

Кинетика и катализ, 2023, T. 64, № 2, стр. 189-202
Исследование кинетических особенностей реакций гидродесульфуризации, гидродеазотирования и гидрирования соединений тяжелого нефтяного сырья на сульфидных Ni6PMonW(12 – n)/Al2O3 катализаторах гидроочистки
А. В. Моисеев a, *, Н. М. Максимов a, П. С. Солманов a, В. А. Тыщенко a
a ФГБОУ ВО Самарский государственный технический университет
443100 Самара, ул. Молодогвардейская, 244, Россия
* E-mail: Moiseev.AV@samgtu.ru
Поступила в редакцию 21.02.2022
После доработки 25.10.2022
Принята к публикации 31.10.2022
- EDN: GNHVEJ
- DOI: 10.31857/S0453881123020053
Аннотация
Проведены кинетические исследования реакций гидродесульфуризации (ГДС), гидродеазотирования (ГДА), а также сравнение гидрирующей активности синтезированных образцов Ni6PMonW(12 – n)/Al2O3 и импортного катализатора гидроочистки вакуумного газойля в процессе гидрооблагораживания тяжелого смесевого нефтяного сырья. Показано, что реакция ГДС описывается уравнением псевдовторого порядка, а реакция ГДА – псевдопервого. Полученные гидрогенизаты отвечают требованиям, предъявляемым к качеству сырья установок каталитического крекинга по коксуемости, содержанию серы и азота.
ВВЕДЕНИЕ
Для углубленной переработки тяжелых нефтей наиболее актуальны схемы, реализующие комплексный подход при выборе деструктивных процессов. На основе проведенного анализа авторы [1] считают, что самыми перспективными являются схемы, сочетающие процессы деасфальтизации, висбрекинга, замедленного коксования и гидрокрекинга; на нефтеперерабатывающих предприятиях Российской Федерации для получения моторных топлив из вакуумного газойля традиционно используется каталитический крекинг. В последние годы сырьевая база каткрекинга расширяется за счет тяжелых продуктов, образовавшихся в процессах деасфальтизации, проводимой с целью удаления асфальтенов, приводящих к быстрой дезактивации катализатора гидроочистки [1], и коксования – самого дешевого деструктивного процесса нефтепереработки [2, 3].
Согласно [4, 5] к качеству сырья установок каталитического крекинга предъявляют достаточно жесткие требования: содержание серы в сырье установки каталитического крекинга не должно превышать 200–400 мг/кг, содержание азота – 73–250 мг/кг, коксуемость по Конрадсону – 0.06–0.10 мас. %. В целях улучшения качества сырья для установки каталитического крекинга используется глубокая гидроочистка, которая включает химические превращения сера-, азоторганических соединений и реакции глубокого гидрирования полициклических ароматических углеводородов (ПАУ). Следовательно, с практической точки зрения, крайне актуально понимание того, как кинетически описываются процессы гидродесульфуризации (ГДС), гидродеазотирования (ГДА) и гидрирования ПАУ (ГИДА) в зависимости от состава катализатора и условий в реакционной системе.
В процессах гидроочистки тяжелых нефтяных фракций традиционно применяются сульфидные NiMo- или NiW-катализаторы, нанесенные на алюмооксидные носители [6]. Известно, что использование фосфора в качестве модифицирующих добавок позволяет существенно увеличить активность катализаторов в реакциях ГДС И ГДА [7–12]. В ряде работ было продемонстрировано, что эффективными предшественниками активной фазы II типа являются PMo12- и PW12-гетерополикислоты структуры Кеггина [13–18]; по мнению авторов [19] гетероатом в катализаторах способствует оптимизации электронной плотности на антисвязывающей d-орбитали Мо в активной смешанной сульфидной фазе и, тем самым, повышает производительность активных центров как в реакциях ГДС, так и ГИДА [19]. Авторы [17, 18] установили, что к росту каталитической активности в реакциях ГДС могут приводить смешаннофазные упаковки сульфидов, сращение аксиально- и базально расположенных плит сульфидов, искривления поверхности кристаллитов, дефекты включения.
Применение лимонной кислоты (ЛК) [20–27] позволяет увеличить активность приготовленных образцов в отношении реакций ГДС стерически затрудненных соединений серы и гидрирования ароматических соединений. Возрастание активности объясняется [20] образованием комплекса никеля, началом его сульфидирования при более высокой температуре и формированием NiMoS-фазы во время активации катализатора. Авторы [24] определили оптимальное количество лимонной кислоты в катализаторах гидроочистки – оно должно соответствовать мольному отношению ЛК : Mo = 1 : 1. В работе [20] оптимальное мольное соотношение Ni : ЛК составляет 1 : 2.
Трехкомпонентные Ni–Mo–W-системы эффективнее, чем биметаллические образцы, для гидрообессеривания дибензотиофена [16] за счет формирования более дефектной фазы. Соотношение Ni/(Mo + W) было исследовано в работе [28], где показано, что наибольшей активностью обладает катализатор состава Ni0.5MoW, а минимальной – Ni1.5MoW. Интересно сравнить поведение катализаторов, отличающихся природой активного компонента, в процессе гидроочистки смесевого сырья.
Кинетика ГДС высококипящих нефтяных фракций обычно описывается степенной зависимостью. Согласно литературным данным [29–38] порядок реакции удаления серы находится в диапазоне 1–3, и, как правило, чем тяжелее сырье, тем он выше. Значение порядка реакции зависит главным образом от типа и концентрации соединений серы, присутствующих в нефтяном дистилляте.
