

Кинетика и катализ, 2023, T. 64, № 2, стр. 121-138
Каталитические методы получения высших 2-кетонов: перспективы Вакер-системы в реакции окисления α-олефинов
Ю. А. Родикова a, *, Е. Г. Жижина a
a ФГБУН ФИЦ Институт катализа им. Г.К. Борескова СО РАН
630090 Новосибирск, просп. Акад. Лаврентьева, 5, Россия
* E-mail: rodikova@catalysis.ru
Поступила в редакцию 26.09.2022
После доработки 21.11.2022
Принята к публикации 02.12.2022
- EDN: GNRGKD
- DOI: 10.31857/S0453881123020065
Аннотация
Проанализированы и обобщены разработанные за последние 60 лет методы получения неразветвленных 2-кетонов С6−С14 каталитическим окислением линейных α-олефинов. Особое внимание уделено рассмотрению каталитической Вакер-системы, важной для промышленного органического синтеза, и предложенных методов ее модификации. Обсуждены методы управления селективностью реакции, рассмотрена роль сокатализаторов, окислителей и лигандов.
Содержание
I. Введение
II. Традиционная Вакер-система и ограничения ее применимости
III. Альтернативные методы окисления α-олефинов
IV. Методы модификации Вакер-системы
Использование бинарных водно-органических сред
Окисление в присутствии катализаторов межфазного переноса
Системы PdCl2 + Ox: переход на альтернативные окислители
Бесхлоридные системы
Пероксосоединения ROOH в качестве стехиометрических окислителей
Использование ex situ Pd-комплексов в качестве катализаторов
Гетерогенные системы
Гетерополисоединения в качестве сокатализаторов
V. Заключение
I. ВВЕДЕНИЕ
Интенсивное развитие науки требует постоянной модернизации “устаревающих” технологий и их переоценки в соответствии с современными мировыми тенденциями. Усовершенствование существующих промышленных производств путем перехода на энерго-, природо- и ресурсосберегающие технологии – одно из перспективных направлений снижения антропогенного воздействия человека на окружающую среду, развиваемых современными исследователями [1, 2].
Неразветвленные 2-кетоны, содержащие от 6 до 14 атомов углерода в цепи, находят широкое применение в различных отраслях промышленности. Они выступают в качестве химических реагентов для проведения органических синтезов, производства спиртов, карбоновых кислот и их производных, используются при получении смазочных материалов, вкусовых и ароматических добавок [3]. Кроме того, они являются компонентами душистых веществ, добавляемых в средства по уходу за тканями для маскировки неприятных запахов [4].
Существующие способы синтеза 2-кетонов многочисленны. Например, в учебной литературе описаны методы гидратации алкинов, ацилирования металлорганических соединений, окисления или дегидрирования вторичных спиртов, присоединения реагентов Гриньяра к нитрилам и другие [5, 6] (схема 1 ). Однако большинство из них основано на использовании труднодоступного сырья, получение которого приводит к огромным шламовым отходам, и агрессивных токсичных реагентов. Лишь немногие – гидратация алкинов и окисление вторичных спиртов – были реализованы в промышленности. Тем не менее, у этих методов также есть свои ограничения, что нацеливает современных исследователей на их модификацию [7].

Схема 1 . Методы получения 2-кетонов.
Переход на каталитические методы получения востребованных органических соединений из доступных предшественников с одновременным снижением числа промежуточных стадий и образующихся отходов – одно из современных направлений эволюции синтетической химии, реализующихся в свете развития “зеленой” химии [8, 9]. По этой причине значительное внимание привлекает синтез 2-кетонов окислением соответствующих 1-алкенов (α-олефинов) в присутствии разнообразных Pd-содержащих каталитических систем, являющихся модификациями традиционной гомогенной хлоридной PdCl2/CuCl2/O2Вакер-системы [10]. В современной литературе доступен ряд обзоров и монографий, посвященных Вакер(Wacker)-окислению α-олефинов, однако они рассматривают эту систему с точки зрения получения различных производных в целом [11–16]. Для полноценного развития данного направления и создания экологичных технологий синтеза высших 2-кетонов необходим комплексный анализ разработанных методов и подходов, выявление их особенностей и недостатков. В связи с этим целью настоящей работы является обзор наиболее эффективных каталитических методов преобразования линейных α-олефинов в востребованные 2-кетоны С6−С14, описанных в литературе. Основное внимание уделено важной для промышленного органического синтеза и металлокомплексного катализа Вакер-системе, методам повышения ее активности и селективности (S).
II. ТРАДИЦИОННАЯ ВАКЕР-СИСТЕМА И ОГРАНИЧЕНИЯ ЕЕ ПРИМЕНИМОСТИ
Один из перспективных методов синтеза 2-кетонов, который интенсивно исследуется с конца 50-х гг. XX века, – стехиометрическая реакция хлористого палладия с олефинами в водном растворе, при которой PdCl2 восстанавливается до металла (I). В качестве окислителя для восстановленной формы палладия первоначально было предложено использовать соль двухвалентной меди (соли других металлов показали более низкую эффективность), которая регенерирует палладий (II), переходя в одновалентную форму, а затем сама легко окисляется кислородом (III), замыкая каталитический цикл. Такой окислительно-восстановительный процесс (IV), детально разработанный на примере синтеза ацетальдегида из этилена и внедренный в промышленность в 1958–60-х гг. фирмами “Wacker-Chemie” и “Farbwerke Hoechst”, получил название Вакер-процесса11 [10, 17].
(I)
$\begin{gathered} {\text{AlkCH = C}}{{{\text{H}}}_{{\text{2}}}}\,{\text{ + PdC}}{{{\text{l}}}_{{\text{2}}}}{\text{ + }}{{{\text{H}}}_{{\text{2}}}}{\text{O}} \to \\ \to {\text{AlkC}}\left( {{\text{ = O}}} \right){\text{C}}{{{\text{H}}}_{{\text{3}}}}\,{\text{ + P}}{{{\text{d}}}^{{\text{0}}}}{\text{ + 2HCl}}, \\ \end{gathered} $(II)
${\text{P}}{{{\text{d}}}^{{\text{0}}}}{\text{ + 2CuC}}{{{\text{l}}}_{{\text{2}}}} \to {\text{PdC}}{{{\text{l}}}_{{\text{2}}}}{\text{ + 2CuCl}},$(III)
${\text{2CuCl + 1/2}}{{{\text{O}}}_{{\text{2}}}}{\text{ + 2HCl}} \to {\text{2CuC}}{{{\text{l}}}_{{\text{2}}}}{\text{ + }}{{{\text{H}}}_{{\text{2}}}}{\text{O}},$(IV)
$\begin{gathered} {\text{AlkCH = C}}{{{\text{H}}}_{{\text{2}}}}{\text{\; + \;1/2}}{{{\text{O}}}_{{\text{2}}}}\begin{array}{*{20}{c}} {\xrightarrow{{{\text{PdC}}{{{\text{l}}}_{{\text{2}}}}{\text{,\;CuC}}{{{\text{l}}}_{{\text{2}}}}}}} \end{array} \\ \to {\text{AlkC( = O)C}}{{{\text{H}}}_{{\text{3}}}}{\text{.}} \\ \end{gathered} $В первой пилотной установке окисление олефинов исследовали в гетерогенном варианте путем пропускания смеси сырого этилена и кислорода над твердотельным катализатором Pd/C. Однако дезактивация и короткий срок службы катализатора привели к дальнейшему изучению процесса в гомогенном варианте [18]. В настоящее время Вакер-окисление этилена по реакции (IV) успешно реализуется с использованием барботажных колонн в двух- и одностадийном исполнениях (рис. 1) при температуре 100–130°С и давлении 0.3–1.0 МПа, обеспечивая образование ацетальдегида с выходом выше 95% [19]. Данный процесс пришел на смену гидратации ацетилена по М.Г. Кучерову (V) [20] и методу А.Е. Фаворского и М.Ф. Шостаковского (VI) [21], применение которых стало сокращаться в связи с их меньшей экологичностью и более высокой стоимостью ацетилена при его меньшей доступности [22]:
(V)
${\text{CH}}{\kern 1pt} \equiv {\kern 1pt} {\text{CH + \;}}{{{\text{H}}}_{{\text{2}}}}{\text{O\;}}\xrightarrow{{{\text{HgS}}{{{\text{O}}}_{{\text{4}}}},{\text{\;}}{{{\text{H}}}_{{\text{2}}}}{\text{S}}{{{\text{O}}}_{{\text{4}}}}}}{\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{C}}\left( {{\text{ = O}}} \right){\text{H,}}$(VI)
${\text{CH}}{\kern 1pt} \equiv {\kern 1pt} {\text{CH\; + \;}}{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{5}}}}{\text{OH}}\xrightarrow{{{\text{КOH}}}}{\text{\;C}}{{{\text{H}}}_{{\text{2}}}} = {\text{CH}} - \,{\text{O}}{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{5}}}}\xrightarrow{{{{{\text{H}}}_{{\text{3}}}}{{{\text{O}}}^{{\text{ + }}}}}}{\text{\;\;C}}{{{\text{H}}}_{{\text{3}}}}{\text{C}}\left( {{\text{ = O}}} \right){\text{H\; + \;}}{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{5}}}}{\text{OH}}{\text{.}}$Рис. 1.
Схема одностадийного (а) и двухстадийного (б) процесса окисления этилена в ацетальдегид: 1 – реактор, 2 – холодильник, 3 – абсорбер, 4 – отпарная колонна, 5 – ректификационная колонна, 6 – регенератор.

