

Кинетика и катализ, 2023, T. 64, № 2, стр. 139-152
Машинное обучение и анализ больших данных в области катализа
В. Г. Филиппов a, Я. А. Михайлов a, *, А. В. Елышев a
a Тюменский государственный университет, группа TsyfroCatLab
625003 Тюмень, ул. Володарского, 6, Россия
* E-mail: y.a.mikhajlov@utmn.ru
Поступила в редакцию 11.06.2022
После доработки 14.09.2022
Принята к публикации 19.10.2022
- EDN: GMSQSK
- DOI: 10.31857/S0453881123020028
Аннотация
Быстрое развитие экспериментальных методов в каталитических исследованиях в последнее время позволяет получать большие объемы данных. Использование новых статистических и расчетных методов обработки, включающих в себя извлечение информации из экспериментальных данных и их непредвзятую интерпретацию, важно для ускорения развития и внедрения каталитических технологий. Извлечение информации может быть достигнуто с применением статистических подходов: PCA, MCR, ALS. В то же время алгоритмы машинного обучения начинают активно использоваться для интерпретации и построения описательных моделей. В настоящей статье рассматриваются основные методы машинного обучения и примеры их успешного применения для анализа данных инфракрасной и рентгеновской абсорбционной спектроскопии.
ВВЕДЕНИЕ
Большинство химических процессов, используемых в индустрии, осуществляется в присутствии катализаторов. Интенсивное развитие новых каталитических технологий и их внедрение в практику, без сомнения, являются одними из важнейших компонентов устойчивого развития. Несмотря на быстро растущее число исследовательских работ в области катализа, понимание механизмов многих каталитических процессов остается сложной задачей. Часто это связано с тем, что целевой результат исследования – описательная модель каталитической системы. Такая эмпирическая модель связывает параметры или условия процесса и активность катализатора. Хотя данный подход дает возможность решать задачу оптимизации, он не является полностью предсказательным. Иными словами, он не может прогнозировать поведение системы для расширенного поля параметров. Кроме того, описательные модели не позволяют связать процессы, происходящие на молекулярном уровне, например структуру активных центров в ходе реакции и каталитическую активность.
В последнее время наука о данных активно внедряется в различные области естественных наук в качестве универсального инструмента для обработки и интерпретации результатов исследований (рис. 1). Развитие и стандартизация экспериментальных методов позволяет генерировать большое количество параметров, что делает возможным применение алгоритмов работы с большими данными. В области катализа этот подход в перспективе может объединить разные аспекты каталитического процесса от молекулярного до реакторного уровня и значительно ускорить внедрение новых технологий и решений в химическую индустрию. Более того, с помощью неэмпирических квантово-механических расчетов можно осуществлять предсказательное моделирование и верифицировать кинетические схемы, используемые при проведении экспериментов и обработке получаемых данных [1, 2].

Первые работы, посвященные применению методов машинного обучения в катализе, появились в начале 2000-х гг. [3]. Вместе с ростом вычислительной мощности микропроцессоров и доступности программного обеспечения для работы с алгоритмами количество исследований с использованием машинного обучения как в катализе, так и в других областях науки значительно возросло. Тенденция применения operando методов для анализа каталитических систем, комплексность получаемых экспериментальных данных, а также необходимость их быстрой обработки в реальном времени привели к значительной заинтересованности в машинном обучении как инструменте для быстрой и эффективной интерпретации данных в последние несколько лет (рис. 2).
Рис. 2.
Результаты запроса в Scopus: катализ, катализ + машинное обучение, катализ + машинное обучение + спектроскопия + микроскопия. Поиск осуществляли с учетом следующих критериев: scopus (catalysis + machine learning + + spectroscopy microscopy) TITLE-ABS-KEY (catalysis AND machine AND learning AND spectroscopy) AND (LIMIT-TO (DOCTYPE, “ar”) OR LIMIT-TO (DOCTYPE, “re”).

В настоящей работе мы представляем обзор современных методов машинного обучения с целью ознакомления с ними читателей, а также приводим отдельные примеры их совместного применения с инструментами исследования в области гетерогенного катализа. Наша главная задача – привлечь широкое внимание химиков-каталитиков к возможностям использования машинного обучения в области характеризации катализаторов и связи их физико-химических свойств с активностью.
