставала меняться.
Реакцию дегидрирования этана исследовали при постоянной температуре в зависимости от времени, а также в нескольких циклах дегидрирования/регенерации. В первом случае эксперименты вели при температуре 625°C. Механическую смесь, содержащую 100 мг катализатора V–Mg–O и 100 мг сокатализатора Ni–Mg–O, загружали в реактор. Реактор нагревали до 625°C со скоростью 10°C/мин в потоке гелия. После выхода температуры на заданное значение в реактор подавали реакционную смесь (5% этана в гелии, скорость потока – 100 мл/мин). Состав газовой смеси на выходе из реактора определяли с помощью газового хроматографа Кристалл-2000М (ЗАО СКБ “Хроматэк”, Россия). Пробы реакционной смеси отбирали каждые 6 мин. Время эксперимента составляло 9 ч. В каждой точке были рассчитаны величины конверсии этана и выхода этилена, а также селективности образования этилена и метана. Аналогично были исследованы однокомпонентные системы V–Mg–O и Ni–Mg–O в смеси с чистым оксидом магния.
Образцы Ni–Mg–O/V–Mg–O испытывали в циклах дегидрирования/регенерации при температуре 650°C. Стадию дегидрирования этана проводили в течение 5 ч, после чего подачу этана в реактор прекращали. Реактор охлаждали до комнатной температуры. Для регенерации катализатора в реактор подавали смесь воздуха (10 мл/мин) и азота (5 мл/мин). Реактор нагревали до 650°C и выдерживали при этой температуре в течение 1 ч. Циклы дегидрирования/регенерации повторяли 4 раза.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В первой части представленной работы было показано, что использование каталитической системы Ni–Mg–O/V–Mg–O приводит к существенному повышению выхода этилена в реакции окислительного дегидрирования этана в диапазоне температур 600–650°C по сравнению с отдельно взятыми образцами V–Mg–O и Ni–Mg–O [23]. Преимуществом смешанной оксидной системы является также отсутствие значительного уменьшения селективности образования этилена, хотя для образца Ni–Mg–O она была достаточно низка. С другой стороны, селективность по метану в температурном интервале 600–650°C для смешанной системы ниже таковой для катализатора V–Mg–O. При более высоких температурах (675–700°C) наблюдается рост концентрации метана с превышением значений, полученных для однокомпонентной ванадиевой системы.
Схематическое представление реакций, которые могут протекать в ходе процесса дегидрировании этана на системе Ni–Mg–O/V–Mg–O, показано на схеме 1 . Водород, выделяющийся в ходе реакции дегидрирования этана (I) на катализаторе V–Mg–O, реагирует с оксидной системой Ni–Mg–O (II) с образованием воды. В соответствии с принципом Ле Шателье, удаление одного из продуктов реакции сдвигает равновесие в сторону продуктов, увеличивая тем самым конверсию этана и выход этилена. Кроме того, нельзя исключать и возможность восстановления оксида никеля этаном (III). В результате этих двух реакций образуются частицы металлического никеля, также способные катализировать реакцию дегидрирования этана. Учитывая существенное содержание метана в составе реакционной смеси на выходе из реактора, особый интерес представляет выяснение вклада вероятных побочных реакций в общий процесс дегидрирования этана. Металлический никель, как известно, склонен катализировать образование углеродных отложений, как структурированных, так и аморфных (кокс). Углеродные отложения блокируют поверхность катализатора, что, как следствие, приводит к снижению эффективности никеля в целевой реакции. Упомянутое выше увеличение селективности по метану может происходить в результате взаимодействия углерода с водородом по реакции (IV) [26]. Другой возможный маршрут образования метана – реакция гидрогенолиза этана (V), которая также может протекать на частицах металлического никеля [27]. Рассмотрим реакцию каталитического разложения этилена (VI) как наиболее предпочтительную для отложения углерода и реакцию гидрогенолиза этана на системе Ni–Mg–O более подробно.

Схема 1 . Предполагаемая схема реакций, протекающих в ходе процесса дегидрирования этана на системе Ni–Mg–O/V–Mg–O.