Первая стадия Вакер-процесса (IV), разработанного на примере этилена, представляет собой образование комплекса между этиленом и Pd-катализатором, находящимся в растворе в виде тетрахлоропалладата [PdCl4]2–, с отщеплением Cl–. Последующие стадии, как и их описание в виде кинетических уравнений, сильно зависят от реакционных условий и используемого модельного субстрата [15, 18].
Так, согласно ряду исследований, значительный вклад в которые внесла группа акад. И.И. Моисеева [23–25], следующим этапом процесса является замещение молекулой воды второго хлоридного лиганда (L) в координационной сфере палладия, находящегося в транс-положении по отношению к координированному этилену. Последующее отщепление протона Н+ приводит к формированию транс-комплекса [(С2Н4)PdCl2(OH)]– (I), трансформация которого с образованием частиц β-гидроксиэтилпалладия [HOСН2СН2PdCl2]– (II) через внедрение α-олефина по связи Pd–OH требует цис-расположения данных лигандов. Для объяснения возможности дальнейшего протекания процесса было выдвинуто предположение о существовании этапа цис-транс-изомеризации транс-комплекса I, предшествующего образованию II. Один из предложенных механизмов этой стадии рассматривает возможность образования тригональной бипирамиды в результате атаки молекулы воды по пятой координате квадратного транс-комплекса I [26, 27]. По другому механизму [18] в координационной сфере транс-комплекса I происходит замещение второй молекулой воды хлоридного лиганда в цис-положении по отношению к α-олефину с последующей диссоциацией Н+ иона. Получившийся комплекс затем претерпевает замещение транс-ОН-лиганда обратно на хлорид-ион в присутствии Н+ с отщеплением молекулы воды и появлением комплекса I с цис-расположением лигандов. Трансформация последнего через реакцию цис-гидроксипалладирования дает комплекс II, внутри которого происходит 1,2-миграция гидрид-иона с образованием [СН3СН(OH)PdCl2]– (III). Точный механизм этапов формирования I и II, а также завершающего этапа – восстановительного элиминирования комплекса III с выделением ацетальдегида – до сих пор остается дискуссионным.
С другой стороны, использование высоких концентраций хлорид-ионов, стерически затрудненных субстратов, реакционных сред с низким содержанием воды в присутствии конкурентных лигандов и т.д. может затруднять стадию цис-транс-изомеризации транс-комплекса I, приводя к протеканию процесса через транс-гидроксипалладирование. Существующие гипотезы о механизмах реакций Pd-катализируемого окисления детально рассмотрены в работах [15, 28].
Реакция Вакер-окисления сыграла значительную роль в развитии гомогенного металлокомплексного катализа. Показав высокую эффективность, она стала отправной точкой для разработки различных Pd-катализируемых процессов, в том числе окисления высших линейных 1-алкенов с получением карбонильных соединений [29, 30]. Традиционное Вакер-окисление α-олефинов по наименее гидрированному атому углерода двойной связи с образованием 2-кетонов открывало широкую перспективу разработки эффективных методов синтеза таких соединений по аналогии с ацетальдегидом. Однако использование системы PdCl2/CuCl2/O2 для окисления неразветвленных 1‑алкенов, содержащих более трех атомов С в цепи, показало, что ее активность и селективность падают с ростом количества углерода. Так, в случае пропилена его окисление при 50–120°С и давлении 9.8 МПа приводило к образованию ацетона с выходом 99%. Двухстадийная технология была разработана фирмой “Hoechst AG” [31]. Окисление же н-бутенов при 100°С и давлении до 4.5 МПа давало метилэтилкетон уже с S ≈ 80% [19].
Анализ полученных данных выявил ряд недостатков традиционной Вакер-системы при ее адаптации к окислению высших α-олефинов, наиболее существенными из которых стали высокая коррозионная активность, низкие скорость реакции и выход целевого кетона, осаждение металлического палладия и CuCl в ходе реакции, образование хлорированных производных, а также Pd-катализируемая изомеризация двойной связи.
Первоначально попытки увеличения активности каталитической системы были основаны на повышении концентрации медных солей, необходимых для эффективного протекания реакции (II). Однако это приводило к росту коррозионной активности гомогенной системы и количества образующихся хлоркетонов (при окислении н-бутенов, например, выход хлоркетонов достигал 10–30%). С другой стороны, для большей стабильности системы и предотвращения кластеризации переходных атомов палладия использовали кислоту HCl, что оказывало благоприятное влияние на реакции (II) и (III), а также способствовало возрастанию устойчивости хлоридных комплексов Cu(I). Тем не менее, присутствие HCl снижало скорость реакции (I), имеющую обратную пропорциональность по отношению к [Н+], и вызывало рост хлорирующей способности Cu(II).
Таким образом, возможные методы контроля активности и селективности гомогенной хлоридной Вакер-системы без изменения ее состава оказались малоэффективными из-за их противоположного влияния на этапы (I)–(III) и отдельные ее параметры. Это ограничило распространение традиционной Вакер-системы на окисление других 1-алкенов помимо этилена и пропилена.
III. АЛЬТЕРНАТИВНЫЕ МЕТОДЫ ОКИСЛЕНИЯ α-ОЛЕФИНОВ
В 1975 г. H. Rogers и соавт. [32] предприняли попытку разработать метод окисления терминальных олефинов, включающий реакции, отличные от традиционного Вакер-процесса. В качестве объекта для исследований была выбрана система HgII-содержащий компонент/реагент Джонса. В присутствии предложенной системы (EtCO2)2Hg/CrO3–H2SO4–H2O на первом этапе происходит оксимеркурирование олефина с образованием производного 2-гидроксиалкилртути, которое затем окисляется под действием реагента Джонса в кето-производное. Кислотный гидролиз последнего приводит к соответствующему 2-кетону и исходному HgII-содержащему компоненту. За 8 ч реакции при 25°С в среде ацетон/H2O выход 2-октанона в такой системе составил 82%.
Позднее, в 1978 г., H. Mimoun и соавт. показали [33], что изолированные или in situ образовавшиеся частицы Rh-O2 способны окислять терминальные олефины в 2-кетоны в мягких условиях по аналогии с Вакер-системой. Ряд высших 2-кетонов с S ≥ 98% при умеренной конверсии был получен за 4 ч при использовании трехкомпонентной каталитической системы RhCl3·3Н2О/Cu(ClO4)2([(CH3)2N]3PO)4/О2 (схема 2 а).
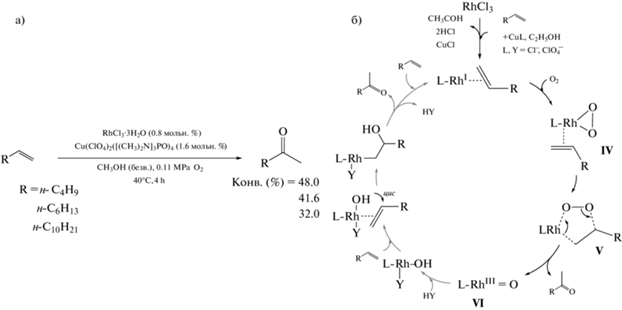
Схема 2 . Окисление α-олефинов в присутствии каталитической системы RhCl3·3Н2О/Cu(ClO4)2([(CH3)2N]3PO)4/О2 (а) и схема каталитического цикла (б).
Авторы предложили механизм (схема 2 б), согласно которому на первом этапе происходит окисление растворителя до ацетальдегида с одновременным формированием комплекса RhI–олефин. Следующий этап – активация молекулярного кислорода – протекает посредством образования пероксокомплекса IV, превращающегося в металлоцикл V. Возможность возникновения подобного металлоцикла была исследована в работе [34] на примере окисления 1-алкенов и тетрацианоэтилена в присутствии [Rh(AsPh3)4O2]+A– (A = ${\text{ClO}}_{4}^{ - }$, ${\text{PF}}_{6}^{ - }$). Разложение металлоцикла дает целевой кетон и оксо-производное VI, участвующее в окислении второй молекулы субстрата по классическому Вакер-механизму. Было установлено, что увеличение объема воды отрицательно влияет на активность каталитической системы, в то время как введение дегидратирующего агента (2,2-диметоксипропана) и небольших количеств HCl производит противоположный эффект. Показано, что содержание продуктов изомеризации двойной связи не превышает 10% от непрореагировавшего субстрата, причем их окисления до соответствующих кетонов не наблюдается. Тем не менее, ряд недостатков, среди которых низкая конверсия субстрата (менее 60%), окисление растворителя до ацетальдегида, а также осаждение CuCl в ходе реакции (до 85%), были выявлены для предложенной системы.
IV. МЕТОДЫ МОДИФИКАЦИИ ВАКЕР-СИСТЕМЫ
В последующие годы большинство предложенных методов получения высших 2-кетонов было основано на реакциях Pd-катализируемого окисления α-олефинов с применением различных добавок и модификаций, нацеленных на решение обнаруженных для традиционной системы проблем.
Использование бинарных водно-органических сред
Ограничения традиционной хлоридной Вакер-системы привели к интенсивному исследованию различных методов ее модификации. Одна из причин снижения активности и селективности системы PdCl2/CuCl2/O2 заключается в ухудшении растворимости субстрата в водном растворе катализатора с ростом длины углеродной цепи. Для решения проблемы уменьшения растворимости высших α-олефинов в воде и, как следствие, падения скорости и эффективности взаимодействия субстрата с водным раствором катализатора было предложено использовать бинарные водно-органические среды.