Первая часть статьи сконцентрирована на базовых принципах работы алгоритмов машинного обучения, успешно применяемых для исследования катализаторов. Вторая часть фокусируется на разборе примеров использования этих методов для интерпретации данных о свойствах каталитической системы, получаемых с помощью инфракрасной (ИК) и рентгеновской адсорбционной (XAS) спектроскопии, являющихся одними из главных источников информации о структуре катализатора, которая часто может служить в качестве параметров каталитической активности. При этом возможность модифицирования рассматриваемых спектроскопических методов для operando исследования с непрерывным извлечением больших массивов данных о каталитической системе повышает интерес к их совместному применению с алгоритмами обработки данных.
МЕТОДЫ МАШИННОГО ОБУЧЕНИЯ
В последние несколько лет тема искусственного интеллекта (ИИ) обрела значительную популярность. Увеличение вычислительной мощности компьютерных комплектующих и появление в них отдельных элементов для работы с ИИ, например тензорных ядер в видеокартах компании NVIDIA, обеспечили возникновение и широкое распространение таких технологий, как компьютерное зрение, автопилотируемые автомобили и т.д. Одной из областей технологии искусственного интеллекта является машинное обучение (МО). В отличии от традиционного программирования, где набор данных и алгоритм для его обработки поступают одновременно в компьютер для получения результата, в схеме с применением машинного обучения такой набор вместе с результатом исследования загружается в компьютер, который, в свою очередь, генерирует алгоритм, связывающий входящие параметры между собой. Для корректной работы МО необходим большой объем эмпирической или теоретической информации. В зависимости от способа их обработки методы машинного обучения условно разделяют на неконтролируемые (НМО) и контролируемые (КМО). Алгоритмы, наиболее часто используемые в физико-химическом анализе, представлены на рис. 3. Целью КМО является установление отношений между спектром и параметрами структуры, основанных на наборе маркированных тренирующих данных, которые, например, включают спектры стандартных веществ с известной точной структурой. Для НМО задача заключается в выявлении паттернов в больших наборах экспериментальных данных без использования каких-либо маркеров и их кластеризации, которая представляет собой группировку отдельных значений в наборе данных в отдельные кластеры на основе конкретных комбинаций принадлежащих им характеристик. Выбор конкретного метода определяется в каждом конкретном случае и зависит от количества данных, их качества и поставленной задачи.

Алгоритмы неконтролируемого машинного обучения
Метод k-средних. Метод k-средних представляет собой один из наиболее популярных методов машинного обучения благодаря простоте и скорости применения на больших массивах данных. Принципиальной его задачей является распределение полученных данных по группировкам на основе их схожести между собой. Такие данные включают спектры сложных каталитических систем, в которых однозначно отнести спектроскопический сигнал к конкретному компоненту представляется затруднительным. Кроме того, такие кластеры могут быть использованы в контролируемых методах машинного обучения (рис. 4). Несмотря на простоту работы алгоритма, следует отметить, что применение метода требует предварительной нормализации поступающих данных, а также определения степени сходства между отдельными точками для формирования центров кластеризации. Выбор оптимального числа групп “k” для массива данных зависит от поставленной задачи, так как при небольшом значении получить локальную информацию становится проблематично, а существенное количество малых групп “k” усложняет его обобщение [4].

Метод главных компонент. Метод главных компонент (МГК, PCA (Principal Component Analysis)) позволяет уменьшить размерность баз данных, увеличивая интерпретируемость, и минимизировать потерю информации (рис. 5) [5].

Иными словами, использование МГК сокращает число переменных, необходимых для описания системы. МГК предусматривает несколько этапов:
1) нормализацию данных;
2) расчет ковариационной матрицы;
3) расчет собственных векторов и значений для определения главных компонентов;
4) выбор характеристического вектора;
5) выстраивание данных вдоль оси главных компонентов.
Результаты МГК не всегда имеют интуитивную физическую интерпретацию, поэтому применение данного метода сводятся к поиску параметров системы с последующим их использованием в других методах. Например, с помощью МГК осуществляют анализ изображений энергодисперсионной рентгеновской спектроскопии наночастиц для определения количества фаз в анализируемом объеме.
Алгоритмы контролируемого машинного обучения
Метод k-ближайших соседей. Метод k-ближайших соседей – непараметрический классификационный алгоритм, т.е. он не делает каких-либо предположений на элементарном наборе данных. Для классификации объект присваивается тому классу, который является наиболее распространенным среди k-соседей данного элемента, классы которых уже известны [6]. Например, новые данные должны быть отнесены к классу А или В (рис. 6). Если k = 2, то данные присваивают классу А, поскольку в круге два треугольника и один квадрат, если k = 5, данные относят к классу В, так как квадратов во внешнем круге больше, чем треугольников. Таким образом в исследованиях органических катализаторов для гомогенного катализа методом k-ближайших соседей было установлено, что лиганды с похожей активностью имеют тенденцию группироваться вместе [7].