Температурная зависимость конверсии этана в реакции гидрогенолиза (маршрут V) представлена на рис. 1. Никель в исходном образце находится в виде оксида NiO, но так как реакционная смесь содержит избыток водорода, то в ходе эксперимента происходит восстановление никеля до металлического состояния. Реакция гидрогенолиза этана протекает на образующихся металлических частицах. Проведение реакции в избытке водорода также препятствует образованию кокса. Как видно из рис. 1, реакция происходит в интервале температур 360–625°C, а наибольшие значения конверсии этана (83–95%) наблюдаются в интервале 440–560°C. Стоит отметить, что образец малоактивен в реакции гидрогенолиза этана при температурах 625–700°C, которые наиболее интересны для проведения реакции дегидрирования этана.

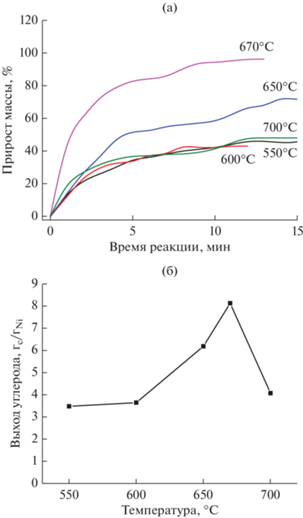
Кинетические кривые отложения углерода при каталитическом разложении этилена на образце Ni–Mg–O приведены на рис. 2а. Видно, что реакция начинается практически сразу после подачи реакционной смеси. Разложение происходит на образующихся при восстановлении NiO частицах металлического никеля. На представленных графиках для всех температур можно выделить две области. В течение первых 2–5 мин наблюдается интенсивное отложение углерода, а в течение последующих 5–15 мин образец постепенно дезактивируется. Таким образом, количество образующихся углеродных отложений в основном определяется первыми минутами реакции. Наиболее интенсивно процесс разложения этилена протекает при температуре 670°C. Для этой же температуры зафиксирован наибольший выход углерода – 8.14 гС/гNi. Как было показано ранее, система Ni–Mg–O характеризуется сильным взаимодействием дисперсных форм оксида никеля с носителем с образованием фазы твердого раствора NixMg1 –xO. В экспериментах ТПВ-H2 пик, соответствующий восстановлению этих частиц, имеет максимум как раз при 670°C [22]. При температурах ниже 670°C восстановление сильносвязанных форм NiO происходит не полностью. При 700°C реакция замедляется, предположительно, из-за диффузионных ограничений. Соответствующая зависимость выхода углерода от температуры реакции показана на рис. 2б. Стоит отметить, что полученные значения выхода углерода превосходят таковые для систем Ni/MgO, приготовленных методами соосаждения [28] и пропитки [29].
Рис. 2.
Кинетические кривые накопления углерода при разложении этилена на образце Ni–Mg–O при различных температурах (а) и зависимость максимального выхода углерода от температуры (б).
Смешанная система Ni–Mg–O/V–Mg–O, а также однокомпонентные системы V–Mg–O и Ni–Mg–O были изучены в процессе дегидрировании этана. Для объяснения вклада никельсодержащей компоненты особый интерес представляет поведение образцов во времени. Для этих экспериментов была выбрана температура 625°C. Соответствующие временные зависимости конверсии этана, селективности образования метана и этилена и выхода этилена представлены на рис. 3.
Рис. 3.
Зависимости конверсии этана (а), выхода этилена (б), селективности по этилену (в) и селективности по метану (г) от времени для образцов V–Mg–O, Ni–Mg–O и совместной системы Ni–Mg–O/V–Mg–O.

Для катализатора V–Mg–O максимальная конверсия 5.5% наблюдается уже через 1 мин после начала подачи этана (рис. 3а), а далее происходит постепенное снижение активности. Селективность по этилену в начальный момент реакции низкая (рис. 3в), но через 20–25 мин составляет порядка 70–80%. Таким образом, наибольшее значение выхода этилена достигается только через 25 мин (рис. 3б). Можно предположить, что в начальный период реакции наиболее активные формы ванадия участвуют в разрыве связи C–C в молекуле этана с последующим образованием метана и окисленных форм COx.
