

Кинетика и катализ, 2023, T. 64, № 2, стр. 203-215
Разложение аммиака на кобальт-силикагелевых катализаторах синтеза Фишера–Тропша
Р. Е. Яковенко a, Т. В. Краснякова a, b, А. Н. Салиев a, М. А. Шилов a, А. В. Волик a, А. П. Савостьянов a, С. А. Митченко a, b, *
a Южно-Российский государственный политехнический университет (НПИ) им. М.И. Платова
346128 Новочеркасск, ул. Просвещения, 132, Россия
b Институт физико-органической химии и углехимии им. Л.М. Литвиненко
283114 Донецк, ул. Р. Люксембург, 70, Россия
* E-mail: samit_rpt@mail.ru
Поступила в редакцию 01.10.2022
После доработки 25.11.2022
Принята к публикации 29.11.2022
- EDN: GNTPBZ
- DOI: 10.31857/S0453881123020090
Аннотация
Показана возможность применения кобальт-силикагелевых катализаторов синтеза Фишера–Тропша для разложения аммиака в проточном режиме в трубчатом реакторе со стационарным слоем катализатора при давлении 0.1 МПа, объемной скорости газа 1000–6000 ч–1 в интервале температур 400–650°С. Активность и производительность по водороду уменьшаются в ряду Co–Ru/SiO2 ≥ ≥ Co–Al2O3/SiO2 > Ru/SiO2 > Co–Al2O3/SiO2(35%)/ZSM-5(30%)/Al2O3(35%). Относительно небольшие эффективные энергии активации, оцененные для всех катализаторов, позволяют проводить реакцию с приемлемой конверсией при умеренных температурах.
ВВЕДЕНИЕ
В последнее столетие перед человечеством остро стоит необходимость выработки все большего количества энергии для удовлетворения растущих потребностей общества. Фундаментальной проблемой современной энергетической системы является негативное влияние на окружающую среду последствий получения энергии из ископаемого топлива [1, 2]. Одним из путей ее решения могут быть возобновляемые источники, такие как энергия солнца и ветра. Однако их применение осложнено периодическим и/или сезонным действием, а долговременное накопление произведенной энергии ограничено для масштабного использования малой энергетической емкостью и скоростью зарядки–разрядки существующих систем ее хранения [3, 4].
В рамках перехода к экологически чистой энергетике ориентация на водород представляется перспективным направлением, не связанным с выбросами СОх и NОх в окружающую среду [5]. Однако хранение и транспортировка водорода сопряжены с рядом сложностей. Например, невозможность обеспечения достаточно хорошей теплоизоляции систем хранения жидкого водорода в сочетании с его низкой температурой кипения приводят к неминуемым потерям Н2 в результате испарения. Помимо этого, выкипание Н2 сопровождается возрастанием взрывоопасности. Еще одной проблемой долговременного хранения водорода является охрупчивание [6–8] емкостей для транспортировки: благодаря высокой диффузионной подвижности водород легко проникает в различные материалы, меняя их прочностные свойства. Кроме того, Н2 имеет низкую объемную плотность как в сжатом газообразном, так и в жидком состояниях. Увеличение объемной плотности жидкого водорода возможно путем криокомпрессии: при 20 К повышение давления от 1 бар до 240 бар приводит к возрастанию плотности Н2 с 70 г/л до 87 г/л [9]. Такое хранение снижает потери водорода при испарении, но сам процесс требует больших энергетических затрат [2].
Концептуально иной подход представляет хранение водорода в виде какого-нибудь легко транспортируемого водородсодержащего соединения, получение Н2 из которого осуществляется непосредственно на месте. Потенциальными претендентами на роль такого “хранилища” могут быть сжиженный нефтяной газ, спирты, бензиновая и дизельная углеводородные фракции, метанол, гидриды металлов и др. Основным недостатком переработки в водород углеродсодержащих соединений является так называемый “углеродный след”. При использовании гидридов металлов в качестве накопителей водорода зачастую требуются высокие температуры для адсорбции/десорбции. Кроме того, сорбционная емкость таких материалов мала, а энтальпия десорбции высока [10].
Применение NH3 для хранения и транспортировки H2 в химически связанном состоянии обладает рядом преимуществ. В настоящее время мировое производство аммиака составляет ~180 Мт в год [11] и имеет тенденцию к росту. Он находится в жидком состоянии при более низком давлении и высокой температуре, чем Н2, ожижение NH3 не так энергозатратно, средства для транспортировки и хранения меньше и легче [12]. Экономичность аккумулирования химически связанного в аммиаке водорода заключается еще и в возможности использования аммиачного трубопровода вместо водородного [13].
Для разложения аммиака в качестве катализаторов применяют различные моно- и биметаллические системы. Основное внимание исследователей сосредоточено на рутениевых катализаторах как наиболее активных [14]. Высокая стоимость и ограниченная доступность Ru в сочетании с быстрой дезактивацией сдерживают его широкое использование в промышленности [12, 15–17]. Следующими по интенсивности изучения являются альтернативные дорогостоящему рутению переходные металлы Fe, Co и Ni [14]. В этой триаде никель и железо дешевле кобальта, однако применение Со эффективнее при меньших температурах [15, 18–21]. Кроме того, в Fe-содержащих системах при низких температурах образуются неактивные нитриды железа [16, 22]. Все это делает привлекательным применение кобальтовых катализаторов для разложения аммиака.