Так, в 1964 г. W. Clement и C. Selwitz изучали окисление 1-додецена в присутствии традиционной Вакер-системы PdCl2/CuCl2/O2, но применяли в качестве растворителя смесь Н2О/N,N-диметилформамид (ДМФА) [35]. В результате прикапывания субстрата в течение 3.5 ч при температуре 60–70°С и объемном содержании воды в смеси 12–17% авторам удалось получить выход 2-деканона 87%. Было отмечено, что использование других смешивающихся с водой растворителей, таких как диметилсульфоксид, ацетон, уксусная кислота (АсОН), тетрагидрофуран (ТГФ), диоксан и ацетонитрил (CH3CN), приводит к уменьшению наблюдаемого выхода 2-кетона по сравнению с ДМФА. Введение субстрата прикапыванием позволило авторам значительно снизить скорость конкурирующей реакции изомеризации 1-алкена, а применение бинарной водно-органической системы улучшило массоперенос между субстратом и катализатором.
При окислении 1-децена в присутствии аналогичной системы в среде Н2О/сульфолан (1 : 1), описанном в патенте компании “Phillips Petroleum Company” [36], был получен 2-деканон с S ≈ ≈ 96.1–97.8% за 4 ч при 80°С и давлении кислорода 0.83–1.03 МПа. Конверсия субстрата составила ≤56%.
Таким образом, использование бинарных водно-органических сред с подходящим коэффициентом взаимной растворимости обеспечило улучшение массообмена между средами, сократив тем самым время реакции и повысив выходы целевых кетонов. Это закрепило применение бинарных сред практически во всех последующих работах. Однако другие проблемы, среди которых образование побочных хлорпроизводных и высокая коррозионная активность, по-прежнему сохранялись.
Окисление в присутствии катализаторов межфазного переноса
Наряду с проведением процесса окисления в бинарных средах в 70–90-х гг. ХХ в. ряд исследователей также предпринял попытку решить проблему низкой скорости реакции окисления высших α-олефинов в водной среде путем использования катализаторов межфазного переноса (КМП), призванных облегчать взаимодействие между компонентами, находящимися в разных фазах. Различные соединения, такие как циклодекстрины (ЦД), полиэтиленгликоли (ПЭГ), каликс[4]арены и четвертичные аммониевые соли, были изучены с этой целью (схема 3 ).

Схема 3 . Соединения, применяемые в качестве КМП в реакциях окисления α-олефинов: а – α-циклодекстрин (α-ЦД); б – β-циклодекстрин (β-ЦД); в – γ-циклодекстрин (γ-ЦД); г – полиэтиленгликоль (ПЭГ-n); д – модифицированный каликс[4]арен; е – бромид гексадецилтриметиламмония (ЦТАБ).
Так, в 1986 г. авторы [37] показали возможность получения 2-кетонов С8–С10 с выходом 62–76%, окисляя соответствующие 1-алкены с помощью каталитической системы PdCl2/CuCl2/O2/α-ЦД при температуре 60–75°С в течение 8–10 ч. Увеличение длины углеродной цепи выше С10 привело к резкому снижению выхода кетонов до значений ниже 15%.
В том же году H. Zahalka и соавт. исследовали окисление ряда диенов и терминальных алкенов С4+ в водной среде в присутствии аналогичной системы с добавлением β-ЦД [38]. Окисляя 1-децен в течение 48 ч при 65°С и атмосферном давлении, авторы получили 2-деканон с выходом 61%. В качестве побочных продуктов наблюдалось образование изомерных деценов, количество которых значительно увеличивалось с ростом длины углеродной цепи субстрата.
В отличие от систем, содержащих ЦД в роли КМП, использование ПЭГ оказалось менее эффективным. Так, авторы [39] получили смесь 2-, 3- и 4,5-деканонов с выходом 68% при селективности образования 2-деканона 88%, окисляя 1-децен в водной среде при 65°С системой PdCl2/CuCl2/O2 в присутствии ПЭГ-400 как КМП. Было отмечено, что замена PdCl2 на Pd(OAc)2 приводит к аналогичному результату, в то время как рост (ПЭГ-1000) или снижение (ПЭГ-200) длины цепи полиэтиленгликоля способствует снижению суммарного выхода кетонов.
Позднее А. Максимов и соавт. в своей работе [40] изучили окисление линейных 1-алкенов С6–С12 в водной среде, применяя CuCl2, O2 и водорастворимый комплекс палладия(II) с каликс[4]ареном, модифицированным пара-бромметилбензонитрилом и 1,3-пропансультоном (VII). Синтез комплекса с соотношением палладий : лиганд = = 1 : 2 проводили интенсивным перемешиванием смеси PdCl2 и лиганда VII при 60°С в течение 2 ч. С участием такой системы при 50°С и давлении кислорода 0.5 МПа за 2 ч реакции выход 2-гептанона на 1-ом и 2-ом циклах реакции составил 89 и 87% соответственно. Достижение близких значений выхода 2-гексанона и 2-октанона (88 и 83%) потребовало увеличения времени реакции до 8 ч.
Как видно из описанных примеров, использование ЦД и каликс[4]аренов в составе Вакер-системы в качестве КМП позволяет осуществлять окисление 1-алкенов в отсутствие органического растворителя. Такой подход обеспечивает образование ряда высших 2-кетонов со значениями конверсии и селективности более 80%, а также облегчает выделение продуктов. Следует отметить, что применение подобных КМП ограничивается размерами переносимой молекулы (наблюдается субстратная селективность). Это ведет к резкому снижению выхода продукта и/или значительному увеличению времени реакции при окислении 1-алкенов, не соответствующих полости “хозяина”. Также в таких системах происходит конкурентное заполнение полости ЦД при накоплении продукта (2-кетона), что требует введения в систему повышенных количеств КМП. Потенциальным решением подобной проблемы является модификация гидроксильных групп ЦД, приводящая к снижению их взаимодействия с карбонильными производными и увеличению стабильности комплекса “гость–хозяин”. Однако это устраняет проблему лишь частично.
В патенте [41] 1979 г., принадлежащем компании “Phillips Petroleum Company”, показана возможность окисления олефинов кислородом в двухфазной водно-органической среде состава Н2О/хлорбензол (1 : 1) при использовании каталитической системы PdCl2/CuCl2/О2 в присутствии добавок хлорида щелочного или щелочноземельного металла (NaCl, LiCl, KCl, CaCl2 и др.) в качестве источника Cl–-ионов и бромида гексадецилтриметиламмония (ЦТАБ) в качестве сурфактанта. В такой системе 2-гексанон образовывался с S > 98% при температуре 105°С и давлении кислорода 0.55 МПа за 5–6 ч. Конверсия 1-гексена при этом варьировалась в интервале 67–75%, превышая в 3.5 раза значения, достигаемые в отсутствие сурфактанта.
В последующих патентах, принадлежащих той же компании, была заявлена возможность получения различных кетонов, содержащих от 3 до 20 атомов углерода, в том числе 2-кетонов, окислением олефинов кислородом или кислородсодержащим газом в присутствии каталитической системы Pd-содержащий компонент/Cu-содержащий компонент/сурфактант и смеси растворителей состава вода/фторуглеродное соединение/органический сорастворитель [42, 43]. В качестве примера в патентах фигурирует реакция окисления 1-додецена с селективностью образования 2-додеканона выше 99% при конверсии субстрата 98%, которую проводили при температуре 120°С и давлении кислорода 2.76 МПа в течение 2 ч в присутствии каталитической системы PdCl2/CuCl2/О2/ЦТАБ в среде Н2О/перфторпентан/декан. В отсутствие перфторпентана в составе растворителей наблюдается снижении конверсии субстрата на 13%.
Описанные примеры показывают, что КМП играют важную роль в системах, компоненты которых находятся в разных фазах. В ряде случаев они способны интенсифицировать взаимодействие органического субстрата с водным раствором каталитической системы без использования дополнительных сорастворителей. Следует отметить, что разработка катализаторов, объединяющих в одной молекуле свойства металлокомплекса и поверхностно-активного вещества со способностью к образованию включений типа “гость–хозяин”, представляется весьма перспективной с точки зрения достижения кооперативного связывания и возможности предориентации субстрата. Тем не менее, требуются дальнейшие исследования в данной области.
Системы PdCl2 + Ox: переход на альтернативные окислители
Важная особенность традиционной Вакер-системы – присутствие хлорид-ионов Cl– в высоких концентрациях, которые не только являются лигандами для Pd2+ и Cu+, стабилизируя их в растворе в виде ${\text{P}}{{{\text{d}}}^{{{\text{II}}}}}{\text{Cl}}_{4}^{{2 - }}$ и ${\text{C}}{{{\text{u}}}^{{\text{I}}}}{\text{Cl}}_{2}^{ - }$, но и обеспечивают с термодинамической и кинетической точек зрения возможность регенерации Pd0 → Pd2+ и функционирования ионов Cu2+ в качестве обратимо действующих окислителей (ОДО) по уравнениям (II) и (III) за счет достижения подходящих значений окислительно-восстановительных потенциалов пар PdII/Pd0 и CuII/CuI [44]
В то же время комплексообразование α-олефина с Pd-катализатором с появлением [(олефин)PdCl3]– и последующая координация Н2О с переходом к комплексу [(олефин)PdCl2(Н2О)] приводят к отщеплению двух хлорид-ионов. Но именно присутствие Cl– одновременно служит причиной и ряда значимых недостатков хлоридной Вакер-системы при окислении олефинов > С4, среди которых образование хлорированных производных и снижение скорости реакции, имеющей обратную квадратичную зависимость по отношению к концентрации хлорид-ионов [18, 45]:
Для снижения количества хлорпроизводных в ряде работ были исследованы системы PdCl2 + + окислитель (Ox), в которых в качестве Ox использовали бесхлоридные соединения.