Ozaki [30] использовал простую степенную кинетическую модель для интерпретации кинетических данных ГДС и обнаружил, что наблюдаемый порядок реакции претерпевает изменения по мере повышения температуры реакции. Авторами [35] был получен порядок реакции, равный 2, однако они отмечали, что константа скорости реакции номинально уменьшалась при увеличении объемной скорости подачи сырья (ОСПС). В работе [38] был изучен процесс гидроочистки вакуумного газойля и показано, что реакции ГДС и ГДА подчиняются описанию степенной формой кинетического уравнения с порядками реакции 2.0–2.2 и 1.5–1.8 соответственно. Авторы также приводят обобщенные результаты других групп: порядок реакции ГДС варьировался от 1.0 до 2.68, а ГДА – от 1.0 до 2.02.
Работы по изучению кинетических характеристик ГДС тяжелых фракций, известные в литературе, крайне ограничены и противоречивы, а для смеси вакуумного газойля с тяжелым газойлем коксования и деасфальтизатом такие исследования не проводились.
Для описания кинетики реакций гидродеазотирования использовали как простые степенные законы первого порядка реакции, так и уравнения Ленгмюра–Хиншельвуда [39–42]. Кинетические исследования реакций ГДА [43] показали, что реакции оснóвных и нейтральных соединений описываются кинетической моделью псевдопервого порядка. Азоторганические соединения в вакуумных нефтяных фракциях представлены производными пиррола и хинолина [44]. Их превращение требует от катализатора высокой гидрирующей активности, поскольку гидродеазотирование таких соединений преимущественно протекает по маршруту гидрирования и гидрогенолиза до амина, замещения аминогруппы на сульфогидрильную группу и элиминирования последней.
Целью настоящей работы является изучение кинетических особенностей протекания реакций гидрогенолиза сера- и азотсодержащих компонентов и гидрирования ненасыщенных соединений в смеси вакуумного газойля с деасфальтизатом и тяжелым газойлем коксования в присутствии сульфидных катализаторов Ni6PMonW(12 – n)/Al2O3 разного состава.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Носитель для синтеза катализаторов гидроочистки готовили на основе переосажденной гидроокиси алюминия AlOOH (псевдобемит), в которую приливали HNO3 (х. ч.) до тех пор, пока pH среды не достигнет 3. Затем на гидротермальной стадии синтеза носителя добавляли 30% триэтиленгликоля для регулирования пористой структуры. Полученный псевдозоль AlOOH формовали методом экструзии. Экструдаты сушили на воздухе при температурах 60, 80, 110°C в течение 2 ч на каждой стадии, далее проводили нагрев со скоростью 1°C/мин до 550°C и прокаливали 2 ч. Пористую структуру носителя определяли в модели БЭТ методом низкотемпературной адсорбции N2 на приборе Autosorb-1 (“Quantochrome”, США). Носитель имел следующие характеристики: удельная площадь поверхности – 345 м2/г, объем пор – 0.52 см3/г, эффективный радиус пор – 55 Å. После прокаливания экструдаты измельчали и отбирали фракцию 0.5–0.25 мм.
Синтез оксидных катализаторов гидроочистки проводили методом однократной пропитки носителя (Al2O3) по влагоемкости, который заключается в пропитке совместным водным раствором соединений-предшественников активных компонентов. В качестве предшественника молибдена была использована фосфорномолибденовая кислота Н3РМо12О40·17Н2О (х. ч.), предшественника вольфрама – фосфорновольфрамовая кислота Н3РW12О40·29Н2О (х. ч.), предшественника никеля – карбонат никеля (ч. д. а.). Комплексообразователем в синтезе оксидных катализаторов гидроочистки служила лимонная кислота. Суммарное мольное количество оксида молибдена(VI) и оксида вольфрама(VI) принималось постоянным. Полученные катализаторы сушили при 60, 80, 110°С в течение 2 ч на каждой стадии.
Содержание активных компонентов – оксидов никеля, молибдена (VI) и вольфрама (VI) – в оксидных катализаторах определяли с помощью рентгенофлюоресцентного анализатора EDX-800HS (“Shimadzu”, Япония) по предварительно построенным калибровочным зависимостям (табл. 1).
Таблица 1.
Результаты исследований физико-химических характеристик оксидной формы синтезированных и промышленного катализаторов
| Катализатор* | Содержание, мас. % | ||
|---|---|---|---|
| NiO | MoO3 | WO3 | |
| Ni6PW12 | 2.6 | – | 20.0 |
| Ni6PMo4W8 | 2.8 | 4.1 | 13.3 |
| Ni6PMo6W6 | 2.7 | 6.2 | 9.9 |
| Ni6PMo12 | 2.6 | 12.4 | – |
| Промышленный образец | 3.8 | 19.0 | – |
Сульфидирование оксидных катализаторов проводили в смеси сероводорода и водорода при соотношениях (об. %) H2S : H2 = 70 : 30 непосредственно в реакторе гидрогенизационной микропроточной установки. Процесс сульфидирования осуществляли под давлением 0.11 МПа при температуре 500°C в течение 2 ч.
Каталитическую активность определяли на проточной лабораторной установке при загрузке 15 см3 катализатора в виде фракции 0.5–0.25 мм, послойно чередующейся с фракцией SiC для исключения локальных перегревов в реакторе. Температура проведения процесса – 360, 390, 420°C, давление – 5.0 МПа, ОСПС – 0.5, 1.0, 2.0, 4.0 ч–1, КВСГ – 1000 нл/л сырья.
В качестве реального нефтяного сырья (табл. 2) для испытаний была использована смесь вакуумного газойля (ВГ), тяжелого газойля коксования (ТГК) и деасфальтизата (ДА) в соотношениях (мас. %) 85 : 10 : 5 соответственно, сырье предоставлено АО “НК НПЗ”. Полученную смесь после проведения процесса гидроочистки можно применять как сырье установки каталитического крекинга.