В случае оксидной системы Ni–Mg–O в течение первых 20 мин отмечены сравнительно высокие значения конверсии этана. Однако низкая селективность образования этилена приводит к меньшему его выходу в сравнении с образцом V–Mg–O. В присутствии смешанной системы Ni–Mg–O/V–Mg–O выход этилена больше, что указывает на эффект совместного действия катализатора и сокатализатора. Максимальная конверсия этана (18%) и выход этилена (12%) достигаются только через 30 мин, после чего активность падает. Среднее значение селективности для системы V–Mg–O/MgO за время эксперимента составляет 71%, а для смешанной системы Ni–Mg–O/V–Mg–O – 62%. Снижение селективности по целевому продукту предположительно связано с протеканием процесса коксообразования на Ni–Mg–O.
Рассмотрим происходящие процессы более подробно. За первый час реакции производительность по этилену составила 0.11 и 0.30 г/гкат для образцов V–Mg–O и Ni–Mg–O/V–Mg–O соответственно. Как уже отмечалось, увеличение выхода целевого продукта может происходить за счет смещения локального равновесия вблизи активного центра в сторону продуктов вследствие расходования водорода на восстановление оксида никеля. К другим процессам, повышающим выход этилена, можно отнести восстановление оксида никеля этаном, а также дегидрирование этана на образующемся металлическом никеле. Следует заметить, что первые два процесса расходуют решеточный кислород оксида никеля. Рост производительности составляет 0.19 г/гкат (0.0068 моль/гкат), что почти в пять раз выше кислородной емкости системы Ni–Mg–O (0.0014 моль/г), определенной методом ТПВ-H2. Можно сделать вывод о том, что поглощение водорода оксидной системой Ni–Mg–O, несомненно, влияет на параметры процесса, но основной причиной возрастания выхода этилена все-таки является дегидрирование на частицах Ni0. Увеличение выхода этилена в результате восстановления всего оксида никеля этаном достигается примерно на 25 мин реакции. Стоит также отметить, что чистая система Ni–Mg–O не может быть использована для дегидрирования этана даже при условии предварительного восстановления, так как на каждой стадии регенерации металлический никель будет переходить в окисленное состояние. Производительность оксидной системы Ni–Mg–O в дегидрировании этана за 1 ч реакции составляет всего 0.064 г/гкат. Для активации такого катализатора потребуется проводить процедуру восстановления после каждого этапа регенерации, что приведет к существенному удорожанию процесса в целом. С другой стороны, в случае смешанной каталитической системы Ni–Mg–O/V–Mg–O восстановление оксида никеля происходит в результате взаимодействия с водородом, выделяющимся в ходе реакции дегидрирования этана на ванадийсодержащей компоненте, либо в результате взаимодействия с этаном. Определение вклада процессов окислительного и неокислительного дегидрирования в конверсию этана, а также разделение процессов восстановления оксида никеля этаном и образовавшимся водородом представляются отдельными задачами, решить которые планируется на следующих этапах работы.
В рамках настоящей работы представляется возможным проанализировать вклад обеих систем (Ni–Mg–O и V–Mg–O) в процесс образования метана (рис. 3г). Для катализатора V–Mg–O наблюдается плавное падение селективности по метану во времени. Добавление Ni–Mg–O в качестве сокатализатора приводит к уменьшению количества метана, образующегося в течение первых 95 мин реакции. Можно предположить, что это связано с поглощением водорода оксидом никеля. По мере израсходования решеточного кислорода и восстановления NiO до металлического состояния концентрация метана на выходе из реактора возрастает. Учитывая тот факт, что образец Ni–Mg–O практически неактивен в реакции гидрогенолиза этана при высоких температурах, увеличение селективности по метану может быть обусловлено газификацией углеродных отложений в результате их взаимодействия с водородом по маршруту (IV).