Так, T. Mitsudome и соавт. предложили простую и регенерируемую систему для окисления терминальных алкенов в соответствующие 2-кетоны без применения сокатализаторов или восстанавливающих агентов, состоящую из PdCl2 и молекулярного кислорода в качестве единственного реокислителя Pd0 [46]. В такой системе в среде N,N-диметилацетамид (ДМАА)/Н2О за 3 ч реакции при 80°С и давлении кислорода 0.61 МПа авторам удалось получить ряд высших 2-кетонов С6–С20 с выходом 81–88% при конверсии соответствующих 1-алкенов 82–88%. Повторное использование каталитической системы на 2-ом и 3-ем циклах в реакции окисления 1-децена показало умеренное снижение выхода 2-деканона до ~80% по сравнению с первым циклом (85%). Отделение непрореагировавшего субстрата и продуктов реакции проводили экстракцией н-гептаном с последующей декантацией полученного раствора. Было показано, что начальная скорость реакции зависит от давления кислорода и концентрации катализатора и не зависит от концентрации субстрата. Применение ДМАА в качестве растворителя, по предположению авторов, облегчает регенерацию частиц Pd0 кислородом, а также препятствует их агрегации.
R. Fernandes и соавт. уделили значительное внимание поиску альтернативного окислителя для регенерации палладия, способного заменить хлорид меди(II) и предотвратить появление различных хлорпроизводных. В качестве таких соединений были предложены сульфат железа(III) [47] и оксид хрома(VI) [48], которые при окислении 1-алкенов С8, С10 и С14 обеспечивали образование соответствующих 2-кетонов в присутствии PdCl2 с выходом до 96% в среде CH3CN/H2O (7 : 1) за 1–6.5 ч реакции при умеренном нагревании. Следует отметить, что в случае Fe2(SO4)3 окисление проводили в атмосфере N2, поскольку в кислородной атмосфере процесс протекал с более низкой эффективностью. Так, за 30 ч реакции выход 2-деканона в атмосфере О2 составил 75%.
A. Smith и соавт. исследовали окисление 1-децена в мягких условиях в присутствии системы PdCl2/Cu(OAc)2/О2 в среде AcNMe2/H2O (7 : 1) [49]. Выход 2-деканона при этом составил 84%.
Приведенные примеры показывают, что существуют различные альтернативные окислители, способные обеспечивать регенерацию Pd0 по уравнению (II) с достаточной эффективностью. Однако вопрос регенерации предлагаемых Ox по уравнению (III) в реакционных условиях с замыканием каталитического цикла реакции (IV) остается во многих работах малоизученным.
Бесхлоридные системы
В традиционной Вакер-системе регенерация палладия по уравнению (II) в действительности протекает с возникновением активных частиц ${\text{PdCl}}_{4}^{{2 - }}$, которые в ходе реакции (I) диссоциируют с образованием двух эквивалентов Cl–, снижающих скорость реакции. Переход на системы PdCl2/бесхлоридный Ox не позволяет полностью преодолеть недостатки системы PdCl2/CuCl2/O2, поскольку в отсутствие достаточного количества хлорид-ионов нарушается термодинамическое равновесие между комплексами Pd2+ и Pd0, что приводит к осаждению последнего. В связи с этим целью многочисленных исследований стал поиск альтернативных компонентов каталитической системы PdX2 + Ox – лигандов, обладающих возможностью обеспечить аналогичную хлорид-ионам функцию и эффективность протекания реакций (I) и (II), а также окислителей Ox, способных регенерировать палладий, превращая стехиометрическую реакцию взаимодействия палладия с олефинами в каталитическую. Разработка стабильной бесхлоридной системы, столь же эффективной, как Вакер-система в реакции получения ацетальдегида, стала приоритетной задачей.
Компания “Catalytica Associates” предложила гомогенную каталитическую систему [50, 51], содержащую помимо PdX2 и CuY2 соединение из класса нитрилов RC≡N в качестве лиганда. Различные анионы, такие как ${\text{BF}}_{4}^{ - }$, CF3COO–, CH3COO–, ${\text{SO}}_{4}^{{2 - }}$ и ${\text{NO}}_{3}^{ - }$, исследовались как X и Y. Было установлено, что основными побочными продуктами в такой системе являются изомерные 2- и 3-алкены, которые затем превращаются в соответствующие кетоны. В отсутствие нитрила регенерация Pd0 → Pd2+ не наблюдается, тогда как увеличение его содержания в системе не оказывает влияния на конверсию, но способствует росту селективности получения целевого 2-кетона. Полная конверсия гексена-1 при образовании 2-гексанона с S = 53.4% была достигнута за 8 ч реакции при температуре 60°С и давлении кислорода 0.55 МПа в присутствии системы Pd(CF3COO)2/Cu(BF4)2/CH3CN.
Позднее, в 1990 г., D. Miller и D. Wayner исследовали возможность ускорения реакции окисления циклических и терминальных олефинов введением добавок неорганических кислот с некомплексообразующими анионами [52]. Авторы показали, что окисление 1-децена с помощью каталитической системы Pd(OAc)2/п-бензохинон (БХ) в присутствии сильных минеральных кислот приводит к многократному ускорению реакции (~42 раза) при высоких значениях конверсии и селективности: $\begin{array}{*{20}{c}} {{\text{HB}}{{{\text{F}}}_{{\text{4}}}}} \\ {{\text{99\;и\;84\% }}} \end{array}$ ≥ $\begin{array}{*{20}{c}} {{\text{HN}}{{{\text{O}}}_{{\text{3}}}}} \\ {{\text{93\;и\;83\% }}} \end{array}$ > $\begin{array}{*{20}{c}} {{\text{HCl}}{{{\text{O}}}_{{\text{4}}}}} \\ {{\text{98\;и\;79\% }}} \end{array}$ > > $\begin{array}{*{20}{c}} {{{{\text{H}}}_{{\text{2}}}}{\text{S}}{{{\text{O}}}_{{\text{4}}}}} \\ {{\text{93\;и\;64\% }}} \end{array} \gg $ $\begin{array}{*{20}{c}} {{\text{HCl}}} \\ {{\text{0\;и\;--}}} \end{array}$. Реакцию проводили в среде ацетонитрил/Н2О (7 : 1) при 60°С в течение 10 мин при мольном отношении субстрат/Pd2+/БХ/кислота равном 1 : 0.02 : 0.9 : 1.25. В качестве побочных продуктов наблюдалось образование 5–10% 3-деканона и 3–4% деканаля, а также 4-деканона (для H2SO4, 14%). Было установлено, что замена Pd(OAc)2 на Pd(NO3)2 или PdSO4 не влияет на активность каталитической системы и суммарный выход кетонов, в то время как повышение концентрации кислоты (HClO4) способствует монотонному росту содержания кетонов с перегибом в интервале концентраций кислоты 0.30–0.45 М. Наблюдаемое ускорение реакции авторы связывают с возможным возрастанием электрофильности PdII и его реакционной способности по отношению к олефинам, которое может достигаться за счет протонирования координированного лиганда, легкости удаления некомплексообразующих анионов или увеличения скорости обмена лигандами в присутствии Н+.
Данный метод, несмотря на высокую эффективность, имеет ряд ограничений, связанных со стабильностью реагентов и продуктов реакции в кислых средах, коррозионной активностью сильных минеральных кислот, а также трудностями выделения продуктов реакций.
Похожая система была представлена B. Morandi и соавт. в патенте [53]. Авторы получили 86% выход 2-додеканона с S = 97.5%, окисляя 1-додецен при комнатной температуре в течение 16 ч с использованием каталитической системы Pd(CH3CN)4(BF4)2/БХ. Дикатионный комплекс палладия синтезировали смешением Pd(OAc)2 c HBF4 в системе растворителей ДМАА/CH3CN/H2O с соотношением 3.5 : 3.5 : 1.
Окисление 1-октена с выходом соответствующего кетона 83% за 6 ч было выполнено авторами [54] с применением системы Pd(OAc)2/О2 (0.1 МПа) в среде ДМСО/Н2О (10 : 1) в присутствии трифторуксусной кислоты при 70°С.
Позднее D. Chaudhari и R. Fernandes исследовали систему Pd(OAc)2/периодинан Десса–Мартина (VIII) в среде CH3CN/H2O (7 : 1) при температуре 50°С в атмосфере азота [55] (схема 4 ). За 1 ч реакции такая система обеспечила выход 2-тетрадеканона 95%, однако выходы кетонов С8 и С10 составили 74 и 79% соответственно.
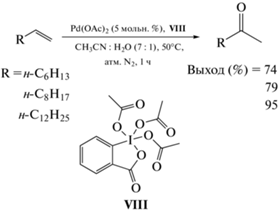
Схема 4 . Окисление α-олефинов в присутствии каталитической системы Pd(OAc)2/периодинан Десса–Мартина.