Таблица 2.
Характеристика тяжелого нефтяного сырья
| Сырье | Содержание | ||||
|---|---|---|---|---|---|
| S, мас. % | N, мг/кг | МАУ, мас. % | БАУ, мас. % | ПАУ, мас. % | |
| ВГ | 2.097 | 2300 | 20.1 | 12.0 | 10.6 |
| ТГК | 2.405 | 900 | 17.3 | 9.1 | 2.0 |
| ДА | 1.596 | 300 | 27.7 | 11.8 | 49.8 |
| Смесевое | 2.120 | 1900 | 20.2 | 11.7 | 11.7 |
Плотность нефтяных фракций и гидрогенизатов определяли в соответствии с [45]. Содержание общей серы измеряли с помощью рентгенофлуоресцентного анализатора EDX800HS (“Shimadzu”, Япония) по предварительно полученным калибровочным зависимостям. Данные о содержании азота в гидрогенизатах получали с использованием элементного анализатора Multi EA 5000 (“Analytik Jena”, Германия) методом некаталитического сжигания с последующим детектированием на хемилюминесцентном детекторе. Коэффициент рефракции находили при температуре 70°C на рефрактометре ИРФ-454 (АО “КОМЗ”, Россия) [46].
Количественное определение ароматических углеводородов (АУВ) проводили с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ) в изотермическом режиме на жидкостном хроматографе Prominence (“Shimadzu”, Япония), снабженном полупрепаративным градиентным насосом LC-20AD, дегазатором подвижной фазы DGU-20A3, спектрофотометрическим детектором с термостатируемой ячейкой SPD-20A и термостатом колонок CTO-20A. Детектирование элюата осуществляли при длине волны 280 нм.
Термоаналитические исследования (дифференциальный термический анализ и термогравиметрический анализ (ДТА-ТГА)) отработанных катализаторов гидроочистки выполняли методом дифференциального термического-термогравиметрического анализа на дериватографе STA 449F3 (“Netzsch”, Германия) в температурном диапазоне 30–800°С и при скорости нагрева 10°С/мин в среде воздуха.
Для Ni6PMo12-образца был проведен анализ гидрогенизатов методами определения плотности и коэффициента преломления, по полученным значениям рассчитан интерцепт рефракции (табл. 3) согласно формуле [47]:
где n – показатель преломления нефтепродукта; d – плотность нефтепродукта при 20°C.РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В настоящей работе выполнены кинетические исследования реакций гидродесульфуризации, гидродеазотирования, а также изучены закономерности реакций гидрирования ароматических углеводородов на триметаллических катализаторах гидроочистки Ni6MonW12 – n(S)/Al2O3 с разным мольным отношением Mo/W (табл. 1). В качестве образца сравнения использовали промышленный NiMo/Al2O3-катализатор гидроочистки вакуумного газойля.
Физико-химические характеристики катализаторов
Катализаторы в оксидной форме анализировали методом термопрограммируемой десорбции аммиака (рис. 1). Было найдено три типа кислотных центров, отличающихся интервалами температур десорбции с них NH3: максимуму десорбции при 180–250°С согласно [48] соответствуют льюисовские кислотные центры, принадлежащие носителю [49], при температурах порядка 400–500°С (плечо) наблюдается десорбция аммиака с бренстедовских кислотных центров [50], а при 600–800°С – с кислотных центров, принадлежащих предшественникам активной фазы катализатора [51].
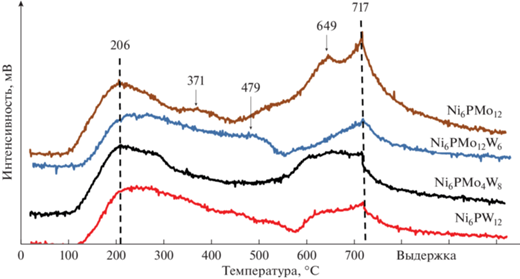
Введение W приводит к возрастанию силы льюисовских кислотных центров (смещение низкотемпературного пика при 200°С в более высокотемпературную область), что согласуется с данными о кислотности гетерополикислот (ГПК) [52] и может свидетельствовать о взаимодействии ГПК с гидроксильными группами носителя, которое ведет к изменению кислотных свойств всей поверхности носителя [53].
Кинетические исследования реакций гидродесульфуризации
Для описания кинетических моделей эксперименты осуществляли при различных объемных скоростях подачи сырья. При поиске оптимальной модели был проведен расчет значений константы скорости и их линеаризация в Аррениусовских координатах для различных степенных моделей, моделей Ленгмюра–Хиншельвуда.
Линеаризация, выполненная в координатах “$\frac{1}{{n - 1}}\left( {c_{{\text{п}}}^{{1 - n}} - c_{{\text{с}}}^{{1 - n}}} \right)$–время контакта”, (где cс – концентрация серы (азота) в сырье, мас. %; cп – концентрация серы (азота) в гидрогенизатах; n – порядок реакции) (рис. 2), для всех температур и времен контакта в настоящем исследовании демонстрирует высокую адекватность модели псевдовторого порядка с квадратом критерия Пирсона 0.9906–0.9998. Для изученных систем также были рассчитаны наблюдаемые константы скорости реакций ГДС (рис. 3) в модели псевдовторого порядка.
Рис. 2.