Активные состояния кобальта в этой реакции однозначно не идентифицированы. Имеются сведения, что в окисленной форме катализатор не демонстрирует высокой активности [2, 17], активным является металлический кобальт [23, 24]. Максимальную производительность демонстрируют катализаторы с размерами наночастиц металла в диапазоне 10–20 нм [2, 23, 25, 26]. Указанные характеристики близки к присущим кобальтовым каталитическим системам синтеза Фишера–Тропша (СФТ), где активная форма катализатора представляет собой металлический кобальт с оптимальными размерами наночастиц 8 ± 2 нм [27]. Все это дает основания полагать, что исследованные нами ранее [28–31] высокопроизводительные кобальт-силикагелевые катализаторы СФТ будут активны также и в реакции разложения аммиака. Проверка такого предположения и составила цель настоящей работы.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приготовление катализаторов
Катализатор Co–Al2O3/SiO2 готовили методом пропитки носителя водным раствором нитрата кобальта и нитрата алюминия по методике, описанной в [30]. В качестве носителя использовали силикагель марки КСКГ (ГОСТ 3956-76) производства ООО “Салаватский катализаторный завод”. Пропитку носителя осуществляли при температуре 75–80°С в течение 0.5 ч, после чего сушили в интервале температур 80–150°С. Катализатор Co‒Ru/SiO2 получали аналогично, применяя вместо нитрата алюминия гидроксотрихлорид рутения. Количество добавляемого Al(NO3)3 или Ru(OH)Cl3 рассчитывали так, чтобы содержание соответствующего металла в катализаторе составляло 1 мас. %.
Гибридный катализатор Co–Al2O3/SiO2(35%)/ ZSM-5(30%)/Al2O3(35%) готовили путем смешения Co–Al2O3/SiO2, бемита и цеолита ZSM-5 с последующим формованием экструзией, затем сушили при 80°С [31].
Ru/SiO2 получали пропиткой SiO2 водным раствором гидроксотрихлорида рутения с последующей сушкой в интервале температур 80–140°С. После сушки все катализаторы прокаливали при 400°С в течение 4 ч.
Методика экспериментальных исследований
Определение активности катализаторов осуществляли на проточной установке (рис. 1) с интегральным реактором в непрерывном проточном режиме. Установка снабжена системой для подачи газа, регулирования расхода, температуры и давления.
Рис. 1.
Технологическая схема установки: 1 – газовый баллон с аммиаком; 2 – регулятор давления в системе; 3, 11, 13 – газовый кран; 4 – ротаметр; 5 – обратный клапан; 6 – манометр давления в системе; 7 – трубчатый реактор; 8 – электрическая печь; 9 – катализатор; 10 – тройник; 12 – газовые часы; 14 – склянка Дрекселя; 15 – аспиратор; 16 – мерная емкость; 17 – регулятор температуры “Термодат”; 18, 19 – термопары.
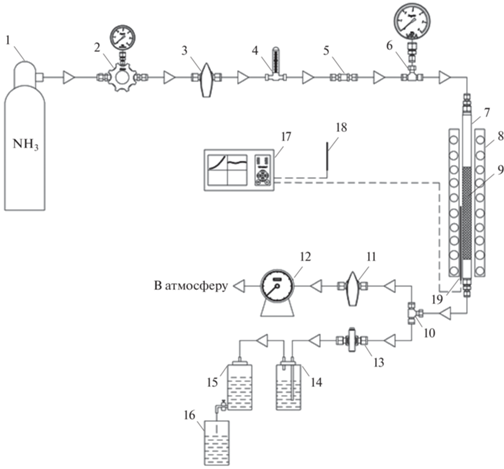
При испытании катализаторов (фракция 1–2 мм) в реактор загружали 3 см3 образца, предварительно восстанавливали водородом в течение 1 ч при температуре 400°С и объемной скорости газа (ОСГ) 3000 ч–1. Об активности катализаторов судили по конверсии NH3 и производительности по водороду.
Конверсию NH3 определяли с учетом изменения объема газов в ходе реакции 2NH3 = 3H2 + N2 по формуле (1):
(1)
${{X}_{{{\text{N}}{{{\text{H}}}_{{\text{3}}}}}}} = \frac{{100 - {{C}_{{{\text{N}}{{{\text{H}}}_{{\text{3}}}}}}}}}{{100 + {{C}_{{{\text{N}}{{{\text{H}}}_{{\text{3}}}}}}}}} \times 100\% ,$Производительность по водороду определяли согласно выражению (2):
(2)
${{G}_{{{{{\text{H}}}_{2}}}}} = \frac{{1.5{{V}_{{{\text{вх}}}}}{{X}_{{{\text{N}}{{{\text{H}}}_{{\text{3}}}}}}}}}{{100{{V}_{{{\text{кат}}}}}}},$Методы анализа и определение конечной концентрации NH3
Газовую смесь анализировали на содержание NH3 пропусканием газового потока при помощи аспиратора и крана (рис. 1) через поглотительную склянку Дрекселя, которая содержала 50 см3 воды, 10 см3 0.1 н H2SO4 и несколько капель метилового оранжевого. Поглотительная склянка соединялась с аспиратором, создающим разрежение для прохождения газа через нее. Газ пропускали через поглотитель до тех пор, пока красная окраска раствора не переходила в желтую. Количество азота и водорода определяли по объему воды, вытекшей из аспиратора.
Содержание аммиака в газе на выходе из реактора ${{С}_{{{\text{N}}{{{\text{H}}}_{{\text{3}}}}}}}$ (об. %) рассчитывали по формуле [32]:
где a – объем NH3 (10 cм3 0.1 н. H2SO4 соответствует 22.4 см3 NH3 при 0°С и 1.01 × 105 Па), см3; V – объем вытекшей воды из аспиратора, см3; P – давление по барометру, Па; Т – температура в лаборатории, К; p – парциальное давление водяных паров при температуре Т, Па.Исследование физико-химических свойств катализаторов
Температурно-программированное восстановление (ТПВ) катализаторов выполняли на анализаторе ChemiSorb 2750 (“Micromeritics”, США). После 1 ч дегазации образцы восстанавливали водородно-азотной смесью (10% H2 по объему, 20 мл мин-1) в интервале температур 20–800°С со скоростью 20°С/мин. Степень восстановления кобальта в катализаторах Co–Ru/SiO2 и Co–Al2O3/SiO2 находили путем импульсного окисления восстановленных катализаторов 30% смесью кислорода в гелии при 550°С.