В 2019 г. авторы [56] описали модифицированную Вакер-систему, состоящую из комплекса Pd(CH3CN)2Cl2 и фоторедокс-катализатора [Ir(ppy)2(bpy)]PF6, способного окислять Pd0 обратно в активную форму Pd2+ под действием света в присутствии О2 и Н2О. При облучении такой системы при 120°С в среде ДМФА/Н2О (6 : 1) в течение 36 ч с использованием люминесцентной лампы мощностью 11 Вт выход 2-тетрадеканона составил 87%.
Из представленных примеров видно, что большинство предложенных систем обеспечивают сохранение или улучшение показателей хлоридной системы PdCl2/CuCl2/O2 при переходе к высшим α-олефинам, решая при этом проблему высокой коррозионной активности и образования хлорпроизводных. Замещение хлорид-ионов нитрильными лигандами при одновременном применении бесхлоридных окислителей является в целом эффективным методом модификации традиционной Вакер-системы, способствующим сохранению ее активности и стабильности в ходе реакции. При этом используемый лиганд должен соответствовать ряду требований: 1) связывать Pd0, препятствуя его осаждению в ходе реакции; 2) обладать умеренной комплексообразующей силой и беспрепятственно отщепляться при образовании комплекса PdII-олефин; 3) иметь низкую способность к смещению положительного заряда Pd2+. Наиболее предпочтительны монодентатные электронодефицитные лиганды.
Пероксосоединения ROOH в качестве стехиометрических окислителей
Экологическая безопасность, отсутствие побочных продуктов и, как следствие, дополнительных процедур очистки целевого соединения и утилизации отходов выгодно отличают пероксид водорода от других окислителей, содержащих активный кислород. В связи с этим возможность использования Н2О2 в качестве безотходного “зеленого” окислителя исследуется едва ли не во всех областях синтетической химии, в том числе и в реакциях Pd-катализируемого окисления α-олефинов [57].
Так, авторы [58] предложили окислять терминальные олефины в соответствующие кетоны с помощью Pd(OAc)2 и 30% раствора H2O2 в среде АсОН в отсутствие галогенид-ионов и сокатализаторов. Применение описанной системы обеспечило образование ряда 2-кетонов с S ≈ 90–95% и конверсией 1-алкенов 92–96% при 80°С за 6 ч (схема 5 а).

Схема 5 . Окисление α-олефинов в присутствии каталитической системы Pd(OAc)2/Н2О2 (а) и схема каталитического цикла (б).
Согласно предложенному механизму (схема 5 б), в такой системе на первой стадии образуется активный интермедиат Pd–OOH, который затем координируется с субстратом, превращаясь в псевдоциклический пероксидный интермедиат IX, внутри которого происходит перенос кислорода. В то время как источником кислорода в молекуле кетона в случае традиционной Вакер-системы служит вода, а кислород выступает в качестве терминального окислителя (см. уравнения (I)–(III)), в H2O2-опосредованном окислении источником кислорода является терминальный окислитель – H2O2.
Следует отметить, что данная система требует использования избытка пероксида водорода ([Н2О2]/[субстрат] ≥ 5) для компенсации непродуктивного разложения Н2О2 и обеспечения полноты протекания реакции, а также для предотвращения осаждения палладия. Изомерные кетоны образуются в качестве побочных продуктов.
В том же году в работе [59] H. Mimoum и соавт. показали, что селективное стехиометрическое окисление терминальных олефинов в метилкетоны в мягких условиях может быть выполнено в присутствии тетрамерных комплексов [RCO2Pd–OOt-Bu]4 (R = –CCl3, –CF3, –CH3, –C5F11), осаждение которых наблюдается в результате замещения карбоксилат-аниона группой t-BuОО–– при перемешивании смеси Pd(RCO2)2 и 80% t-BuООН при комнатной температуре в течение 2 ч. Наиболее активным среди синтезированных оказался комплекс CF3CO2Pd–OOt-Bu, в присутствии которого за 10 мин реакции при 20°С в безводном бензоле выход С6 и С8 кетонов составил 98% в расчете на Pd. На основании полученных данных авторы предложили механизм, включающий аналогично схеме 5 б следующие этапы: 1) координирование терминального олефина с образованием пероксидного π-олефинового комплекса; 2) нуклеофильное цис-пероксипалладирование в координационной сфере металла с появлением псевдоциклического пятичленного аддукта; 3) разложение псевдоциклического комплекса посредством разрыва связи О–О с последующим β-гидридным сдвигом с выделением кетона и трет-бутоксипалладиевого комплекса; 4) регенерация исходного катализатора а) в присутствии избытка t-BuООН замещением группы t-BuО–– на более нуклеофильную t-BuОО–– с возникновением t-BuОН или б) в отсутствие избытка t-BuООН замещением t-BuО–– второй молекулой олефина с формированием π-аллильного комплекса и t-BuОН.
В 2009 г. B. Michel и соавт. описали высокоактивную каталитическую систему Pd(хинолин-2-оксазолин)Cl2/AgSbF6/70% раствор t-BuOOH, обеспечивающую превращение 1-децена в 2-деканон с выходом 86% за 20 мин в мягких условиях [60]. Реакцию проводили в темноте в среде CH2Cl2, постепенно нагревая предварительно охлажденную в ледяной бане смесь до комнатной температуры. Катализатор получали в виде оранжевого порошка за 16 ч при перемешивании хинолин-2-оксазолина (X) в качестве бидентатного лиганда и Pd(CH3CN)2Cl2 в среде CH2Cl2 в атмосфере N2. Метод отличается высокой скоростью целевой реакции, однако требует предварительной активации Pd-катализатора избытком 70% раствора t-BuOOH.

Позднее авторы [61] исследовали окисление 1-октена в качестве модельного субстрата 50%-ным раствором Н2О2 (10 экв.) в присутствии комплекса L–Pd(MeCN)2(OSO2CF3), синтезированного ex situ смешением лиганда и Pd(OAc)2 в подходящем растворителе при комнатной температуре с последующим добавлением трифторметансульфоновой кислоты. В такой системе наибольший выход 2-октанона 80% при конверсии субстрата >99% достигался за 24 ч при температуре 27°С в ацетоне с батофенантролином (Bphen) в качестве лиганда.
Приведенные примеры показывают, что системы Pd-катализатор/пероксосоединение в явном или модифицированном виде имеют высокий потенциал для разработки “зеленых” процессов окисления α-олефинов, обеспечивая в ряде случаев достаточно короткие времена реакций при больших значениях выхода продукта в мягких условиях. Однако сверхстехиометрическое применение ROOH является общим недостатком подобных систем.
Использование ex situ Pd-комплексов в качестве катализаторов
В 1981 г. M. Andrews и K. Kelly предложили проводить окисление олефинов в кетоны кислородом в присутствии комплекса Pd(CH3CN)2ClNO2 (XI), способного претерпевать окислительно-восстановительные превращения с образованием полимерного нитрозопроизводного [PdCl(NO)]n [62]. Такой комплекс, по сути, объединил в себе функции активного центра катализатора (PdII), лиганда, стабилизирующего палладий, ОДО, исключив тем самым необходимость использования дополнительного сокатализатора. Применяя методы ИК- и ЯМР-спектроскопии, авторы показали, что окисление протекает через быструю и обратимую координацию алкена с комплексом XI с последующим образованием пятичленного металлоцикла XII, внутри которого происходит перенос кислорода с нитрогруппы на α-олефин. Такая система обеспечила селективное окисление децена-1 в 2-деканон при комнатной температуре в толуоле, однако достигаемые выходы оказались невысокими.

Позднее, в 1998 г., авторам [63] удалось выделить и охарактеризовать гетерометаллический полимерный комплекс состава [(PdCl2)2CuCl2(ДМФА)4]n (XIII), полученный в мягких условиях взаимодействием PdCl2(CH3CN)2 и CuCl с ДМФА в среде 1,2-дихлорэтана в атмосфере О2. Было показано, что такой комплекс может катализировать окисление 1-децена в среде ДМФА/Н2О при 50°С, приводя к 2-деканону с умеренными выходами. Авторы полагают, что формирование XIII может протекать в системе PdCl2/CuCl/O2/ДМФА и вносить свой вклад в окисление алкенов наряду с традиционным рассмотрением процесса по уравнениям (I)–(III).
Умеренные значения конверсии в интервале 25–50% при S образования 2-кетонов 99% получили авторы [64, 65] при окислении 1-алкенов в присутствии водорастворимого комплекса, синтезированного взаимодействием Pd(OAc)2 с дисульфонатом батофенантролина (BPS) в Н2О (схема 6 а). Реакцию проводили в течение 10 ч при температуре 100°С и давлении кислорода 3 МПа в водной среде. Согласно экспериментальным данным, использование устойчивого к окислению бидентатного диамина BPS в качестве лиганда стабилизирует Pd0, предотвращая его кластеризацию и осаждение в виде палладиевой черни, а также снижает редокс-потенциал пары Pd2+/Pd0, делая регенерацию Pd0 в присутствии О2 более благоприятной. Найденный дробный порядок по палладию, равный ½, свидетельствует о возникновении димерных частиц Pd с двумя мостиковыми гидроксильными лигандами (XIV), которые диссоциируют при координации α-олефина с образованием комплекса XV типа цис-I, претерпевающего цис-гидроксипалладирование с формированием комплекса β-гидроксиалкилпалладия XVI. Разложение последнего приводит к генерации кетона и Pd0, стабилизированного BPS (схема 6 б).