Линеаризация кинетических данных реакций ГДС в степенной модели 2.0-порядка, полученных в процессе гидроочистки для Ni6PMo12 (а), Ni6PMo6W6 (б), Ni6PMo4W8 (в), Ni6PW12 (г) и промышленного катализатора (д), в зависимости от условного времени контакта в Аррениусовских координатах. Условия эксперимента: проточный реактор, температура 360, 390, 420°C, P = 5.0 МПа, ОСПС = 0.5, 1.0, 2.0, 4.0 ч–1, соотношение Н2/сырье = 1000 нм3/м3 сырья.
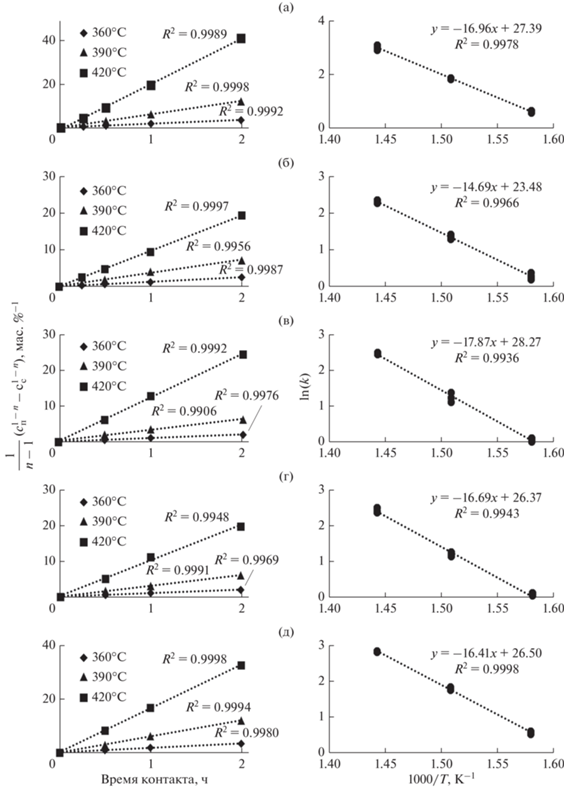
Рис. 3.
Константы скорости реакций гидродесульфуризации. Для промышленного катализатора рассчитаны приведенные константы скорости.
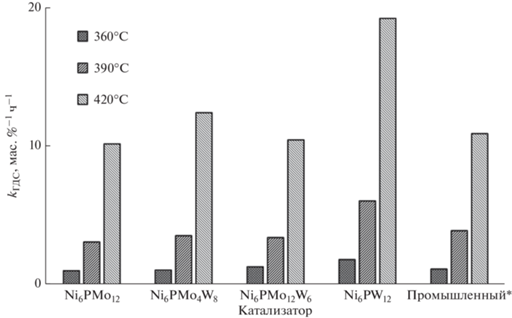
Поскольку во всех синтезированных катализаторах одинаковое общее количество (моль) металлов, а в образце сравнения оно выше, то для корректного сравнения активности для образца сравнения были рассчитаны приведенные константы скорости реакции ГДС:
Самой высокой гидродесульфуризующей активностью обладает образец Ni6PMo12, превосходя промышленный образец при всех температурах испытания. Среди триметаллических образцов бóльшую активность при высокой температуре проявляет Ni6PMo4W8. Эти результаты согласуются с данными, полученными для модельных систем в более ранних работах [54].
Для образцов с бóльшим количеством (или более сильных) льюисовских кислотных центров, что характерно для всех W-содержащих систем, как это следует из данных ТПД, ДТА-ТГА и активности катализаторов в реакциях гидрирования, выше вероятность прочной адсорбции тяжелых конденсированных компонентов сырья (смолы, частично асфальтены, асфальтогеновые кислоты и т.д.). Эти соединения допируют электронную плотность на активную фазу катализатора и подавляют ее гидродесульфуризующую активность [55]. Возможность таких взаимодействий показана на примере модифицирования носителя фосфором [56]. Переносить водород данные соединения, по-видимому, не могут по двум причинам. Во-первых, они достаточно прочно адсорбированы, поскольку являются гетероатомными и включают, как правило, несколько ароматических фрагментов [57]; во-вторых, не исключена и полицентровая адсорбция, а перемещение по поверхности носителя для этих соединений проблематично, т.к. допирование электронной плотности на носитель приводит к снижению подвижности SH-групп и невозможности перегруппировок “SH-группа–льюисовский кислотный центр”. Таким образом, для катализаторов с меньшим количеством льюисовских кислотных центров (или меньшей силы) в условиях гидроочистки реального нефтяного сырья реализуются меньшие по своей величине эффекты ингибирования за счет адсорбции соединений на носителе и на активных центрах катализатора, что и приводит к изменению положения катализаторов в ряду активности [54].
Кинетические исследования реакций гидродеазотирования
Линеаризация, выполненная в координатах “$\ln \left( {\frac{{{{c}_{с}}}}{{{{c}_{п}}}}} \right)$–время контакта” (рис. 4) для всех температур и времен контакта, исследованных в настоящей работе, демонстрирует высокую адекватность модели псевдопервого порядка с квадратом критерия Пирсона на уровне 0.9692–0.9992. Для исследованных систем рассчитаны наблюдаемые константы скорости реакций гидродеазотирования (рис. 5) в модели псевдопервого порядка.
Рис. 4.
Линеаризация кинетических данных реакции ГДА в степенной модели 1.0-порядка, полученных в процессе гидроочистки для Ni6PMo12 (а), Ni6PMo6W6 (б), Ni6PMo4W8 (в), Ni6PW12 (г) и промышленного катализатора (д), в зависимости от условного времени контакта в Аррениусовских координатах. Условия эксперимента: проточный реактор, температура 360, 390, 420°C, P = 5.0 МПа, ОСПС = 0.5, 1.0, 2.0, 4.0 ч–1, соотношение Н2/сырье = 1000 нм3/м3 сырья.