Удельную поверхность определяли методом БЭТ по тепловой десорбции аргона с использованием анализатора ChemiSorb 2750. Предварительно образцы выдерживали в токе гелия в течение 1 ч при температуре 200°С.
Микроструктуру и элементный состав поверхности катализаторов изучали методами сканирующей электронной микроскопии (СЭМ) и энергодисперсионного микроанализа (ЭДА) с помощью растрового электронного микроскопа Quanta 200 (“FEI”, США), оснащенного системой энергодисперсионного анализа EDAX Genesis при ускоряющем напряжении 30 кВ.
Рентгенофазовый анализ (РФА) проводили на дифрактометре ARL X’TRA (“Thermo Fisher Scientific”, Швейцария) в CuКα-излучении методом сканирования по точкам (шаг – 0.03°, время накопления в точке – 1 с) в интервале 2θ от 5° до 90°. Определение фазового состава осуществляли с помощью базы данных PDF-2 в программном комплексе Crystallographica. Размеры областей когерентного рассеяния (ОКР) Со3О4 оценивали для рефлекса при 2θ ≈ 37.1° по уравнению Шеррера [33]:
(4)
$d({\text{C}}{{{\text{o}}}_{{\text{3}}}}{{{\text{O}}}_{{\text{4}}}}) = \frac{{K{{\lambda }}}}{{{{\beta }}\cos {{\theta }}}},$Соответствующие им размеры кристаллитов металлического кобальта определяли согласно [34] как:
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Характеристика катализаторов
По данным микрорентгеноспектрального анализа распределение элементов, входящих в состав кобальтовых катализаторов, по поверхности образцов равномерное (рис. 2). Для не содержащих рутений образцов поверхность представлена микрочастицами сферической формы диаметром до 5 мкм (рис. 2а и 2б). В образце гибридного катализатора Co–Al2O3/SiO2(35%)/ZSM-5(30%)/ Al2O3(35%) (рис. 2б) такие частицы разрознены, а для Co–Al2O3/SiO2 (рис. 2а) микросферы агломерированы в более крупные образования с линейными размерами до 50 мкм. Поверхность кобальт-рутениевого катализатора представлена (рис. 2в) блоками частиц, не имеющими правильной геометрической формы, размером ~10 мкм. Содержание рутения в катализаторах близко к расчетному: 1.5 ± 0.4 и 1.0 ± 0.3 мас. % для Co–Ru/SiO2 и Ru/SiO2 соответственно.
Рис. 2.
СЭМ-микрофотографии поверхности и картографирование активных элементов катализаторов: (а) – Co‒Al2O3/SiO2, (б) – Co–Al2O3/SiO2(35%)/ZSM-5(30%)/Al2O3(35%), (в) – Co–Ru/SiO2, г – Ru/SiO2.

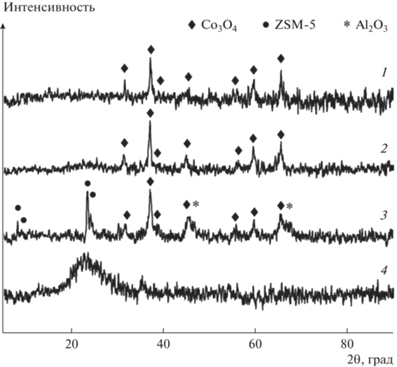
РФА свежеприготовленных кобальтовых катализаторов в окисленной форме свидетельствует о наличии рефлексов фазы Со3О4 со структурой шпинели [35] (рис. 3, кривые 1, 2, 3). Для Co–Al2O3/SiO2 (рис. 3, кривая 2) рентгеновские максимумы оксида алюминия зафиксированы не были, очевидно, в силу малого (1%) содержания этой фазы. На дифрактограмме Co–Al2O3/SiO2(35%)/ZSM-5(30%)/ Al2O3(35%) (рис. 3, кривая 3) кроме линий Со3О4 наблюдаются рефлексы цеолита ZSM-5 [36] и нестехиометрического оксида алюминия (Al2O3)1.333 [37] с кубической структурой шпинели дефектного типа. Наличие линий (Al2O3)1.333 является следствием начала образования фазы Al2O3 в результате термической обработки добавленного в катализатор связующего бемита. Поскольку прокаливание в ходе приготовления осуществлялось при температуре 400°С, меньшей, чем необходимо для кристаллизации Al2O3 (450–600°С), эта фаза окончательно не сформировалась [38]. По данным рентгеновской дифракции в Ru-содержащих образцах фаза оксида рутения не обнаружена (рис. 3, кривая 4). Причиной отсутствия рефлексов кристаллической фазы RuOх может быть малое содержание Ru в образцах, а также его высокая степень дисперсности.
Рис. 3.
Дифрактограммы катализаторов: (1) – Co–Ru/SiO2; (2) – Co–Al2O3/SiO2; (3) – Co–Al2O3/SiO2(35%)/ZSM-5(30%)/Al2O3(35%); (4) – Ru/SiO2.
Оцененные по формуле Шеррера величины области когерентного рассеяния (ОКР) оксида кобальта, а также соответствующие им размеры кристаллитов Со0 приведены в табл. 1. Размеры кристаллитов металлического кобальта для Co‒Al2O3/SiO2 и гибридного катализаторов в пределах экспериментальных погрешностей совпадают с определенными методом ПЭМ [27, 28] диаметрами наночастиц 8 ± 2 для обоих катализаторов.