Дерево принятия решений. Метод “дерево принятия решений” является типичным индукционным алгоритмом и представляет собой функцию в форме данных (х) и соответствующий результат f(х), используется для нахождения функции новой порции данных (х), сфокусированной на правилах классификации, отображаемых в виде дерева решений (рис. 7). Убывающим рекурсивным способом алгоритм сравнивает атрибуты между внутренними узлами дерева решений, оценивает убывающие ветви в соответствии с различными атрибутами узла и позволяет сделать вывод, исходя из узлов листьев. На протяжении от корня до листового узла существует конъюнктивное правило, и все дерево решений отвечает группе дизъюнктивных правил выражения.

Популярность применения метода “дерево принятия решений” для классификации данных обуславливает наличие большого количества разработанных алгоритмов, таких как: ID3, C4.5, PUBLIC, CART, CN2, SLIQ, SPRINT и т.д. [8]. К достоинствам метода относят скорость классификации, а также высокие результаты при наличии шумов. Недостатком может быть изменение общего вида дерева решений при внесении небольших корректировок в исследуемые данные [9].
Линейная регрессия. Линейная регрессия относится к статистическому методу, в котором независимые переменные x используются для предсказания зависимых переменных у (рис. 8). Различают несколько видов линейной регрессии: простую, в которой значение зависимой переменной определяется как y = β0+ β1x + ε, и многомерную (MLR), где y = β0 + β1x1 + … + βmxm + ε, где β – коэффициент регрессии, ε – ненаблюдаемая случайная величина, которая добавляет “шум” к линейной зависимости между зависимой переменной и регрессорами. Также существуют более сложные модели, которые включают квадратичные параметры и взаимодействие между ними [3]. Линейная регрессия часто применяется для анализа теоретических данных, полученных в ходе эксперимента, например, совместно с теорией функционала плотности (DFT). Авторы работы [10] предсказывают методами линейной регрессии значения энергии адсорбции ключевых интермедиатов в реакции разложения оксида азота (NO) на металлических наночастицах.

Нейронные сети. Нейронные сети объединяют группу методов машинного обучения, в основе которых лежит имитация функционирования центральной нервной системы (головного мозга) человека для решения поставленных задач. В нейросетях общепринято выделять три главных слоя связанных между собой нейронов: входной, содержащий независимые переменные (изображения, числовые и текстовые данные); скрытый с активирующей функцией (сигмоидой) [11]; выходной с полученным результатом (рис. 9). Для корректной работы нейронных сетей предварительно проводят их обучение на тренировочных массивах данных с последующей проверкой адекватности построенной модели. Отметим, что подробный разбор применения нейронных сетей в области катализа рассматривается в статье авторов H. Li, Z. Zhang, Z. Liu [12].

РЕНТГЕНОВСКАЯ АБСОРБЦИОННАЯ СПЕКТРОСКОПИЯ (XAS)
Традиционно рентгеновскую абсорбционную спектроскопию можно разделить на два класса – тонкую структуру поглощения рентгеновского излучения вблизи края (XANES) и расширенную тонкую структуру поглощения рентгеновских лучей (EXAFS) – в зависимости от положения рассматриваемой области относительно белой линии спектра [13]. Обе области содержат ценную информацию о степени окисления и координации атомов и часто используются в каталитических исследованиях, в том числе в исполнении operando, когда измерения соответствующих параметров проводятся на активном катализаторе. Широкое применение XAS ограничивалось необходимостью применения дорогостоящих синхротронов и отсутствием математического аппарата для количественного анализа полученных спектров. Оба метода в течение длительного времени использовались лишь для качественного анализа, например измерения края, наличия сдвига в зависимости от присутствия или отсутствия конкретных отпечатков. Однако с появлением первых алгоритмов для расчета теоретических спектров на основе локальных гипотетических структур популярность XANES и EXAFS значительно выросла.
Необходимость быстрой обработки больших массивов теоретических и экспериментальных данных рентгеновской абсорбционной спектроскопии, а также усложнение каталитических моделей привели к активному развитию и распространению алгоритмов машинного обучения в этой области.