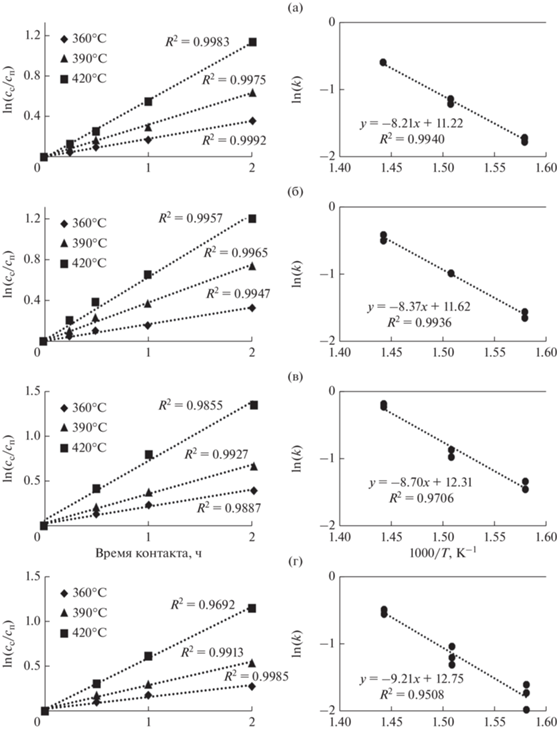
Рис. 4.
Окончание
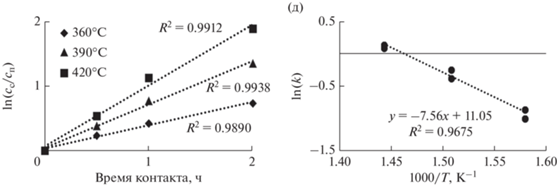
Рис. 5.
Константы скорости реакций гидродеазотирования. Для промышленного катализатора рассчитаны приведенные константы скорости.
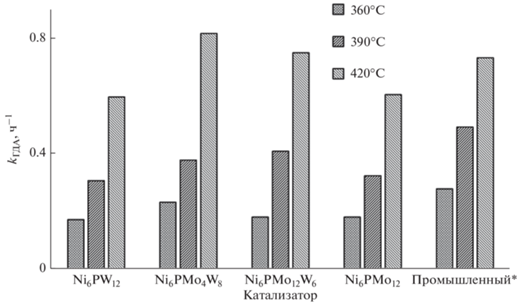
Наиболее высокой гидродеазотирующей активностью, сопоставимой с таковой промышленного катализатора, обладает Ni6PMo4W8. Образцы триметаллических катализаторов проявили бóльшую активность, чем биметаллические Ni6PMo12 и Ni6PW12, во всем интервале температур. Эти результаты согласуются с данными о гидрировании хинолина, полученными ранее для модельных систем [54], что отвечает и соответствует механизму реакции азоторганических соединений, в котором первой стадией является гидрирование ароматического кольца с последующим деазотированием образовавшегося интермедиата.
Исследование закономерностей реакций гидрирования ароматических углеводородов
Результаты исследования активности в реакциях гидрирования ПАУ синтезированных образцов катализаторов представлены на рис. 6.
Рис. 6.
Содержание ПАУ в гидрогенизатах, полученных в процессе гидроочистки в присутствии Ni6PMo12 (а), Ni6PMo6W6 (б), Ni6PMo4W8 (в), Ni6PW12 (г) и промышленного катализатора (д). Условия эксперимента: проточный реактор, температура 360, 390, 420°C, P = 5.0 МПа, ОСПС = 0.5, 1.0, 2.0 ч–1, соотношение Н2/сырье = 1000 нм3/м3 сырья.
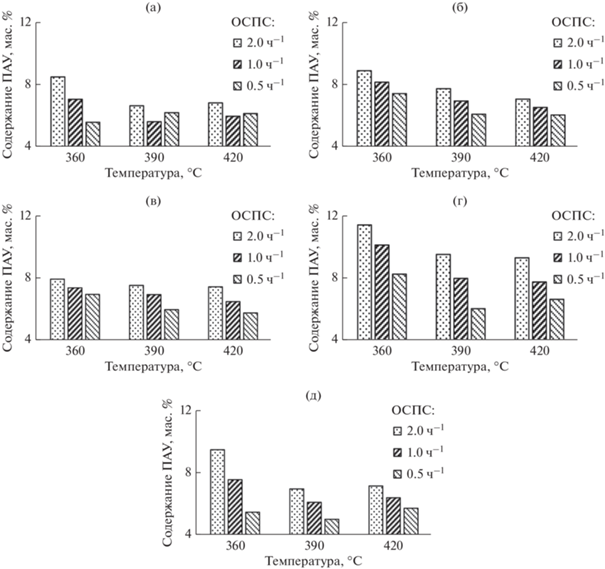
Ni6PW12 проявляет наименьшую гидрирующую активность по сравнению с остальными катализаторами. Максимальное содержание ПАУ в гидрогенизатах этого образца составляет 11 мас. %, минимальное – 6 мас. %. Следует отметить, что содержание ПАУ в гидрогенизате достаточно близко к такому в исходном сырье (11.7 мас. %), что может быть объяснено деструкцией тяжелых компонентов сырья (смол, частично асфальтенов, асфальтогеновых кислот) с образованием дополнительного количества ПАУ [58]. Таким образом, количество ПАУ, обнаруживаемое в катализатах, будет являться результатом двух классов реакций – гидрирования и гидрогенолиза. В свою очередь результирующая будет определяться как ОСПС, так и температурой процесса.