Таблица 1.
Размер кристаллитов Со3О4 для катализаторов в окисленной форме, d(Co3O4), и оценка ожидаемых размеров ОКР металлического кобальта в восстановленных катализаторах, d(Co0)
| Катализатор | d(Co3O4), нм | d(Co0), нм |
|---|---|---|
| Co–Ru/SiO2 | 15 ± 3 | 11 ± 2 |
| Co–Al2O3/SiO2 | 13 ± 3 | 10 ± 2 |
| Co–Al2O3/SiO2(35%)/ZSM-5(30%)/Al2O3(35%) | 11 ± 3 | 8 ± 2 |
Размеры наночастиц металлического кобальта в пределах экспериментальных ошибок попадают в интервал 10–20 нм, оптимальный для высокопроизводительных катализаторов разложения аммиака.
Профили ТПВ кобальтсодержащих катализаторов (рис. 4, кривые 1, 2, 3) в целом однотипны. В интервале температур 200–600°С зафиксированы два интенсивных максимума, связанных со стадийным восстановлением Co3O4 [39, 40]:
(I)
${\text{C}}{{{\text{o}}}_{{\text{3}}}}{{{\text{O}}}_{{\text{4}}}}{\text{ + }}{{{\text{H}}}_{{\text{2}}}} \to 3{\text{CoO + }}{{{\text{H}}}_{{\text{2}}}}{\text{O}},$Рис. 4.
Профили ТПВ катализаторов: (1) – Co–Ru/SiO2; (2) – Co–Al2O3/SiO2; (3) – Co–Al2O3/SiO2(35%)/ZSM-5(30%)/Al2O3(35%); (4) – Ru/SiO2.
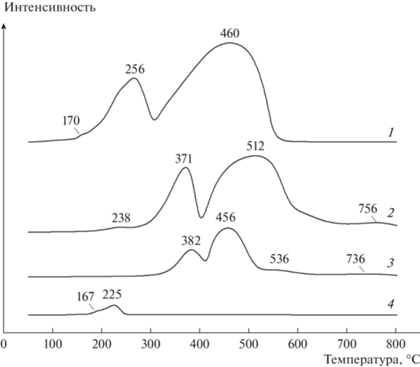
Для образца Ru/SiO2 (рис. 4, кривая 4) наблюдаются максимумы при 167 и 225°С, которые можно отнести к восстановлению RuOх: согласно [41, 42] оксид рутения восстанавливается при более низких температурах, чем оксиды Со. Профиль ТПВ катализатора Co–Ru/SiO2 (рис. 4, кривая 1) также содержит аналогичный максимум при 170°С.
На профиле ТПВ образца Co–Ru/SiO2 (рис. 4, кривая 1) имеется смещение максимумов восстановления оксида кобальта в низкотемпературную область, связанное, очевидно, с введением Ru в состав катализатора. Появление металлического Ru в процессе восстановления способствует активации водорода путем диссоциативной адсорбции. Последующий спилловер водорода с рутения на оксид кобальта [43] облегчает восстановление последнего при более низких температурах по сравнению с катализаторами без рутения.
Профили ТПВ катализаторов Co–Al2O3/SiO2 и Co–Al2O3SiO2(35%)/ZSM-5(30%)/Al2O3(35%) содержат еще и низкоинтенсивные максимумы. Для Co–Al2O3/SiO2 небольшой пик в области низких температур при 238°С может быть обусловлен разложением остаточного Co(NO3)2 – предшественника оксида кобальта [44–46].
Максимумы при температуре выше 700°С обычно связывают с восстановлением трудновосстановимых соединений, например, алюминатов кобальта [39].
Небольшой пик при 536°С на кривой 3 может быть вызван остаточным разложением бемита, использованного в качестве связующего для приготовления гибридного катализатора: при термообработке бемит превращается в оксид алюминия.
Степень восстановления кобальта в Co–Ru/SiO2 и Co–Al2O3/SiO2 практически одинакова: 85 и 86% соответственно.
Удельная поверхность катализаторов приведена в табл. 2. Невысокие значения удельной поверхности для Co–Ru/SiO2 и Co–Al2O3/SiO2 обусловлены, вероятно, блокированием пор носителя SiO2 кобальтом, массовая доля которого в катализаторе составляет 20%, а также уменьшением массовой доли носителя в катализаторе при большой загрузке активного металла. Существенная величина удельной поверхности гибридного катализатора, очевидно, связана с наличием в его составе высокопористого цеолита.
Каталитические испытания
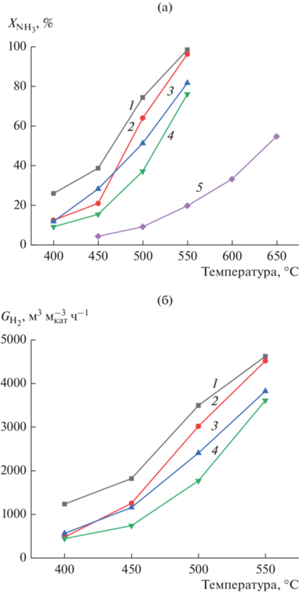
Каталитические испытания нанесенных на силикагель катализаторов СФТ показали, что все они проявляют активность в реакции разложения аммиака. В реакторе, содержащем только носитель SiO2 без активной составляющей катализатора, конверсия аммиака становится значимой только начиная с 550°С, при более низких температурах вкладом носителя в разложение NH3 можно пренебречь. Степень превращения во всех каталитических системах увеличивается с повышением температуры (рис. 5а). Для исследуемых катализаторов в изученном температурном интервале активность убывает в следующем порядке: Co–Ru/SiO2 ≥ Co–Al2O3/SiO2 > Ru/SiO2 > Co–Al2O3/ SiO2(35%)/ZSM-5(30%)/Al2O3(35%). Производительность по водороду также зависит от температуры (рис. 5б) и повторяет ряд активности катализаторов.