Базы данных для алгоритмов машинного обучения в XAS
Корректная работа нейронных сетей и контролируемого машинного обучения обусловлены использованием большого количества качественных обучающих данных. Эти два подхода решают задачи применения доступных баз данных экспериментальных спектров XAS [14] и получения моделированных спектров. Так, спектры XANES можно смоделировать в программном обеспечении FEFF, не требующем большой вычислительной мощности [15], а для EXAFS таким решением может стать IFEFFIT [16]. Создание базы данных спектров XANES и EXAFS в каталитических системах предполагает прямое моделирование спектра индивидуального катализатора, наночастиц, кластеров и т.д. Поскольку существующие базы данных [17] сведены к спектрам четко определенных порошков, их применимость к анализу каталитических систем (гетерогенных катализаторов, наночастиц, биметаллических сплавов и т.д.) ограничена информацией о состоянии окисления и координационном окружении.
Применение контролируемого машинного обучения
Перед применением обученной нейронной сети для обработки реальных экспериментальных данных необходимо удостовериться в адекватности построенного алгоритма с помощью референсных материалов, спектры которых известны или могут быть достоверно смоделированы. В работе Timoshenko J. et al. [18] предсказанные нейросетью структурные модели наночастиц сравнивали с теоретическими EXAFS-спектрами, полученными методом RMC (Reverse Monte Carlo)–EXAFS. В этом же исследовании авторы продемонстрировали возможность быстрой обработки большого количества экспериментальных спектров биметаллического CuZn-нанокатализатора, что позволило наблюдать времязависимое изменение локальной структуры вокруг каталитически активных компонентов в реакционных условиях (рис. 10).
Рис. 10.
(a) – Развитие неполной функции парной корреляции (RDF) для наночастицы Cu50Zn50, полученной нейросетью на основе времязависимых данных EXAFS k-края цинка (Zn); (б–д) – временные зависимости интегрированных областей под первым Zn–O RDF-пиком (координационное число первой оболочки Zn–O) и под первым Zn–M RDF-пиком (координационное число первой оболочки Zn–M) для медно-цинкового нанокатализатора и для Zn100-наночастиц. Адаптировано из [18]. Распространяется на условиях лицензии Creative Commons Attribution 3.0 Unported Licence. Adapted from [18]. Licensed under a Creative Commons Attribution 3.0 Unported Licence.

Успешное использование нейронной сети для метода XANES можно найти в работе Timoshenko J. et al. [19]. Нейросеть была обучена для построения 3D-модели металлического платинового нанокатализатора, нахождения скрытых связей между особенностями спектров XANES и геометрией катализатора, что привело к увеличению чувствительности XANES-спектров металлических наночастиц до четвертой координационной оболочки и позволило определять размер и форму частицы (рис. 11).
Рис. 11.
Координационные числа (КЧ), предсказанные нейросетью по экспериментальным данным XANES. (а) – Сравнение координационных чисел первой оболочки, предсказанных нейросетью, и результатов традиционного EXAFS-анализа для платиновой наночастицы на γ-Al2O3. (б) – КЧ, предсказанные нейросетью для четвертой координационной оболочки. Серые пустые круги соответствуют КЧ для платиновых модельных кластеров с разными размерами и формами. Кластеры получены с применением кубической гранецентрированной (КГЦ) структурной платины, усеченной вдоль плоскостей (100) или (111), а также для кластеров типа “икосаэдр” и “ГПУ” (гексагональная плотноупакованная решетка). Пурпурный прямоугольник на рис. 11б показывает доверительную область для КЧ, полученных для образца S3 с помощью анализа MS-EXAFS. (в) – Соответствующие возможные 3D-модели частиц. Рисунок адаптирован с разрешения из [19] (Copyright 2017 American Chemical Society). Adapted with permission from [19]. Copyright 2017 American Chemical Society.

Стоит отметить, что для наиболее эффективного применения КМО необходим выбор алгоритма, подходящий для решения специфических задач. Одной из них является определение оксидов металлов по спектру XANES. В общем случае выбор формы оксид/не оксид производится путем сравнения степени сдвига в пике XANES, полученного в ходе эксперимента или теоретической симуляции, с референтным пиком XANES чистого вещества; при этом высок риск недостоверного отнесения, связанный с человеческим фактором. В работе [20] оценивали восемь алгоритмов КМО для установления параметров состояний оксидов десятью XANES спектрами, которые были отобраны из SPring-8 Experimental Data Repository System Portal. По результатам исследований авторы пришли к выводу – логистическая регрессия (LR) имеет наибольшую точность, равную 80%, и это единственный алгоритм из всех протестированных, который может успешно различить оксидное состояние и не оксидное. Ошибочный выбор в случае CuO и NbO связан с их слабой окисленностью. Несмотря на это, машинное обучение может надежно прогнозировать степень окисления металлов в оксидах по данным XAS. Таким образом, подобный алгоритм упрощает определение оксидного состояния в отсутствие эталонных спектров.