Ранее в [54] на модельных соединениях было показано, что азоторганические соединения адсорбируются на поверхности катализатора сильнее, чем ароматические. При повышении температуры с 390 до 420°C в нефтяном сырье образуется большее количество гидрированных азоторганических соединений, которые прочнее адсорбируются на носителе катализатора и активной фазе [59–62], чем ПАУ, и понижают активность сульфидной фазы в реакциях гидродесульфуризации [63] и гидрирования [64] как по механизму конкурентной адсорбции, так и путем снижения электрондефицитности активной фазы.
Сравнение значения интерцепта рефракции с литературными данными [65] позволяет заключить, что преимущественно в составе полученных гидрогенизатов находятся нафтеновые и нафтено-ароматические углеводороды. Очевидно, что в соответствии с температурами выкипания фракции (вакуумные и остаточные фракции с температурой кипения выше 350°С), а также реакциями гидрирования ПАУ данные углеводороды преимущественно должны иметь конденсированные циклы. Это обуславливает обратимость их гидрирования при повышенных температурах. Аналогичные закономерности были найдены ранее для масляных дистиллятов в процессе гидроочистки [66].
Для определения коксуемости отобраны гидрогенизаты, полученные при температуре процесса гидроочистки 360°С. Такой выбор объясняется предельным значением коксуемости (0.05 мас. %), определение которой возможно по методу Рамсботтома [67].
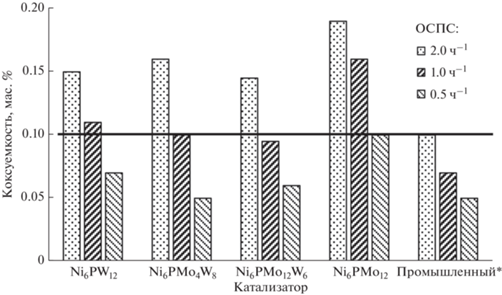
Все образцы гидрогенизатов обладают сравнимой (в пределах погрешности определения метода) коксуемостью, рис. 7. В присутствии промышленного образца образуется гидрогенизат с наименьшим значением коксуемости во всем интервале значений ОСПС. Учитывая промышленные требования к качеству сырья процесса каталитического крекинга по коксуемости, все гидрогенизаты, полученные при ОСПС = 0.5 ч–1, 360°С и более высоких температурах, могут перерабатываться на установках [68–70].
Рис. 7.
Коксуемость гидрогенизатов, полученных в процессе гидроочистки. Условия эксперимента: проточный реактор, Т = 360°C, P = 5.0 МПа, ОСПС = 0.5, 1.0, 2.0 ч–1, соотношение Н2/сырье = 1000 нм3/м3 сырья.
Наибольшей гидрирующей активностью среди представленных образцов обладает Ni6PMo4W8. Максимальное содержание ПАУ на данном катализаторе составляет порядка 8 мас. %, минимальное – 6 мас. %. Гидрирующая активность этого образца сравнима с таковой промышленного, при этом содержание металлов в промышленном образце выше.
Отработанные катализаторы после проведения процесса гидроочистки тяжелого нефтяного сырья были исследованы методом дифференциального термического-термогравиметрического анализа (рис. 8).
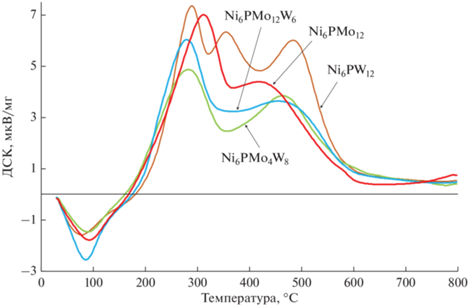
На кривой наблюдаются 2 пика в области температур 290–320 и 470–495°C. Первый относят к окислению анионов серы сульфидной фазы. Для образца Ni6PMo12 данный пик располагается в области более высоких температур, что свидетельствует о менее подвижной сульфидной сере и большей активности этого катализатора в реакциях ГДС при повышенных температурах [71].
Второй пик относят к удалению углеродных отложений. Считается [72], что процесс окисления коксовых отложений начинается с разрушения связей в их конденсированных структурах, поэтому легкость окисления кокса обратно пропорциональна степени конденсированности этих структур: чем она выше, тем бóльшие температуры необходимы для удаления кокса. В свою очередь, конденсированность отложений кокса зависит от гидрирующей активности катализатора. Таким образом, на образце Ni6PMo4W8 отложились наименее конденсированные коксовые структуры, а на Ni6PW12 – наиболее конденсированные. Полученные результаты согласуются с данными об активности этих катализаторов в реакциях гидрирования ПАУ.
ЗАКЛЮЧЕНИЕ
Исследованы кинетические особенности процесса гидроочистки высококипящего смесевого нефтяного сырья в широком интервале параметров (Т = 360–420°С, ОСПС = 4.0, 2.0, 1.0 и 0.5 ч–1) на катализаторах Ni6PMo12, Ni6PMo4W8, Ni6PMo6W6, Ni6PW12, а также на промышленном образце сравнения.
Выбраны и экспериментально подтверждены кинетические модели реакций ГДС и ГДА. На основе проведенного математического анализа показано, что кинетическими моделями, адекватно описывающими протекание реакций ГДС и ГДА процесса, являются модели псевдовторого порядка для процесса гидродесульфуризации и псевдопервого порядка для процесса гидродеазотирования.
Исследована активность образцов катализаторов в реакциях ГИДА. Показано, что наибольшую активность проявляет Ni6PMo4W8. Данные об активности в ГИДА полностью согласуются с результатами определения коксуемости гидрогенизатов, ТПД-анализа и ДТА-ТГА.