Все образцы после тестирования в реакции дегидрирования этана были исследованы методом ТПВ-H2. Для сравнения также были изучены исходные образцы Ni–Mg–O и V–Mg–O. Методика эксперимента включала стадию восстановления в водороде, стадию реокисления кислородом воздуха и еще одну стадию восстановления. Профили ТПВ-H2 исходных и реокисленных образцов представлены на рис. 4а. Следует отметить, что для системы Ni–Mg–O характерно наличие несколько форм оксида никеля, отличающихся по размеру частиц и силе взаимодействия с матрицей MgO [20, 22]. На стадии реокисления образуются частицы NiO, пик восстановления которых виден на профилях ТПВ-H2 при ~360°C. Для исходного образца V–Mg–O в первом цикле восстановления наблюдаются два пика при 335 и 530°C. Согласно литературным данным, эти пики соответствуют разным окисленным формам ванадия [30, 31]. Если предположить, что весь ванадий находится в степени окисления +5, то величина поглощения водорода составляет 38.6% от максимально возможного в случае полного восстановления. Таким образом, можно заключить, что два вышеуказанных пика соответствуют восстановлению V+5 в V+3. Во втором и третьем цикле на профилях ТПВ-H2 присутствует один пик, обычно относимый к восстановлению изолированных тетраэдров ${\text{VO}}_{4}^{{3 - }}$ или дисперсных частиц Mg3(VO4)2 [32]. Известно, что более крупные частицы ванадатов Mg3(VO4)2 и Mg2V2O7 восстанавливаются при более высоких температурах (700–900°C) [32, 33]. В нашем случае при нагревании образца до 700°C происходит восстановление примерно 70% ванадия, что позволяет сделать предположение о нахождении остальных 30% ванадия в виде грубодисперсных кристаллитов ванадатов магния. Наблюдаемые закономерности указывают на значительное усиление взаимодействия MgO с V2O5 в ходе первого цикла восстановления/окисления.
Рис. 4.
Профили ТПВ-H2 образцов в исходном состоянии и после одного цикла восстановления/реокисления (а). Профили ТПВ-H2 образцов после испытаний в реакции дегидрирования этана (б).

Образцы после испытаний в реакции дегидрирования этана перед тестированием методом ТПВ-H2 были регенерированы в потоке воздуха при 500°C. Кривые ТПВ-H2 приведены на рис. 4б. Профиль образца Ni–Mg–O после реакции напоминает профиль исходного образца (рис. 4а). Пик при 380°C соответствует восстановлению частиц NiO, не взаимодействующих с носителем, а пики при 450 и 670°C можно отнести к крупным и малым частицам твердого раствора NixMg1– xO. Таким образом, характер взаимодействия NiO с MgO не претерпевает существенных изменений в ходе реакции дегидрирования и последующей реактивации. Профиль катализатора V–Mg–O после реакции схож с профилем этого же образца после стадий восстановления/реокисления. Дегидрирование и последующая регенерация влияют на систему V–Mg–O таким же образом, что и восстановление в атмосфере водорода с последующим реокислением. В случае смешанной системы Ni–Mg–O/V–Mg–O от компоненты V–Mg–O можно ожидать подобного поведения. Пик при 575°C в профиле ТПВ-H2 смешанной системы связан с восстановлением окисленных форм ванадия V+5. Следовательно, пик при 345°C должен соответствовать восстановлению частиц оксида никеля, слабо взаимодействующих с MgO. Можно заключить, что определяющий вклад в восстановление никеля и его выход из состава твердого раствора NixMg1– xO вносит водород, выделяющийся в результате дегидрировании этана на катализаторе V–Mg–O.
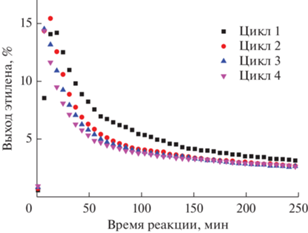
На финальном этапе исследования система Ni–Mg–O/V–Mg–O была испытана в нескольких циклах дегидрирования/регенерации. Из рис. 5 хорошо видно, что в каждом цикле достигаются практически одинаковые значения максимального выхода этилена. При этом характер снижения активности во времени несколько отличается. Начиная со второго цикла, падение активности происходит быстрее. С уменьшением конверсии этана наблюдается увеличение селективности по этилену и уменьшение селективности по метану. Как было показано методом ТПВ-H2, после первого цикла дегидрирования/регенерации никель находится в системе Ni–Mg–O в виде оксида и практически не образует твердый раствор NixMg1– xO. В результате этого скорость восстановления оксида никеля до металлического состояния во втором и последующих циклах дегидрирования выше. Более быстрое формирование частиц металлического никеля ускоряет процесс закоксовывания и вызванную этим дезактивацию каталитической системы.