Использование неконтролируемого машинного обучения
Изучение механизмов каталитических реакций с помощью operando спектроскопии может быть осложнено одновременным наличием наблюдательных компонентов (spectator species), промежуточных продуктов реакции и активных центров. В таких случаях применение контролируемых методов машинного обучения и нейронных сетей ограничивается ввиду отсутствия однозначной модели, соответствующей полученному ансамблю спектров [21]. В этих ситуациях предпочтительно использование неконтролируемого машинного обучения, которое дает возможность решить две основные задачи: группирование и уменьшение размерности. Задача кластеризации заключается в поиске нескольких спектров, описывающих весь набор данных и сведения об образце, в то время как уменьшение размерности помогает определить параметры в наборе данных, которые являются наиболее “очевидными” или вариативными, а также позволяет избежать предвзятости наблюдателя [22]. В EXAFS в качестве таких параметров могут выступать амплитуда, форма, фаза, частота колебаний или позиция главных пиков в Фурье-преобразованных спектрах, в то время как для XANES это позиция края поглощения и интенсивность белой линии [23].
В исследовании степени окисления железа в комплексных катализаторах FeMoBi в процессе аммоксидирования пропилена [24] для анализа спектров XANES авторы отдали предпочтение использованию PCA, поскольку линейный комбинированный анализ не мог предложить независимое от модели определение числа независимых компонентов. Авторы показали, что число главных компонентов, необходимых для воспроизведения всех 6 экспериментальных спектров, равно 2. Стандартные соединения Fe2(MoO4)3 и Li2Fe(MoO4)3 были хорошо воспроизведены путем комбинирования этих двух главных компонентов, и целевое преобразование было выполнено с основы абстрактных компонентов до основы, соответствующей двум стандартам.
Повышение скорости и доступности рентгеновской спектроскопии, включая возможность ее применения с методами operando, в сочетании с достаточно прямой интерпретацией приводят к увеличению популярности использования указанного метода в исследованиях гетерогенных каталитических систем. Стоит отметить, что этот метод позволит получить большой набор качественных и количественных данных, расширить возможности самого метода, а также провести быструю и непредвзятую интерпретацию результатов. При этом расширение существующих и создание новых баз данных, содержащих экспериментальные и теоретические спектры реальных катализаторов, могут ускорить интегрирование рентгеновской спектроскопии с теорией.
ИК-СПЕКТРОСКОПИЯ
ИК-спектроскопия является одним из самых распространенных методов молекулярной спектроскопии и занимается изучением взаимодействия инфракрасного излучения с веществом путем поглощения, излучения или отражения. Эти взаимодействия определяются строением молекулы и связаны с переходами между колебательными энергетическими состояниями или, в классической интерпретации, с колебаниями атомных ядер относительно равновесных положений. Число и частоты полос зависят, во-первых, от числа образующих молекулу атомов, масс атомных ядер, геометрии и симметрии равновесной ядерной конфигурации, и во-вторых, от потенциального поля внутримолекулярных сил. Таким образом, колебательные спектры представляют собой чрезвычайно специфические и чувствительные характеристики молекул, чем и объясняется широкое применение их в химических исследованиях [25].
Позволяя напрямую наблюдать взаимодействия между сорбированными молекулами и катализаторами, ИК-спектроскопия также является одним из наиболее эффективных спектроскопических методов анализа химии поверхности гетерогенных катализаторов. Основные преимущества ИК-спектроскопии в гетерогенном катализе – высокая чувствительность метода по отношению к адсорбатам на поверхности катализатора и их структуре, возможность применения ИК в различных конфигурациях (ИК-спектроскопия с преобразованием Фурье с диффузным отражением (DRIFT) и ИК-спектроскопия ослабленного полного отражения (ATR)), а также относительная доступность и экспрессность метода.
Зачастую отнесение сигналов ИК-спектроскопии – эмпирический процесс, применимый только к относительно простым спектрам и веществам. Интерпретация ИК-спектров осложняется тем, что их аккуратное квантово-химическое моделирование все еще довольно затратно. Результатом анализа, особенно для сложных систем, которыми и являются большинство катализаторов, становятся неразрешенные пики, и перед исследователями встает сложный вопрос о моделировании не отдельных веществ, а целого ансамбля того, что находится в катализаторе.