По итогам исследований найдено, что по активности в реакциях ГДС и ГИДА катализатор Ni6PMo4W8 не уступает промышленному образцу, а в реакциях ГДА превосходит его. Полученные на образце Ni6PMo4W8 гидрогенизаты отвечают требованиям, предъявляемым к качеству сырья установок каталитического крекинга по коксуемости, содержанию серы и азота.
Список литературы
Castañeda L.C., Muñoz J.A.D., Ancheyta J. // Fuel. 2012. V. 100. P. 110.
Ершов Д.С., Хафизов А.Р., Мустафин И.А. // Фундаментальные исследования. 2017. № 12. С. 282.
Rana M.S., Samano V., Ancheyta J., Diaz J.A.I. // Fuel. 2007. V. 86. P. 1216.
Проектная документация “Комплекс каталитического крекинга на ОАО “СНПЗ”. Разработан ОАО “Самаранефтехимпроект”, 2011. 190 с.
Проектная документация “Комплекс установки гидроочистки вакуумного газойля на ОАО “КНПЗ”. Разработан ЗАО “Нефтехимпроект”, 2013. 213 с.
Topsøe H., Clausen B.S., Massoth F.E. // Catal. Sci. Technol. 1996. V. 11. P. 269.
Achchar M., Lamonier C., Ezzamarty A., Lakhdar M., Leglise J., Payen E. // C. R. Chim. 2009. V. 12. I. 6–7. P. 677.
Ferdous D., Dalai A.K., Adjaye J. // Appl. Catal. A: Gen. 2004.V. 260. P. 153.
Maity S.K., Ancheyta J., Rana M.S., Rayo P. // Catal. Today. 2005. V. 109. P. 42.
Rayo P., Ramírez J., Torres-Mancera P., Marroquin G., Maity K., Ancheyta J. // Fuel. 2012. V. 100. P. 34.
García-Vila A., Cuevas-García R., Ramíreza J. // Catal. Today. 2014. V. 220–222. P. 310.
Nava R., Infantes-Molina A., Castaño P. // Fuel. 2011. V. 90. P. 2726.
Soni K., Boahene P.E., Mouli K.Ch., Dalai A.K., Adjaye J. // Appl. Catal. A: Gen. 2011. V. 398. P. 27.
North J., Poole O., Alotaibi A., Bayahia H., Kozhevnikova E.F., Alsalme A., Siddiqui M.R.H., Kozhevnikov I. // Appl. Catal. A: Gen. 2015. V. 508. P. 16.
Méndez F.J., Llanos A., Echeverría M., Jáuregui R., Villasana Y., Díaz Y., Liendo-Polanco G., Ramos-García M.A., Zoltan T., Brito J.L. // Fuel. 2013. V. 110. P. 249.
Amaya S.L., Alonso-Núnez G., Zepeda T.A., Fuentes S., Echavarría A. // Appl. Catal. B: Env. 2014. V. 148–149. P. 221.
Zhang B.S., Yi Y.J., Zhang W., Liang C.H., Su D.S. // Mater. Charact. 2011. V. 62. P. 684.
Nikulshina M., Mozhaev A., Lancelot Ch., Marinova M., Blanchard P., Payen E., Lamonier C., Nikulshin P. // Appl. Catal. B: Env. 2018. V. 224. P. 951.
Nikulshin P.A., Tomina N.N., Pimerzin A.A., Mashkovsky I.S., Stakheev A.Yu., Kogan V.M. // Appl. Catal. A: Env. 2011. V. 393. P. 146.
Escobar J., Barrera M.C., Reyes J.A., Toledo J.A., Santés V., Colín J.A. // J. Mol. Catal. A: Chem. 2008. V. 287. P. 33.
Tao X., Zhou Y., Wei Q., Yu G., Cui Q., Liu J., Liu T. // Fuel Process. Technol. 2014. V. 118. P. 200.
Li H., Li M., Chu Y., Liu F., Nie H. // Appl. Catal. A: Gen. 2011. V. 403. P. 75.
Calderón-Magdaleno M.Á., Mendoza-Nieto J.A., Klimova T.E. // Catal. Today. 2014. V. 220–222. P. 78.
Escobar J., Barrera M.C., Gutiérrez A.W., Terrazas J.E. // Fuel Proc. Technol. 2017. V. 156. P. 33.
Klimov O.V., Pashigreva A.V., Bukhtiyarova G.A., Budukva S.V., Fedotov M.A., Kochubey D.I., Chesalov Y.A., Zaikovskii V.I., Noskov A.S. // Catal. Today. 2010. V. 150. P. 196.
Pashigreva A.V., Bukhtiyarova G.A., Klimov O.V., Chesalov Yu.A., Litvak G.S., Noskov A.S. // Catal. Today. 2010. V. 149. P. 19.
Pereyma V.Yu., Klimov O.V., Prosvirin I.P., Gerasimov E.Yu., Yashnik S.A., Noskov A.S. // Catal. Today. 2018. V. 305. P. 162.
Yi Y., Zhang B., Jin X., Wang L., Williams C.T., Xiong G., Su D., Liang Ch. // J. Mol. Catal. A: Chem. 2011. V. 351. P. 120.
Michel V., Dorothée L., Christophe G. // Appl. Catal. B: Env. 2012. V. 128. P. 3.
Ozaki H., Satomi Y, Hisamitsi T. // World Petroleum Congress, Tokyo, May 1975. 1975. P. 97.
Kam E.K.T., Al-Bazzaz H., Al-Fadhli J. // Ind. Eng. Chem. Res. 2008. V. 47. I. 3. P. 594.