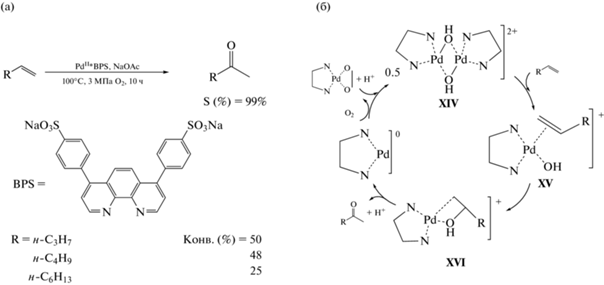
Схема. 6. Окисление α-олефинов в присутствии комплекса PdII*BPS (а) и схема каталитического цикла (б).
Гетерогенные системы
В литературе описано множество каталитических систем, эффективных в реакции окисления α-олефинов. Однако большинство из них страдает от выпадения Pd0 в ходе реакции из-за ее длительности, а также отсутствия эффективных сокатализаторов, способных обеспечивать быструю регенерацию Pd2+. Основной способ достижения стабильности предложенных каталитических систем – использование органических лигандов, к строению и составу которых предъявляется ряд требований, описанных ранее. Это приводит к увеличению количества компонентов каталитической системы и ограничениям, связанным с проведением процесса только с применением раствора каталитического комплекса. В этой связи ряд исследователей поставил своей целью разработку высокоактивных гетерогенных катализаторов для процессов окислительной трансформации высших α-олефинов в 2-кетоны.
Так, в 1992 г. авторы [66] описали гетерогенный катализатор, представляющий собой прослойки модифицированного монтмориллонита, содержащие координированный Pd (схема 7 ). Синтез катализатора осуществляли в толуоле прививкой 3-триэтоксисилильного производного к предварительно полученному Н-монтмориллониту при кипячении в инертной атмосфере с последующим взаимодействием образованного производного с Pd(PhCH2CN)2Cl2 при комнатной температуре в бензоле. Было установлено, что в присутствии избытка 30% раствора Н2О2 при 80°С в среде АсОН такой интеркалят обеспечивает эффективное окисление ряда 1-алкенов С6–С12 в соответствующие 2-кетоны с выходом 90–96% при полной конверсии за 45–60 мин и может быть использован повторно. По мнению авторов, высокая эффективность подобной системы (TON = = 4500 ч–1), превосходящая таковую для гомогенной системы Pd(OAc)2/H2O2 в 10 раз, объясняется изменением электронного окружения PdII в результате комплексообразования с силилалкиламинным бидентатным лигандом, а также поляризацией двойной связи субстрата благодаря кислотности межслоевого пространства монтмориллонита, создаваемой мостиковыми базальными атомами кислорода. При этом ограниченное межслоевое пространство интеркалята требует определенной пространственной ориентации субстрата, что препятствует изомеризации двойной связи и обеспечивает рост селективности образования 2-кетона.

Схема 7 . Окисление α-олефинов в присутствии гетерогенного катализатора Pd/монтмориллонит.
Позднее M. Kulkarni и соавт. [67] описали окисление октена-1 в присутствии катализатора Pd0/C, используя KBrO3 в качестве недорогого и легкодоступного терминального окислителя. Процесс вели в течение 12 ч в среде ТГФ/Н2О (4 : 1) при температуре 65°С с добавлением трехкратного избытка окислителя. Выход 2-октанона составил 84%.
В 2017 г. авторы [68] исследовали окисление ароматических и алифатических терминальных алкенов в соответствующие кетоны в присутствии наночастиц Pd0, нанесенных на ZrO2, в отсутствие сокатализаторов и кислот. В такой системе за 20 ч реакции в атмосфере О2 выход С8- и С10-кетонов составил 90 и 68% соответственно. Окисление вели в среде ДМСО/Н2О при 80°С и давлении кислорода 0.2 МПа, предварительно активировав катализатор с применением О2 (1 МПа) в автоклаве при 80°С в течение 1 ч в среде ДМСО. Возможность многоциклового использования такой системы была продемонстрирована на примере реакции окисления стирола в ацетофенон в 6 циклах. В последующих циклах снижение каталитической активности было более значительным в результате агрегации наночастиц Pd (размер частиц после 1-го и 8-го циклов увеличился с 4.2 до 25.1 нм).
Ряд кетонов С5–С8 с выходом 78–81% был получен авторами [69] при окислении соответствующих 1-алкенов 30% раствором Н2О2 (10 экв.) на гетерогенном катализаторе Pd0/g-C3N4, приготовленном восстановлением с помощью NaBH4 смеси PdCl2 и синтезированного на отдельной стадии графитоподобного нитрида углерода g-C3N4. Реакции проводили в среде CH3CN/H2O при температуре 25–55°С в присутствии оксида графена, обладающего свойством слабой кислоты. Катализатор показал хорошую гидролитическую устойчивость и умеренное снижение выхода ацетофенона при окислении стирола на 7% после пяти рециклов.
Несмотря на существующий в настоящее время высокий интерес исследователей к разработке гетерогенных систем, число публикаций, посвященных их применению для окисления 1-алкенов в 2-кетоны, остается ограниченным.
Гетерополисоединения в качестве сокатализаторов
Гетерополисоединения (ГПС) представляют собой обширный класс металл-кислородных полиядерных кластерных соединений, состав, строение и свойства которых могут варьироваться в широких пределах [70–72]. Еще в 60-х гг. XX в. было показано, что внедрение легко восстанавливаемых атомов ванадия(V) в состав металлатного каркаса различных ГПС приводит к появлению у них окислительных свойств, сила которых зависит от содержания ванадия. Это способствовало пристальному вниманию со стороны различных научных коллективов к данному типу соединений (преимущественно к ГПС структуры Кеггина (рис. 2)) и стало отправной точкой для их всестороннего исследования в качестве ОДО. Сегодня ванадийсодержащие ГПС – ГПС-х (х – число атомов ванадия(V) в составе) – находят применение во многих областях химии в качестве кислотных и окислительных катализаторов для трансформации различных органических соединений [73–78], в том числе в реакциях окисления α-олефинов.

Так, серия патентов 1988-89 гг., принадлежащих компании “Catalytica Associates”, демонстрирует возможность получения высших 2-кетонов одностадийным окислением α-олефинов кислородом под давлением с применением каталитической системы Pd-содержащий компонент/изо- или гетерополианион в присутствии добавок переходного металла, пригодного к окислительно-восстановительным превращениям (Cu, Fe или Mn), и/или комплексообразующего лиганда из класса нитрилов (RC≡N) [79–81]. На примере окисления 1-гексена авторы показали, что введение в систему добавок в ряде случаев увеличивает значения конверсии и S, а также сокращает время реакции. Наилучшее соотношение данных показателей (96.4 и 87%) было достигнуто за 16 мин при использовании каталитической системы Pd(NO3)2/Н9PMo6V6O40/Cu(NO3)2 в среде Н2О/CH3CN/Н2SO4 при температуре 85°С и давлении кислорода 0.69 МПа в реакторе, обеспечивающем эффективное смешение фаз со скоростью до 2500 об/мин. Для ряда систем, в присутствии которых селективность возрастала до значений выше 99%, показатели конверсии снижались до ~30%. Окисление 1-октена в аналогичных условиях в течение 141 мин привело к образованию 2-октанона с S = 65% при конверсии субстрата 86%.
Окисление 1-децена системой PdCl2/O2 в присутствии водного раствора молибдованадофосфорной гетерополикислоты (ГПК), содержащей от 2 до 8 атомов ванадия(V), в среде Н2О/сульфолан (1 : 1), описанное в патенте компании “Phillips Petroleum Company” [36], позволило получить 2-деканон с S в интервале 96.1–97.8% за 4 ч при 80°С и давлении кислорода 0.83–1.03 МПа. Конверсия субстрата при этом составила 31.3–45.1%.
Позднее E. Monflier и соавт. описали многокомпонентную каталитическую систему для окисления терминальных олефинов, включающую PdSO4/H9PMo6V6O40/CuSO4/О2, а также замещенный β-ЦД. Было показано, что каталитические свойства β-ЦД могут быть улучшены подбором подходящих функциональных групп, определяющих его растворимость в водной и органической фазах [82]. Наилучшим среди исследованных оказался гептакис(2,6-ди-O-метил)-β-ЦД, обладающий хорошей растворимостью в водной фазе и частичной растворимостью в органической. В присутствии этой системы при 80°С и атмосферном давлении 100%-ное преобразование 1-децена в водной среде с выходом 2-деканона 98% происходило за 6 ч [83, 84]. Достижение близких значений выхода гомологов 2-деканона, таких как 2-ундеканон и 2-тетрадеканон, потребовало увеличения времени реакции в 4 и 15 раз соответственно [85]. По предположению авторов, в такой системе происходит частичное замещение протонов с формированием Cu–ГПС-х-комплекса, проявляющего высокую активность в реокислении Pd0. Кроме того, обладая липофильной полостью хозяина, β-ЦД образует комплекс включения с субстратом, облегчая тем самым его массоперенос между органической и водной фазами. Изомерные алкены были идентифицированы в качестве побочных продуктов.
T. Yokota и соавт. показали возможность использования гомогенно-гетерогенной каталитической системы, состоящей из Pd(OAc)2/С, раствора ГПС состава (NH4)5H6PMo4V8O40 и О2, для окисления 1-алкенов С6–С14 в соответствующие 2-кетоны с выходами 74–88% [86]. В качестве наиболее предпочтительных условий проведения процесса были выбраны температура 60°С, среда этанол/вода (19 : 1), время реакции 5.5 ч при медленном введении субстрата в течение 5 ч. Реакцию проводили в присутствии добавок NH4Cl и MeSO3H, введение которых, по предположению авторов, обеспечивает окисление изомерных алкенов и препятствует выпадению Pd0 соответственно. Тем не менее, изомерные 2- и 3-алкены с выходом 5–18% все же наблюдались в качестве побочных продуктов.