Эта проблема расхождения теории и эксперимента в науке о материалах известна как “materials gap” и наиболее сильно проявляется при интерпретации ИК-спектров. “Materials gap” описывает пробел в данных между упрощенными модельными системами и сложными реальными катализаторами. В целом теоретические исследования поверхности катализатора ограничиваются модельными системами, такими как поверхности монокристаллов с четко определенными кластерами, в то время как реальные катализаторы обычно представляют собой частицы неправильной формы, которые случайным образом распределены на материалах с высокой удельной поверхностью. Эта проблема может быть решена путем сильного упрощения систем, на которых могут проводиться расчеты, или повышения точности расчетов, что приводит к резкому увеличению затрачиваемых на расчеты ресурсов. Таким образом, идеальным решением является соблюдение баланса точность–размер системы.
Однако сегодня машинное обучение становится новым инструментом во многих областях химической и физической науки и потенциально позволяет преодолеть разрыв между необходимостью высокоточных вычислений и ограниченной вычислительной мощностью. На практике для анализа ИК-спектра уже давно применяются различные методы машинного обучения. Так, Ellis et al. использовали метод множественной линейной регрессии для классификации различных видов мышечной пищи (говядина, баранина, свинина, курица, индейка) на основе соответствующих ИК-спектров [26]. Howley et al. идентифицировали наркотики, содержащие ацетаминофен, по спектрам комбинационного рассеяния, с использованием метода PCA для сокращения многомерных спектральных данных и улучшения прогностической эффективности некоторых известных методов машинного обучения [27]. Zou et al. обучили модель SVM на спектрах ближнего инфракрасного (NIR) диапазона для идентификации порошка окситетрациклина [28].
Недавние успехи в количественной оценке на основе данных ИК-спектроскопии включают в себя возможность определения коэффициентов экстинкции для конкретных участков спектра в сочетании с деконволюцией пиков, интегрированием и априорными предположениями о размерах частиц и распределении покрытия адсорбатом [29].
В работе [30] авторы представили “первые принципы” количественной методологии поверхностно-селективной ИК-спектроскопии и интегрировали их с подходами, основанными на анализе данных, постановкой задач, зависящих от химии, и экспериментальными данными для устранения “materials gap”. Схематическое изображение “materials gap” и общая методология его устранения зоны приведены на рис. 12.
Рис. 12.
Подход, объединяющий экспертные знания и спектроскопические данные для устранения “materials gap”. “Materials gap” (a) и соответствующий рабочий процесс для его устранения (б). Адаптировано из [30]. Распространяется на условиях лицензии Creative Commons CC BY license. Adapted from [30]. Distributed under the terms of the Creative Commons CC BY license.

Процесс преодоления “разрыва в материалах” происходит следующим образом. С применением теории функционала плотности (DFT) генерируются спектры отдельных молекул CO, хемосорбированных на разных участках наночастиц Pt. После удаления нелокальных минимумов (выбросов) каждой точке данных присваиваются метки, которые описывают структуру материала. Координационная среда сайта количественно определяется его значением обобщенного координационного числа (GCN). С помощью кластеризации k-средних присваиваются значения GCN дискретным группам GCN, затем применяется суррогатная модель для расширения отфильтрованного первичного набора данных на основе DFT до вторичного набора данных сложных спектров. Эта суррогатная модель включает коэффициенты масштабирования покрытия, которые количественно связывают сдвиги в частотах и интенсивности первичного набора фильтрованных данных с пространственным покрытием. Последним шагом в суррогатной модели, основанной на физике, является спектральная свертка, выполняемая преобразованием Фурье для генерации синтетических комплексных спектров. Генерируются сотни тысяч сложных спектров, связанных с различным распределением занятых участков, различным покрытием и различной шириной линий. Суррогатная модель, управляемая данными, обучается на этих синтетических сложных спектрах для изучения микроструктуры. После обучения на синтетических комплексных спектрах модель применяют к экспериментальным спектрам [30].
Joshua Lansford и Dionisios Vlachos [30], с помощью синтетических ИК-спектров монооксида углерода на платине, реализовали полиномиальную регрессию с помощью ансамблей нейронных сетей для изучения функций распределения вероятностей (PDF), которые описывают места адсорбции и количественно определяют неопределенность прогноза, являющуюся следствием неточности применения идеальной модели к реальным системам. Они использовали полученные данные, чтобы вывести подробную микроструктуру поверхности из экспериментальных спектров и распространить эту методологию на другие системы.