Рис. 5.
Зависимости выхода этилена от времени в 4 циклах дегидрирования/регенерации для образца Ni–Mg–O/V–Mg–O.
Стоит отметить, что описанные эксперименты по дегидрированию этана проводились в течение достаточно длительного времени до полной дезактивации катализатора. На практике продолжительность циклов может быть оптимизирована для достижения максимальной эффективности. Помимо этого, требуется подбор оптимальных условий регенерации катализатора. Следует также уточнить содержание ванадия и никеля в оксидных системах и массовое соотношение между компонентами Ni–Mg–O и V–Mg–O, что может привести к существенному увеличению стабильности системы. Тем не менее, представленные результаты демонстрируют перспективность применения смешанной каталитической системы Ni–Mg–O/V–Mg–O для окислительного дегидрирования углеводородов.
ЗАКЛЮЧЕНИЕ
В настоящей работе подробно изучен процесс дегидрирования этана в присутствии смешанной системы Ni–Mg–O/V–Mg–O. Оксиды V–Mg–O и Ni–Mg–O были синтезированы с использованием золь-гель методики. Показано, что использование такой смешанной системы приводит к существенному повышению выхода этилена и производительности процесса. В ходе реакции выделяющийся водород расходуется на восстановление оксида никеля в системе Ni–Mg–O, что способствует увеличению конверсии этана. Существенный вклад в повышение выхода этилена вносит реакция неокислительного дегидрирования этана на частицах металлического никеля, образовавшихся в результате восстановления NiO. В ходе длительных экспериментов также наблюдается рост селективности по метану.
С целью исследования возможных маршрутов появления метана изучена активность образца Ni–Mg–O в реакциях гидрогенолиза этана и разложения этилена. Показано, что данный образец практически неактивен в гидрогенолизе этана при температурах 625–700°C. С другой стороны, он проявляет существенную активность в реакции разложения этилена с отложением углерода. Таким образом, наиболее вероятным маршрутом образования метана является газификация углеродных отложений в результате взаимодействия с водородом.
Исследование методом ТПВ-H2 образцов Ni–Mg–O/V–Mg–O после тестирования в реакции дегидрирования этана показало, что на стадии дегидрирования происходит восстановление никеля и его выход из состава твердого раствора NixMg1– xO, а в регенерированном образце присутствуют только частицы NiO. Поведение однокомпонентной системы Ni–Mg–O отличается тем, что твердый раствор в регенерированном образце сохраняется.
Смешанная система Ni–Mg–O/V–Mg–O была также испытана в 4-х циклах дегидрирования/регенерации. Несмотря на наблюдаемое снижение выхода этилена с каждым циклом, можно предположить, что оптимизация таких параметров, как содержание активных компонентов и массовое соотношение между компонентами V–Mg–О и Ni–Mg–О позволит увеличить стабильность катализаторов и повысить эффективность процесса в целом.
Список литературы
Li D., Xu R., Li X., Li Z., Zhu X., Li K. // Energy & Fuels. 2020. V. 34. P. 5381.
Рябов Г.А. // Альтернативная энергетика и экология (ISJAEE). 2021. № 4–6. С. 82.
Li J., Zhang H., Gao Z., Fu J., Ao W., Dai J. // Energy & Fuels. 2017. V. 31. P. 3475.
Iugai I.A., Steksova Y.P., Vedyagin A.A., Mishakov I.V., Bauman Y.I., Belyy V.A., Danilovich D.P., Krivoshapkina E.F., Krivoshapkin P.V. // Surf. Coat. Technol. 2020. V. 400. P. 126208.
Li Y.Y., Dong X.Y.M., Sun X.D., Wang Y., Zhu J.H. // ACS Appl. Mater. Interf. 2016. V. 8. P. 30193.
Imtiaz Q., Armutlulu A., Donat F., Naeem M.A., Müller C.R. // ACS Sustain. Chem. Eng. 2021. V. 9. P. 5972.
Frey F.E., Huppke W.F. // Indust. Eng. Chem. 1933. V. 25. № 1. P. 54.
Champagnie A.M., Tsotsis T.T., Minet R.G., Webster A.I. // Chem. Eng. Sci. 1990. V. 45. № 8. P. 2423.