В 2009 г. J. Ettedgui и R. Neumann описали метод, позволяющий селективно окислять терминальные алкены C6–C9 в соответствующие 2-кетоны, а также гексадиен-1,5 в 3-метилциклопентанон при температуре 150°C в среде ДМАА/Н2О с помощью N2O в качестве терминального окислителя [87]. Реакцию вели в течение 18 ч в присутствии металлорганического полиоксометаллатного гибридного катализатора PdII(15-краун-5-фенантролин)Cl2–H5PV2Mo10O40, синтезированного по схеме 8 . Образование желто-оранжевого нерастворимого комплекса XVII было подтверждено методами элементного анализа, УФ-Вид- и ИК-спектроскопии, электроспрей ионизационной масс-спектрометрии и ЯМР-спектроскопии.

Схема 8 . Схема синтеза металлорганического полиоксометаллатного гибридного катализатора PdII(15-краун-5-фенантролин)Cl2–H5PV2Mo10O40.
Авторами настоящего обзора также ведутся интенсивные исследования реакции окисления высших α-олефинов в присутствии V-содержащих ГПС, нацеленные на реализацию отечественного экологичного процесса синтеза высших 2-кетонов, обеспечивающего получение не только отдельных кетонов, но и всего ряда С6–С14. Прототипом к представленному исследованию стала работа К.И. Матвеева и Г.М. Максимова, опубликованная в 1995 г. [88] (заявка на изобретение от 15.01.1981). В указанном изобретении авторы предложили каталитическую систему, состоящую из PdSO4 и натриевой соли фосфорномолибдованадиевой ГПК структуры Кеггина брутто-состава NayH3 + x – yPMo12 – xVxO40, содержащей от 1 до 4 атомов ванадия(V). ГПС-х получали замещением атомов MoVI в фосфорномолибденовой ГПК состава H3PMo12O40 на VV путем ее растворения в растворе метаванадата натрия и Н3РО4. Окисление 1-алкенов проводили в присутствии указанного катализатора в водно-метанольном растворе (50–90 об. % МеОН) при температуре 50–90°С и давлении кислорода 0.1–0.5 МПа. Такой способ обеспечивал S образования 2-кетонов 95–98% при конверсии 1-алкенов 31−92%. Возможность регенерации каталитической системы в патенте не обсуждалась.
К основным недостаткам предложенной каталитической системы [88] можно отнести низкие значения ее активности и производительности вследствие невысокого содержания атомов ванадия в составе ГПС-х. Так, в 0.20 М растворе Na3H4PMo8V4O40 (раствор с наибольшим содержанием VV из предложенных в прототипе) концентрация ванадия составляет всего 0.80 моль/л, что не обеспечивает полной конверсии исходных 1-алкенов. Как следствие, в условиях реакции происходит быстрое восстановление атомов VV до VIV и резкое снижение редокс-потенциала системы (Е) до значения ~0.6 В, из-за чего раствор ГПС-х теряет свою способность окислять образующуюся в ходе целевой реакции восстановленную форму Pd0 до активной формы PdII. Это провоцирует образование палладиевой черни и постепенную дезактивацию катализатора. Аналогичным недостатком обладают и другие предложенные системы Pd + ГПС-х, основанные на применении ГПС с низким содержанием ванадия(V).
Совершенствование методов синтеза фосфорномолибдованадиевых ГПК позволило сотрудникам ИК СО РАН разработать новые высокованадиевые ГПС-х брутто-состава HaPzMoyVxOb (z = = 1–3, y = 8–18, x = 1–12, a = 2b – 6y – 5(x + z), b = 40–89) [89], использование которых легло в основу создания новой эффективной технологии получения 2-кетонов С5–С10 с S > 97% при конверсии субстрата выше 99% (заявка на изобретение № W22035099 от 21.06.2022). Согласно предложенному способу, синтез 2-кетонов С5–С10 проводится путем каталитического окисления 1-алкенов в двухфазной водно-органической среде при применении смешивающегося с водой органического растворителя в присутствии гомогенной каталитической системы, состоящей из Pd(OAc)2 и 0.20–0.30 М водного раствора кислой натриевой соли ГПК состава NaH10P4Mo18V7O87 (PdII + NaГПК-7P4). Реакция осуществляется в двухстадийном режиме: целевая реакция протекает при температуре 75–95°С и атмосферном давлении в течение 9–10 ч, регенерация катализатора выполняется на отдельной стадии кислородом или кислородсодержащим газом (воздухом) в автоклаве при температуре 150–170°С и парциальном давлении кислорода 0.4–0.5 МПа в течение 15–20 мин.
Предложенная каталитическая система отличается высокой концентрацией ванадия (1.4–2.1 моль/л). Это обеспечивает достаточный запас окислительной емкости для эффективного протекания процесса, позволяя снизить загрузки компонентов катализатора, увеличить эффективность каталитической системы, а также упростить аппаратурное оформление целевой реакции за счет ее проведения при атмосферном давлении. Кроме того, система PdII + NaГПК-7P4 пригодна к многоцикловому использованию по реакциям (I) + (II) и (III) с сохранением конверсии и селективности, не требует дополнительных агентов и КМП. В настоящее время работы в данном направлении продолжаются.
Приведенные исследования показывают, что гомогенные и гетерогенные Pd-содержащие каталитические системы, включающие ГПС-х, имеют хорошую перспективу для разработки эффективных методов окисления высших α-олефинов. В большинстве случаев предложенные процессы отличаются мягкими условиями и не требуют повышенного давления, обеспечивая при этом высокие значения конверсии субстрата и селективности образования 2-кетонов. Отметим, что применяемый ОДО должен не только обладать способностью принимать электроны с Pd0, окисляя его до Pd2+, но и быстро регенерироваться для завершения каталитического цикла. Лишь ограниченное число ГПС-х способно выполнять обе задачи эффективно.
V. ЗАКЛЮЧЕНИЕ
В настоящее время в литературе представлено большое число разработок, позволяющих выполнять селективное окисление терминальных алкенов в соответствующие 2-кетоны как с использованием гомогенных, так в присутствии гетерогенных каталитических систем. Как видно, в целом стратегия перехода на бесхлоридные системы при сохранении PdII в качестве активного компонента каталитической системы оправдывает свое применение. Основными достижениями новых систем Pd + Ox по сравнению с “хлоридными” становятся рост целевых значений конверсии и S, увеличение скорости окисления высших α-олефинов благодаря использованию многофазных систем, добавок и КМП, а также решение проблемы образования хлорированных производных, требующих выделения и утилизации.
Проведенные фундаментальные и прикладные исследования кинетических закономерностей и технологических особенностей превращения линейных α-олефинов в востребованные 2-кетоны открывают реальные перспективы для создания нового поколения относительно простых гомогенных и гетерогенных Pd-катализируемых процессов, пригодных для синтеза востребованных 2-кетонов с учетом современных требований “зеленой” химии. Внедрение подобных процессов позволит переориентировать связанные с применением 2-кетонов промышленные отрасли (фармацевтическая и лакокрасочная отрасли, производство бытовой химии, парфюмерных и косметических изделий) на современные экологичные технологии и будет способствовать снижению себестоимости производимой этими отраслями продукции. Тем не менее, анализ представленных публикаций позволяет сделать вывод, что работы по созданию новых катализаторов и поиску современных технологических решений в данной области все же далеки от завершения. Ряд недостатков, среди которых использование повышенного давления в интервале 0.5–6 МПа, ограниченность информации о стабильности каталитической системы и возможности ее многоциклового применения, недоработанные методы отделения и регенерации каталитической системы, по-прежнему требуют устранения.
Список литературы
Sheldon R.A. // Green. Chem. 2007. V. 9. P. 1273.
Eco-Friendly Synthesis of Fine Chemicals. Ed. Ballini R. RSC Publishing, 2009.
Balakrishnan M., Arab G.E., Kunbargi O.B., Gokhale A.A., Grippo A.M., Toste F.D., Bell A.T. // Green Chem. 2016. V. 18. P. 3577.
Пaт. 2371472 PФ, 2009.
Реутов О.А., Курц А.Л., Бутин К.П. Органическая химия. Т. 3. Москва: Бином. Лаборатория знаний, 2010. С. 20.
Травень В.Ф. Органическая химия: учебное пособие для вузов в 3 т. Т. 2. Москва: Бином. Лаборатория знаний, 2015, 433 с.
Arends I.W.C.E, Sheldon R.A. Modern Oxidation Methods / Ed. Bäckvall J.-E. Weinheim: Wiley-VCH, 2004. P. 83–118.
Anastas P.T., Warner J.C. Green Chemistry: Theory and Practice. New York: Oxford University Press, 1998.
Tang S., Bourne R., Smith R., Poliakoff M. // Green Chem. 2008. V. 10. P. 268.
Smidt J., Hafner W., Jira R., Sedlmeier J., Sieber R., Kojer H., Rüttinger R. // Angew. Chem. 1959. V. 71. P. 176.
Cornell C.N., Sigman M.S. // Inorg. Chem. 2007. V. 46. P. 1903.
Gligorich K.M., Sigman M.S. // Chem. Commun. 2009. P. 3854.