Для обеспечения меры неопределенности в предсказанных PDF-файлах, когда модель применяется к экспериментальным спектрам, ансамбли из 200 нейронных сетей обучали на синтетических спектрах, сгенерированных с применением различных разделов первичных данных DFT. Вследствие этого вышеупомянутые ансамбли улавливают неопределенность модели, возникающую из первичных данных DFT, на основе которых генерируются синтетические ИК-спекты, дисперсии, связанной с коэффициентом масштабирования, и конкретного набора задействованных гиперпараметров.
Таким образом, авторы сгенерировали сотни тысяч сложных синтетических спектров с помощью свертки Фурье, которые адекватно покрывают все пространство состояний спектров CO, адсорбированного на наночастицах Pt.
С применением нейросетевых моделей решается обратная задача, где прогнозируется микроструктура поверхности, совпадающая с синтетическим, а в конечном счете и с экспериментальным спектрами. Занятые места адсорбции могут быть идентифицированы экспериментально только для очень простых поверхностей с упорядоченными слоями адсорбатов на монокристаллах методами сканирующей туннельной микроскопии высокого давления (HPSTM) [31] или дифракции электронов с низкими энергиями (LEED) [32] в сочетании с масс-спектрометрией (MS) и термодесорбционной спектроскопией (TDS). Такая подробная научная характеристика поверхности недоступна для настоящих гетерогенных катализаторов.
Из-за отсутствия достаточного количества экспериментальных ИК-спектров, для которых была выполнена одновременная характеристика мест адсорбции CO и покрытия, Joshua Lansford и Dionisios Vlachos [30] тестировали структурную суррогатную модель с эффектами покрытия на четко определенных литературных экспериментальных данных спектроскопии высокого разрешения характеристических потерь энергии электронами (HREELS) и данных поверхностного комбинационного рассеяния.
На рис. 13a–13в показаны дискретизированные экспериментальные спектры, прогнозы модели обратного машинного обучения для типов и сред сайтов, а также диапазон предсказания 95%. Предполагается, что хемосорбированный CO на Pt(111) займет 70% верхних позиций и 30% мостиковых позиций. Покрытие CO высокое на уровне с низким индексом (группа 10), что соответствует экспериметальным результатам. Это свидетельствует, что большая часть данной поверхности действительно состоит из Pt(111).
Рис. 13.
Литературные данные по спектрам HREEL и поверхностно-усиленной рамановской спектроскопии (SERS) для CO на платине, преобразованные в ИК-спектры, и предсказанная микроструктура: (а) – дискретизированные литературные спектры; (б), (в) – прогнозы модели обратного машинного обучения для типов и сред сайтов. Адаптировано из [30]. Распространяется на условиях лицензии Creative Commons CC BY license. Adapted from [30]. Distributed under the terms of the Creative Commons CC BY license.
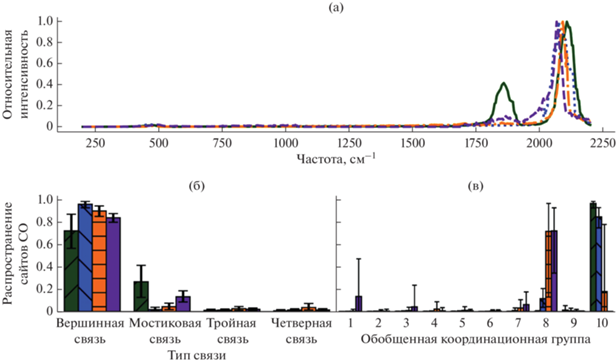
В целом предсказания модели прекрасно согласуются с известной микроструктурой и охватом CO для ограниченного числа хорошо описанных исследований и дают уверенность в том, что предлагаемый метод может быть использован для анализа экспериментальных данных ИК-спектроскопии для устранения “materials gap”. Кроме того, авторы обнаружили, что включение низкочастотного диапазона спектров повышает вероятность совпадений и позволяет также определять NO на наночастицах Pt.