Korsten H., Hoffmann U. // AIChE J. 1996. V. 42. I. 5. P. 1350.
Ross L.D. // Chem. Eng. Prog. 1965. V. 61. P. 77.
Mederos F.S., Ancheyta J. // Energy Fuels. 2017. V. 31. I. 5. P. 5691.
Rodríguez M.A., Elizalde I., Ancheyta J. // Fuel. 2012. V. 100. P. 152.
Satterfield C.N. // AIChE J. 1975. V. 21. P. 209.
Sylvester N.D., Pitayagulsarn P. // Can. J. Chem. Eng. 1974. V. 52. P. 539.
Boahene Ph.E., Soni K.K., Dalaia A.K., Adjaye J. // Catal. Today. 2013. V. 207. P. 101.
McIlvried H.G. // Ind. Eng. Chem. Proc. Des. Dev. 1971. V. 10. P. 125.
Hanlon R.T. // Energy Fuels. 1987. V. 1. P. 424.
Anabtawi J.A., Mann R.S., Khulbe K.C. // J. Catal. 1980. V. 63. P. 456.
Machida M., Sakao Y., Ono S. // Appl. Catal. A: Gen 2000. V. 201. P. 115.
Wei Q., Wen S.-C., Zhou Y.-S. // Fuel Proc. Technol. 2015. V. 129. P. 76.
Martínez J., Ancheyta J. // Fuel. 2014. V. 138. P. 27.
ГОСТ 3900-85. Москва: МЕЖГОСУДАРСТВЕННЫЙ СТАНДАРТ: Издательство стандартов, 1991. 36 с.
Белянин Б.В., Эрих В.Н., Корсаков В.Г. Технический анализ нефтепродуктов и газа. Ленинград: Химия, 1986. 184 с.
Добрянский А.Ф. Химия нефти. Ленинград: Гостоптехиздат, 1961. 224 с.
Li D., Nishijima A., Morris D.E., Guthrie G.D. // J. Catal. 1999. V. 188. P. 111.
Callant M., Holder K.A., Grange P., Delmon B. // Bull. Soc. Chim. Belg. 1995. V. 104. I. 4–5. P. 245.
Xu Y.D., Shu Y.Y., Liu S.T., Huang J.S., Guo X.X. // Catal. Lett. 1995. V. 35. P. 233.
Zhang X., Liu Q., Zhang Q., Liu Q., Chen L., Li Y., Wang C., Ma L. // Energy Procedia. 2019. V. 158. P. 984.
Никитина Е.А. Гетерополисоединения. Москва: Госхимиздат, 1962. 326 с.
Guo X.-M., Song M.-N., Zhao X., Zhao L.-F. // J. Fuel Chem. Technol. 2016. V. 44. P. 1326.
Moiseev A.V., Maximov N.M., Solmanov P.S., Verevkin S.P., Tyshchenko V.A. // React. Kinet. Mechan. Catal. 2022. V. 135. P. 927.
Garcia-Cruz I., Valencia D., Klimova T., Oliedo-Roa R. // J. Mol. Catal. A: Chem. 2008. V. 281. P. 79.
Nikulshina M., Mozhaev A., Lancelot C., Blanchard P., Marinova M., Lamonier C., Nikulshin P. // Catal. Today. 2019. V. 329. P. 24.
Сергиенко С.Р. Высокомолекулярные соединения нефти. Москва: Химия, 1964. 541 с.
Казакова Л.И., Крейн С.Э. Физико-химические основы производства нефтяных масел. Москва: Химия, 1978. 320 с.
Turaga U.T., Ma X., Song Ch. // Catal. Today. 2003. V. 86. I. 1–4. P. 265.
Laredo G.C., Altamirano E., De losReyes J.A. // Appl. Catal. A: Gen. 2003. V. 242. I. 2. P. 311.
Laredo G.C., De los Reyes J.A., Cano J.L., Castillo J.J. // Appl. Catal. A: Gen. 2001. V. 207. I. 1–2. P. 103.
Laredo G.C., Altamirano E., De los Reyes J.A. // Appl. Catal. A: Gen. 2003. V. 243. I. 2. P. 207.
Garcia-Cruz I., Valencia D., Klimova T., Oliedo-Roa R. // J. Mol. Catal. A. 2008. V. 281. P. 79.
Nguyen M.-T., Tayakout-Fayolle M., Chainet F., Pirngruber G.D., Geantet Ch. // Appl. Catal. A: Gen. 2017. V. 530. P. 132.
Проскуряков В.А., Драбкина Е.А. Химия нефти и газа. Санкт-Петербург: Химия, 1995. 448 с.
Томина Н.Н. Дисс. … д.х.н.: Самара, СамГТУ, 2009. 351 с.
ГОСТ 32332-2013. Минск: МЕЖГОСУДАРСТВЕННЫЙ СТАНДАРТ: Издательство стандартов, 2015. 29 с.
Мейерс Р.А. Основные процессы нефтепереработки, под ред. Глаголевой О.Ф., Лыкова О.П. Санкт-Петербург: ЦОП “Профессия”, 2011. 944 с.
Алиев Р.Р. Катализаторы и процессы переработки нефти. Москва: ВНИИНП. 2010. 304 с.
Радченко Е.Д., Нефедов Б.К., Алиев Р.Р. Промышленные катализаторы гидрогенизационных процессов нефтепереработки. Москва: Химия, 1987. 224 с.
Ueda T., Toya T., Hojo M. // Inorg. Chim. Acta. 2004. V. 357. P. 59.
Масагутов Р.М., Морозов Б.Ф., Кутепов Б.И. Регенерация катализаторов в нефтепереработке и нефтехимии. Москва: Химия, 1987. 144 с.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