McDonald R.I., Liu G., Stahl S.S. // Chem. Rev. 2011. V. 111. P. 2981.
Sigman M.S., Werner E.W. // Acc. Chem. Res. 2012. V. 45. № 6. P. 874.
Темкин О.Н. // Кинетика и катализ. 2020. Т. 61. № 5. С. 595.
Гогин Л.Л., Жижина Е.Г. // Катализ в промышленности. 2021. Т. 21. № 1–2. С. 67.
Pat. 1049845 DE, 1959.
Jira R. Applied Homogeneous Catalysis with Organometallic Compounds. V. 1 / Eds. Cornils B., Herrmann W.A. Weinheim: VCH, 1996. P. 374.
Новый справочник химика и технолога. Сырье и продукты промышленности органических и неорганических веществ. Ч. II. Санкт-Петербург: АНО НПО “Профессионал”, 2005, 2007. С. 51.
Jira R., Laib R.J., Bolt H.M. Ulmann’s Encyclopedia of Industrial Chemistry. 1985. V. A1. P. 31.
Трофимов Б.А., Иванов А.В. // Вестник СПбГУ. Сер. 4. 2014. Т. 1. № 59. С. 558.
Органическая химия: Учебн. пособие. 2-е изд., испр. и доп. Новосибирск: Сиб. унив. изд-во, 2001. С. 478.
Варгафтик М.Н., Моисеев И.И., Сыркин Я.К. // Докл. АН СССР. 1962. Т. 147. С. 399.
Моисеев И.И., Варгафтик М.Н., Сыркин Я.К. // Докл. АН СССР. 1963. Т. 153. С. 140.
Варгафтик М.Н., Моисеев И.И., Сыркин Я.К. // Изв. АН СССР. Сер. хим. 1963. С. 1147.
Моисеев И.И. π-Комплексы олефинов в жидкофазном окислении. Москва: Наука, 1970. 270 с.
Henry P.M. // J. Org. Chem. 1973. V. 38. P. 2415.
Keith J.A., Henry P.M. // Angew. Chem. Int. Ed. 2009. V. 48. P. 9038.
Jira R. Applied Homogeneous Catalysis with Organometallic Compounds. V. 1 / Eds. Cornils B., Herrmann W.A. Weinheim: VCH, 2002. P. 386.
Tsuji J. Innovations in Organic Synthesis. Chichester: Wiley, 1995. 549 p.
Smidt J., Krekeler H. // Per. Refiner. 1963. V. 42. P. 149.
Rogers H.R., McDermott J.X., Whitesides G.M. // J. Org. Chem. 1975. V. 40. № 24. P. 3577.
Mimoun H., Machirant M.M.P., Seree de Roch I. // J. Am. Chem. Soc. 1978. V. 100. № 17. P. 5437.
Igersheim F., Mimoum H. // J. Chem. Soc. Chem. Commun. 1978. P. 559.
Clement W.H., Selwitz C.M. // J. Org. Chem. 1964. V. 29. № 1. P. 241.
Pat. 4507506 US, 1985.
Harada A., Hu Y., Takahashi S. // Chem. Lett. 1986. P. 2083.
Zahalka H.A., Januszkiewicz K., Alper H. // J. Mol. Catal. 1986. V. 35. P. 249.
Alper H., Januszkiewicz K., Smith D.J.H. // Tetrahedron Lett. 1985. V. 26. № 19. P. 2263.
Максимов А.Л., Бучнева Т.С., Караханов Э.А. // Нефтехимия. 2003. Т. 43. № 3. С. 173.
Pat. 4152354 US, 1979.
Pat. 4448892 US, 1984.
Pat. 4532362 US, 1985.
Jira R., Freiesleben W. Organometallic Reactions. V. 3. Eds.: Becker E., Tsutsui M. NY: John Wiley, 1972.
Grate J.H., Hamm D.R., Mahajan S. // Mol. Eng. 1993. V. 3. P. 205.
Mitsudome T., Umetani T., Nosaka N., Mori K., Mizugaki T., Ebitani K., Kaneda K. // Angew. Chem. Int. Ed. 2006. V. 45. № 3. P. 481.
Fernandes R.A., Chaudhari D.A. // J. Org. Chem. 2014. V. 79. № 12. P. 5787.
Fernandes R.A., Bethi V. // Tetrahedron. 2014. V. 70. P. 4760.
Smith A.B., Cho Y.S., Friestad G.K. // Tetrahedron Lett. 1998. V. 39. P. 8765.
Pat. 4738943 US, 1988
Pat. 4847421 US, 1989.
Miller D.G., Wayner D.D.M. // J. Org. Chem. 1990. V. 55. P. 2924.
Pat. 9096519 B2 US, 2015.
Wang Y.-F., Gao Y.-R., Mao S., Zhang Y.-L., Guo D.-D., Yan Z.-L., Guo S.-H., Wang Y.-Q. // Org. Lett. 2014. V. 16. № 6. P. 1610.
Chaudhari D.A., Fernandes R.A. // J. Org. Chem. 2016. V. 81. № 5. P. 2113.
Ho Y.A., Paffenholz E., Kim H.J., Orgis B., Rueping M., Fabry D.C. // ChemCatChem. 2019. V. 11. № 7. P. 1889.
Michel B.W., Sigman M.S. // Aldrichimica Acta. 2011. V. 44. № 3. P. 55.
Roussel M., Mimoun H. // J. Org. Chem. 1980. V. 45. № 26. P. 5387.
Mimoum H., Charpentier R., Mitschler A., Fischer J., Weiss R. // J. Am. Chem. Soc. 1980. V. 102. P. 1047.
Michel B.W., Camelio A.M., Cornell C.N., Sigman M.S. // J. Am. Chem. Soc. 2009. V. 131. P. 6076.
Cao Q., Bailie D.S., Fu R., Muldoon M.J. // Green Chem. 2015. V. 17. P. 2750.
Andrews M.A., Kelly K.P. // J. Am. Chem. Soc. 1981. V. 103. P. 2894.
Hosokawa T., Nomura T., Murahashi S.-I. // J. Organomet. Chem. 1998. V. 551. P. 387.
ten Brink G.-J., Arends I.W.C., Papadogianakis G., Sheldon R.A. // Chem. Commun. 1998. P. 2359.
ten Brink G.-J., Arends I.W.C., Papadogianakis G., Sheldon R.A. // Appl. Catal. A: Gen. 2000. V. 194–195. P. 435.
Rao Y.V.S., Rani S.S., Choudary B.M. // J. Mol. Catal. 1992. V. 75. P. 141.
Kulkarni M.G., Shaikh Y.B., Borhade A.S., Chavhan S.W., Dhondge A.P., Gaikwad D.D., Desai M.P., Birhade D.R., Dhatrak N.R. // Tetrahedron Lett. 2013. V. 54. P. 2293.
Zhang Z., Kumamoto Y., Hashiguchi T., Mamba T., Murayama H., Yamamoto E., Ishida T., Honma T., Tokunaga M. // ChemSusChem. 2017. V. 10. № 17. P. 3482.
Gao X., Li Z., Yan W., Peng X. // J. Saudi. Chem. Soc. 2020. V. 24. P. 663.
Поп М.С. Гетерополи- и изополиоксометаллаты: пер. с англ. Новосибирск: Наука. Сиб. отделение, 1990. 232 с.
Максимов Г.М. // Успехи химии. 1995. Т. 64. № 5. С. 480.
Kozhevnikov I.V. // Chem. Rev. 1998. V. 98. № 1–2. P. 171.
Neumann R. // Inorg. Chem. 2010. V. 49. № 8. P. 3594.
Zhizhina E.G., Odyakov V.F. // ChemCatChem. 2012. V. 4. № 9. P. 1405.
Albert J., Lüders D., Bösmann A., Guldi D.M., Wasserscheid P. // Green Chem. 2014. V. 16. № 1. P. 226.
Gromov N.V., Taran O.P., Delidovich I.V., Pestunov A.V., Rodikova Yu.A., Yatsenko D.A., Zhizhina E.G., Parmon V.N. // Catal. Today. 2016. V. 278. № 1. P. 74.
Rodikova Y., Zhizhina E. // React. Kinet. Mech. Catal. 2020. V. 130. № 1. P. 403.
Gromov N.V., Medvedeva T.B., Sorokina K.N., Samoylova Y.V., Rodikova Y.A., Parmon V.N. // ACS Sustain. Chem. Eng. 2020. V.8. № 51. P.18947.
Pat. 4720474 US, 1988.
Pat. 4723041 US, 1988.
Pat. 4853357 US, 1989.
Tilloy S., Bertoux F., Mortreux A., Monflier E. // Catal. Today. 1999. V. 48. P. 245.
Monflier E., Blouet E., Barbaux Y., Mortreux A. // Angew. Chem. Int. Ed. 1994. V. 33. № 20. P. 2100.
Monflier E., Tilloy S., Blouet E., Barbaux Y., Mortreux A. // J. Mol. Catal. A: Chem. 1996. V. 109. P. 27.
Monflier E., Tilloy S., Fremy G., Barbauxa Y., Mortreux A. // Tetrahedron Lett. 1995. V. 36. № 3. P. 387.
Yokota T., Sakakura A., Tani M., Sakaguchi S., Ishii Y. // Tetrahedron Lett. 2002. V. 43. P. 8887.
Ettedgui J., Neumann R. // J. Am. Chem. Soc. 2009. V. 131. № 1. P. 4.
Пат. 1031045 СССР, 1995.
Пaт. 2230612 C1 PФ, 2004.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