Рис. 5.
Зависимости конверсии аммиака ${{X}_{{{\text{N}}{{{\text{H}}}_{{\text{3}}}}}}}$ (а) и производительности по водороду ${{G}_{{{{{\text{H}}}_{{\text{2}}}}}}}$ (б) от температуры при ОСГ 3000 ч–1 для катализаторов: (1) – Co‒Ru/SiO2; (2) – Co–Al2O3/SiO2; (3) – Ru/SiO2; (4) – Co-Al2O3/SiO2(35%)/ZSM-5(30%)/Al2O3(35%); (5) – SiO2.
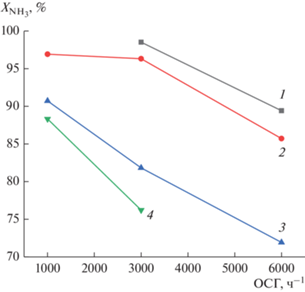
Повышение объемной скорости подаваемого аммиака с 1000 до 6000 ч–1 для всех катализаторов сопровождается снижением конверсии, обусловленным уменьшением времени контакта молекул NH3 с поверхностью катализаторов (рис. 6).
Рис. 6.
Зависимость конверсии аммиака от ОСГ для катализаторов: (1) – Co–Ru/SiO2; (2) – Co–Al2O3/ SiO2; (3) – Ru/SiO2; (4) – Co–Al2O3/SiO2(35%)/ZSM-5(30%)/Al2O3(35%).
Значения эффективной энергии активации, определенные из линейной анаморфозы уравнения Аррениуса (рис. 7), убывают в ряду Co–Al2O3/SiO2 ≈ ≈ Co–Al2O3/SiO2(35%)/ZSM-5(30%)/Al2O3(35%) > > Ru/SiO2 > Co–Ru/SiO2 (табл. 3).
Рис. 7.
Температурные зависимости конверсии аммиака ${{X}_{{{\text{N}}{{{\text{H}}}_{{\text{3}}}}}}}$ в аррениусовских координатах при ОСГ 3000 ч–1 для катализаторов: (1) – Co–Ru/SiO2; (2) – Co–Al2O3/SiO2; (3) – Ru/SiO2; (4) – Co‒Al2O3/ SiO2(35%)/ZSM-5(30%)/Al2O3(35%).
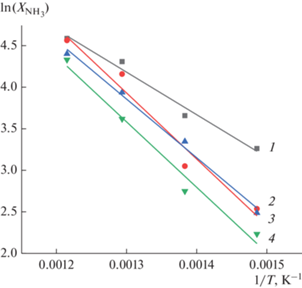
Таблица 3.
Параметры уравнения Аррениуса реакции каталитического разложения аммиака
| Катализатор | Co–Ru/SiO2 | Co–Al2O3/SiO2 | Ru/SiO2 | Co–Al2O3/SiO2(35%)/ ZSM-5(30%)/Al2O3(35%) |
|---|---|---|---|---|
| Ea, кДж/моль | 42.7 ± 4.2 | 66.3 ± 8.7 | 58.8 ± 3.4 | 65.7 ± 6.6 |
| ln(A) | 10.9 ± 0.7 | 14.3 ± 1.4 | 13.1 ± 0.5 | 13.9 ± 1.1 |
Рутениевые катализаторы считаются наиболее эффективными среди каталитических систем на основе переходных металлов. Удельная каталитическая активность (объем аммиака, разложившегося в час на 1 г металла) исследованного нами нанесенного на силикагель Ru (1%) оказалась сравнимой с зафиксированной для других рутенийсодержащих катализаторов (табл. 4): в наших экспериментах при пятикратно меньшей загрузке активного металла и сниженной в 4 раза объемной скорости подаваемого газа степень превращения аммиака составляет 82%, что сопоставимо с литературными данными.
Таблица 4.
Конверсия аммиака ${{X}_{{{\text{N}}{{{\text{H}}}_{{\text{3}}}}}}}$ в Ru-содержащих системах при 550°С
| Загрузка Ru, мас. % | Носитель | Промотор | ОСГ, мл г–1ч–1 |
${{X}_{{{\text{N}}{{{\text{H}}}_{{\text{3}}}}}}}$, % | Источник |
|---|---|---|---|---|---|
| 5 | АУ | 30 000 | 14 | [47] | |
| 5 | Al2O3 | 30 000 | 78 | [48] | |
| 5 | Al2O3 | K | 30 000 | 86 | [47] |
| 5 | Мезопористый уголь СМК-3 | K | 30 000 | 79 | [49] |
| 5 | Углеродные нанотрубки | 30 000 | 85 | [47] | |
| 5 | Графитизированный уголь | 30 000 | 95 | [47] | |
| 1 | SiO2 | 7500 | 82 | Настоящая работа |
Протестированные кобальтовые катализаторы также демонстрируют активность в разложении аммиака, близкую к известной из литературы (табл. 5). Следует отметить, что изученные нами катализаторы для достижения достаточно высокой активности не требуют промотирования и/или использования в качестве носителя коммерчески ограниченно доступных углеродных наноматериалов или оксидов редкоземельных металлов.
Таблица 5.
Конверсия аммиака ${{X}_{{{\text{N}}{{{\text{H}}}_{{\text{3}}}}}}}$в Со-содержащих системах при температуре 500°С и ОСГ 6000 ч–1
В настоящей работе самым активным оказался промотированный рутением кобальтовый катализатор – степень превращения NH3 при 550°С составила 98.5%. При той же температуре близкую активность (96.3%) демонстрирует Со-катализатор, промотированный Al2O3, что обусловлено, очевидно, сходством их характеристик – близостью степени восстановления кобальта, удельной поверхности и размеров кристаллитов Со0. Эффективность указанных катализаторов, вероятно, является следствием высокой степени восстановления Со и оптимального размера наночастиц металлического кобальта, который попадает в интервал 10–20 нм. Причиной большей активности катализатора Co–Ru/SiO2 может быть [54] структурная и электронная модификация каталитически активных центров в результате промотирования рутением.