Shelepova E.V., Vedyagin A.A. // Chem. Eng. Process: Process Intensification. 2020. V. 155. P. 108072.
Shelepova E.V., Vedyagin A.A., Mishakov I.V., Noskov A.S. // Chem. Eng. J. 2011. V. 176–177. P. 151.
Imamura H., Takada T., Kasahara S., Tsuchiya S. // Appl. Catal. 1990. V. 58. № 1. P. 165.
Chetina O.V., Lunin V.V. // Russ. Chem. Rev. 1994. V. 63. № 6. P. 483.
Tian Y., Dudek R.B., Westmoreland P.R., Li F. // Chem. Eng. J. 2020. V. 398. P. 125583.
Yusuf S., Haribal V., Jackson D., Neal L., Li F. // Appl. Catal. B: Env. 2019. V. 257. P. 117885.
Neal L.M., Yusuf S., Sofranko J.A., Li F. // Energy Technol. 2016. V. 4. № 10. P. 1200.
Fairuzov D., Gerzeliev I., Maximov A., Naranov E. // Catalysts. 2021. V. 11. № 7. P. 833.
Wang C., Yang B., Gu Q., Han Y., Tian M., Su Y., Pan X., Kang Y., Huang C., Liu H., Liu X., Li L., Wang X. // Nature Commun. 2021. V. 12. № 5447.
Karnaukhov T.M., Vedyagin A.A., Cherepanova S.V., Rogov V.A., Stoyanovskii V.O., Mishakov I.V. // Int. J. Hydrogen Energy. 2017. V. 42. № 52. P. 30543.
Vedyagin A.A., Karnaukhov T.M., Cherepanova S.V., Stoyanovskii V.O., Rogov V.A., Mishakov I. V. // Int. J. Hydrogen Energy. 2019. P. 20690.
Karnaukhov T.M., Vedyagin A.A., Cherepanova S.V., Rogov V.A., Mishakov I.V. // J. Sol-Gel Sci. Technol. 2019. V. 92. № 1. P. 208.
Karnaukhov T.M., Veselov G.B., Cherepanova S.V., Vedyagin A.A. // Mater. V. 15. № 6. P. 2021.
Veselov G.B., Karnaukhov T.M., Stoyanovskii V.O., Vedyagin A.A. // Nanomater. 2022. V. 12. № 6. P. 952.
Веселов Г.Б., Ильина Е.В., Тренихин М.В., Ведягин А.А. // Кинетика и катализ. 2022. Т.63. № 5. С. 592.
Vedyagin A.A., Mishakov I.V., Karnaukhov T.M., Krivoshapkina E.F., Ilyina E.V., Maksimova T.A., Cherepanova S.V., Krivoshapkin P.V. // J. Sol-Gel Sci. Technol. 2017. V. 82. № 2. P. 611.
Wang C., Bauman Y.I., Mishakov I.V., Stoyanovskii V.O., Shelepova E.V., Vedyagin A.A. // Processes. 2022. V. 10. № 3. P. 506:1-13.
Mishakov I.V., Bauman Y.I., Streltsov I.A., Korneev D.V., Vinokurova O.B., Vedyagin A.A. // Resource-Efficient Technologies. 2016. V. 2. № 2. P. 61.
Liu S., Zhang B., Liu G. // React. Chem. Eng. 2021. V. 6. № 1. P. 9.
Lemesh N.V., Strizhak P.E. // Mater. Sci. Poland. 2018. V. 36. № 4. P. 739.
Park C., Keane M.A. // J. Catal. 2004. V. 221. № 2. P. 386.
Corrna A., Nieto J.M.L., Parades N., Dejoz A., Vazquez I. // Stud. Surf. Sci. Catal. 1994. V. 82. № C. P. 113.
Iwamoto M., Takenaka T., Matsukami K., Hirata J., Kagawa S., Izumi J. // Appl. Catal. 1985. V. 16. № 2. P. 153.
Lee E.K., Jung K.D., Joo O.S., Shul Y.G. // Catal. Lett. 2004. V. 98. № 4. P. 259.
Jin M., Cheng Z.M. // Catal. Lett. 2009. V. 131. № 1–2. P. 266.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