Минимальная в ряду исследованных нами кобальтовых образцов производительность гибридного катализатора, скорее всего, обусловлена меньшей загрузкой активного металла – содержание Со в нем примерно в три раза ниже, чем в Co–Al2O3/SiO2, вследствие его разбавления цеолитом ZSM-5 (30 мас. %) и Al2O3 (35%). При этом, как и следовало ожидать, природа активных центров катализатора Co–Al2O3/SiO2 и его гибридного аналога одинакова – на это указывают совпадающие в пределах экспериментальных погрешностей значения энергии активации диссоциации аммиака (табл. 3).
При выборе для тестирования в разложении аммиака гибридного катализатора СФТ мы полагали, что наличие кислотных центров цеолита будет благоприятствовать адсорбции оснóвных молекул NH3, и, тем самым, увеличению скорости их превращения. Действительно, в изученном температурном интервале удельная каталитическая активность гибридного катализатора Co–Al2O3/SiO2(35%)/ZSM-5(30%)/Al2O3(35%), отнесенная к единице массы активного металла, оказалась выше, чем у Co–Al2O3/SiO2 (табл. 6). Отметим, что кислотные центры цеолита в гибридном катализаторе (т.е. центры преимущественной адсорбции аммиака) не содержат активного в его разложении металлического кобальта, локализованного на силикагеле. Тем не менее, их присутствие способствует увеличению удельной каталитической активности последнего по сравнению с традиционным катализатором СФТ Co–Al2O3/SiO2. Этот факт является косвенным подтверждением того обстоятельства, что активные центры катализатора СФТ и кислотные центры цеолита в зерне гибридного катализатора находятся в наноразмерной близости друг к другу [28, 31].
Таблица 6.
Удельная каталитическая активность кобальтовых катализаторов СФТ в реакции диссоциации аммиака в расчете на единицу массы активного металла в зависимости от температуры при ОСГ 3000 ч–1
| Т, °С | Удельная активность катализаторов, ${\text{ммол}}{{{\text{ь}}}_{{{{{\text{Н}}}_{{\text{2}}}}}}}{\text{ г}}_{{{\text{мет}}}}^{{ - 1}}{\text{ ми}}{{{\text{н}}}^{{ - 1}}}$ | |||
|---|---|---|---|---|
| Co–Ru/SiO2* | Ru/SiO2 | Co–Al2O3/SiO2 | Co–Al2O3/SiO2(35%)/ ZSM-5(30%)/Al2O3(35%) | |
| 400 | 1.51 | 100.5 | 3.7 | 10.6 |
| 450 | 2.23 | 207.4 | 6.2 | 17.7 |
| 500 | 4.27 | 430.2 | 18.8 | 42.4 |
| 550 | 5.65 | 684.7 | 28.3 | 86.7 |
Одна из насущных технологических задач получения водорода из аммиака – снижение температуры осуществления реакции. Оцененная эффективная энергия активации в исследованных нами системах (табл. 3) существенно меньше, чем для известных из литературы катализаторов на основе кобальта и рутения (табл. 7). Небольшая энергия активации позволяет проводить реакцию с приемлемой конверсией при более низких температурах. Минимальная энергия активации присуща катализатору Co–Ru/SiO2, именно этот образец демонстрирует максимальную активность в исследованном нами температурном диапазоне 400–550°С, несмотря на малую величину предэкспоненциального множителя (табл. 3). На основании вышесказанного можно в согласии с данными работы [61] заключить, что энергия активации оказывает большее влияние на эффективность разложения аммиака, чем предэкспоненциальный фактор.
Таблица 7.
Эффективные энергии активации Ea в Со- и Ru-содержащих системах
| Катализатор | Ea, кДж/моль | Источник |
|---|---|---|
| Co(90%)/Al2O3 | 123 | [25] |
| Co(90%)/Al2O3 | 148 | [55] |
| Co(7%)/AX-21 | 89 | [56] |
| Co(5%)/CNTs | 93 | [57] |
| Co(5%)/CNTs | 79 | [58] |
| Co(7%)/CNTs | 94 | [55] |
| Co(20%)/La-MgO | 167 | [59] |
| Co(7%)/MESO-C | 104 | [55] |
| Co(5%)/MgO-Al2O3 | 192 | [26] |
| Co(5%)/MgO-CeO2 | 81 | [26] |
| Co(5%)/MgO-La2O3 | 67 | [18] |
| Co(7%)/MSC-30 | 102 | [55] |
| Co(10%)/MWCNTs | 69 | [43] |
| Co(10%)/Ti-NT | 85 | [60] |
| Co(10%)/Ce0.6Zr0.3Y0.1O2 | 50 | [61] |
| Co/NС | 75 | [62] |
| Co(20%)-Al2O3(1%)/SiO2 | 67 | Настоящая работа |
| Co(20%)-Ru(1%)/SiO2 | 43 | Настоящая работа |
| Ru(4%)/Al2O3 | 125 | [63] |
| Ru(10%)/SiO2 | 82 | [19] |
| Ru(8.5%)/Al2O3 | 117 | [64] |
| Ru(1%)/SiO2 | 59 | Настоящая работа |
ЗАКЛЮЧЕНИЕ
Протестированные нами промышленные катализаторы синтеза Фишера–Тропша демонстрируют достаточно высокую эффективность в разложении аммиака. Активность и производительность по водороду кобальт-силикагелевых катализаторов СФТ уменьшаются в ряду Co–Ru/SiO2 ≥ Co–Al2O3/SiO2 > Ru/SiO2 > Co–Al2O3/ SiO2(35%)/ZSM-5(30%)/Al2O3(35%). Близкая активность Co–Ru/SiO2 и Co–Al2O3/SiO2 может быть следствием сходства основных параметров этих катализаторов, таких как степень восстановления кобальта, удельная поверхность и размеров кристаллитов Со0. Максимальная активность Co-Ru/SiO2 обусловлена, вероятно, синергетическим эффектом, вызванным добавками рутения. Относительно небольшие эффективные энергии активации, оцененные для всех катализаторов, позволяют осуществлять реакцию с приемлемой конверсией при умеренных температурах.
Список литературы
York R., Bell S.E. // Energy Res. Soc. Sci. 2019. V. 51. P. 40.
Lucentini I., Garcia X., Vendrell X., Llorca J. // Ind. Eng. Chem. Res. 2021. V. 60. № 51. P. 18560.
Hameer S., Niekerk J.L. // Int. J. Energy Res. 2015. V. 39. № 9. P. 1179.
Chang F., Gao W., Guo J., Chen P. // Adv. Mater. 2021. V. 33. № 50. P. 2005721.
Mukherjee S., Devaguptapu S.V., Sviripa A., Lund C.R., Wu G. // Appl. Catal. B: Env. 2018. V. 226. P. 162.
Silverstein R., Eliezer D., Glam B. // Energy Procedia. 2017. V. 107. P. 199.
Kobayashi N., Koyama M., Kobayashi K., Hojo T., Akiyama E. // Mater. Transact. 2022. V. 63. № 2. P. 247.
Robertson I.M., Sofronis P., Nagao A., Martin M.L., Wang S., Gross D.W., Nygren K.E. // Metallurg. Mater. Transact. A. 2015. V. 46. № 6. P. 1085.
Hwang H.T., Varma A. // Curr. Opin. Chem. Eng. 2014. V. 5. P. 42.
Фатеев В.Н., Алексеева О.К., Коробцев С.В., Серегина Е.А., Фатеева Т.В., Григорьев А.С., Алиев А.Ш. // Chemical Problems. 2018. № 4. С. 453.
UTL:https://ar2020.phosagro.ru/download/full-reports/ ar_ru_annual-report_pages_phosagro_2020.pdf (дата обращения: 09.09.2022 г.)
Lamb K.E., Dolan M.D., Kennedy D.F. // Int. J. Hydrogen Energy. 2019. V. 44. № 7. P. 3580.
Тарасов Б.П., Лотоцкий М.В., Яртысь В.А. // Российский химический журнал. 2006. Т. 50. № 6. С. 34.
Le T.A., Do Q.C., Kim Y., Kim T.W., Chae H.J. // Korean J. Chem. Eng. 2021. V. 38. № 6. P. 1087.
Lendzion-Bielun Z., Narkiewicz U., Arabczyk W. // Materials. 2013. V. 6. № 6. P. 2400.
Zhang J., Xu H., Ge Q., Li, W. // Catal. Commun. 2006. V. 7. № 3. P. 148.
Li G., Yu X., Yin F., Lei Z., Zhang H., He X. // Catal. Today. 2022. V. 402. P. 45.
Podila S., Driss H., Zaman S.F., Alhamed Y.A., AlZahrani A.A., Daous M.A., Petrov L.A. // J. Mol. Catal. A: Chem. 2016. V. 414. P. 130.
Choudhary T.V., Sivadinarayana C., Goodman D.W. // Catal. Lett. 2001. V. 72. № 3. P. 197.
Li X.K., Ji W.J., Zhao J., Wang S.J., Au C.T. // J. Catal. 2005. V. 236. № 2. P. 181.
Liu H., Wang H., Shen J., Sun Y., Liu Z. // Appl. Catal. A: Gen. 2008. V. 337. № 2. P. 138.
Ertl G., Knözinger H., Weitkamp J. // Weinheim: VCH. 1997. V. 2. P. 427.
Bell T.E. // Top. Catal. 2016. V. 59. № 15. P. 1438.
Lendzion-Bieluń Z., Pelka R., Arabczyk W. // Catal. Lett. 2009. V. 129. № 1. P. 119.
Gu Y.Q., Jin, Z., Zhang H., Xu R.J., Zheng M.J., Guo Y.M., Song Q.S., Jia C.J. // J. Mater. Chem. A. 2015. V. 3. № 33. P. 17172.
Podila S., Alhamed Y.A., AlZahrani A.A., Petrov L.A. // Int. J. Hydrogen Energy. 2015. V. 40. № 45. P. 15411.
Savost’yanov A.P., Yakovenko R.E., Sulima S.I., Bakun V.G., Narochnyi G.B., Chernyshev V.M., Mitchenko S.A. // Catal. Today. 2017. V. 279. P. 107.
Yakovenko R.E., Savost’yanov A.P., Narochniy G.B., Soromotin V.N., Zubkov I.N., Papeta O.P., Svetogorov R.D., Mitchenko S.A. // Catal. Sci. Technol. 2020. V. 10. P. 7613.
Савостьянов А.П., Яковенко Р.Е., Нарочный Г.Б., Бакун В.Г., Сулима С.И., Якуба Э.С., Митченко С.А. // Кинетика и катализ. 2017. Т. 58. № 1. С. 1.
Яковенко Р.Е., Зубков И.Н., Нарочный Г.Б., Папета О.П., Денисов О.Д., Савостьянов А.П. // Кинетика и катализ. 2020. Т. 61. № 2. С. 278.
Яковенко Р.Е., Зубков И.Н., Савостьянов А.П., Соромотин В.Н., Краснякова Т.В., Папета О.П., Митченко С.А. // Кинетика и катализ. 2021. Т. 62. № 1. С. 109.
Авербух А.Я., Тумаркина Е.С., Мухленов И.П., Копылев Б.А, Румянцева Е.С. Практикум по общей химической технологии: Учебное пособие для студентов вузов. Под ред. И.П. Мухленова. Москва: Высшая школа, 1979. 421 с.
Young R.A. The Rietveld Method. Oxford University Press, 1995. 298 p.
Schanke D., Vada S., Blekkan E.A., Hilmen A.M., Hoff A., Holmen A. // J. Catal. 1995. V. 156. № 1. P. 85.
PDF-2. The powder diffraction file TM. International Center for Diffraction Data (ICDD). PDF-2 Release 2012. web site: www.icdd.com. 2014. № (ICDD 42-1467)
PDF-2. The powder diffraction file TM. International Center for Diffraction Data (ICDD). PDF-2 Release 2012. web site: www.icdd.com. 2014. № 000-61-0765
PDF-2. The powder diffraction file TM. International Center for Diffraction Data (ICDD). PDF-2 Release 2012. web site: www.icdd.com. 2014. № 010-75-0921
Липпенс Б.К., Стеггерда И.И. Активная окись алюминия. Строение и свойства адсорбентов и катализаторов. Москва: Мир, 1973. С. 190.
Lin H.Y., Chen Y.W. // Mater. Chem. Phys. 2004. V. 85. № 1. P. 171.
Wong L., Tang L., Scarlett N.V., Chiang K., Patel J., Burke N., Sage V. // Appl. Catal. A: Gen. 2017. V. 537. P. 1.
Wang C., Liu N., Zhang, C., Liu X., Li X., Zhao X.S. // Appl. Surf. Sci. 2019. V. 497. P. 143776.
Li J., Liu Z., Wang R. // J. Colloid Interface Sci. 2018. V. 531. P. 204.
Bruce L.A., Hoang M., Hughes A.E., Turney T.W. // Appl. Catal. A: Gen. 1993. V. 100. № 1. P. 51.
Prieto G., Concepción P., Murciano R., Martínez A. // J. Catal. 2013. V. 302. P. 37.
Pei Y., Ding Y., Zhu H., Zang J., Song X., Dong W., Wang T., Lu Y. // Catal. Lett. 2014. V. 144. № 8. P. 1433.
Steen E., Sewell G.S., Makhothe R.A., Micklethwaite C., Manstein H., Lange M., O’Connor C.T. // J. Catal. 1996. V. 162. № 2. P. 220.
Li L., Zhu Z.H., Yan Z.F., Lu G.Q., Rintoul L. // Appl. Catal. A: Gen. 2007. V. 320. P. 166.
Su Q., Gu L., Yao Y., Zhao J., Ji W., Ding W., Au C.T. // Appl. Catal. B: Env. 2017. V. 201. P. 451.
Li L., Zhu Z.H., Lu G.Q., Yan Z.F., Qiao S.Z. // Carbon. 2007. V. 45. № 1. P. 11.
Zhang H., Alhamed Y.A., Al-Zahrani A., Daous M., Inokawa H., Kojima Y., Petrov L.A. // Int. J. Hydrogen Energy. 2014. V. 39. № 31. P. 17573.
Zhang H., Alhamed Y.A., Chu W., Ye Z., AlZahrani A., Petrov L. // Appl. Catal. A: Gen. 2013. V. 464. P. 156.
Podila S., Driss H., Zaman S.F., Alhamed Y.A., AlZahrani A.A., Daous M.A., Petrov L.A. // J. Mol. Catal. A: Chem. 2016. V. 414. P. 130.
Podila S., Driss H., Zaman S.F., Ali A.M., Al-Zahrani A.A., Daous M.A., Petrov L.A. // Int. J. Hydrogen Energy. 2017. V. 42. № 38. P. 24213.
Khan W.U., Alasiri H.S., Ali S.A., Hossain M.M. // Chem. Rec. 2022. V. 22. № 7. P. e202200030.
Zhang Z.S., Fu X.P., Wang W.W., Jin Z., Song Q.S., Jia C.J. // Sci. China Chem. 2018. V. 61. № 11. P. 1389.
Torrente-Murciano L., Hill A.K., Bell T.E. // Catal. Today. 2017. V. 286. P. 131.
Zhang H., Alhamed Y., Kojima Y., Al-Zahrani A.A., Petrov L.A. // Comptes rendus de l’Acad’emie bulgare des Sciences. 2013. V. 66. P. 519.
Zhang J., Müller J.O., Zheng W., Wang D., Su D., Schlögl R. // Nano Lett. 2008. V. 8. № 9. P. 2738.
Hu X.C., Wang W.W., Jin Z., Wang X., Si R., Jia C.J. // J. Energy Chem. 2019. V. 38. P. 41.
Lara-Garcia H.A., Mendoza-Nieto J.A., Pfeiffer H., Torrente-Murciano L. // Int. J. Hydrogen Energy. 2019. V. 44. № 57. P. 30062.
Huang C., Li H., Yang J., Wang C., Hu F., Wang X., Lu Z.H., Feng G., Zhang R. // Appl. Surf. Sci. 2019. V. 478. P. 708.
Li G., Zhang H., Yu X., Lei Z., Yin F., He X. // Int. J. Hydrogen Energy. 2022. V. 47. № 26. P. 12882.
McCullough K., Chiang P.H., Jimenez J.D., Lauterbach J.A. // Materials. 2020. V. 13. № 8. P. 1869.
Di Carlo A., Vecchione L., Del Prete Z. // Int. J Hydrogen Energy. 2014. V. 39. № 2. P. 808.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