В дальнейшем Joshua Lansford и Dionisios Vlachos разработали физические концепции, включая энергию орбитального взаимодействия и интеграл перекрытия энергии, для объяснения и возможности предсказания способности тест-молекул различать структурные параметры катализатора [33]. Они рассматривали заселенность перекрывания кристаллических орбиталей (COOP) для конкретных молекулярных орбиталей и количественно оценивали характер их связей, который напрямую влияет на частоты колебаний. С использованием только одного расчета адсорбата из теории функционала плотности была вычислена энергия взаимодействия отдельных молекулярных орбиталей адсорбата с атомными орбиталями сайта на различных участках поверхности. Сочетание COOP с разрешенной молекулярной орбиталью и изменением энергии орбитального взаимодействия позволяло выбрать оптимальную молекулу зонда. Эти концепции были рассмотрены на примере трех молекул зонда, а именно CO, NO и C2H4, на поверхностях Pt с различными координационными числами для понимания того, какие орбитали металла взаимодействуют с орбиталями адсорбата и смещают частоты больше всего. Наконец, применяя ранее разработанную структуру машинного обучения [30], авторы показали, что модели, обученные на сотнях тысяч спектров C2H4, вычисленных с помощью DFT, превосходят модели, обученные с помощью спектров CO и NO.
Описанный метод может также использоваться для различных целевых параметров, которые могут быть спроектированы для оптимизации каталитической активности или других свойств химии поверхности, таких как селективность, устойчивость к коррозии и загрязнению, устойчивость к патогенам и грибкам, ширина запрещенной зоны на поверхности и проводимость, емкость и концентрация дефектов полупроводников.
Поскольку, как уже говорилось ранее, большинство катализаторов – это сложные системы, которые являются носителями не отдельных веществ, а целых ансамблей, полученные в ходе исследований результаты можно применять для моделирования отдельных, наиболее вероятных, структур. Принципиальная возможность моделирования подобных структур требует мультимодального подхода и является предметом будущих исследований. Использование методов, основанных на “первых принципах”, поможет смоделировать структуры веществ и процессы, протекающие на катализаторах, что в свою очередь ускорит целевое создание новых каталитических систем.
ОСНОВНЫЕ ТРУДНОСТИ ПРИМЕНЕНИЯ МО В ОБЛАСТИ КАТАЛИЗА
Существует несколько проблем применения машинного обучения в области катализа. Наиболее распространенной проблемой для исследователей является небольшой объем данных. Поскольку абсолютный объем данных, относящихся к текущим исследованиям, невелик, очень часто бывает трудно продолжить МО на их основе. Один из способов решения вопроса нехватки данных – трансферное обучение, при котором информация и обученная модель МО переносятся из задач обучения с большим количеством данных на задачи, страдающие дефицитом данных [34].
Другая не менее важная проблема – это отсутствие общей структурированной базы данных электронных спектров, полученных за последние десятилетия. Наличие такой базы может способствовать применению моделей машинного обучения в области катализа, что не только ускоряет разработку модели, позволяя избежать избыточных дорогостоящих вычислений DFT, но также предполагает появление “эталонов” для тестирования моделей. Недавно был выпущена база данных Open Catalyst Dataset для разработки и тестирования моделей машинного обучения [35], которая обеспечивает траектории оптимизации в отношении многочисленных поверхностей и адсорбатов. Поскольку информация в базы данных попадает из разных источников, эти данные зачастую не согласованы из-за различных параметров расчета.
Стоит также упомянуть проблему предвзятости отношения к некоторым методам. В связи с явным преимуществом одних из них в плане применимости методов больших данных потенциально существует некоторая систематическая предвзятость в построении моделей, которые базируются на таких методах. Есть риск, что такие методы отлично смогут описывать соответствующие экспериментально наблюдаемые параметры модели, полученные с их использованием, но не будут отражать все детали поведения исследуемой системы. В плане выбора инструмента важно не допускать редукционизма и продолжать изучать каталитические системы в разных аспектах.
Машинное обучение способствовало огромному прогрессу во всех областях, особенно в области катализа. Синергетическая комбинация методов ab initio и машинного обучения сформировала мощную парадигму для понимания каталитических реакций и разработки новых катализаторов. С дальнейшим развитием теории катализа и методов МО ожидается, что для исследования и прогнозирования каталитических реакций на поверхностях будет применяться больше подходов, “основанных на данных” (data-driven, data-informed и data-inspired подходы).
ЗАКЛЮЧЕНИЕ
Наука о данных активно внедряется в различные области естественных наук в качестве универсального инструмента для обработки и интерпретации данных. Несмотря на успешное использование методов МО в материаловедении и молекулярных науках, в катализе эти методы пока слабо распространены. Тем не менее, нет никаких сомнений, что МО станет одним из главных способов описания механизмов многих каталитических процессов.
Для применения методов МО в разработке новых катализаторов необходимы не только расчетные, но и экспериментальные данные для конкретных каталитических реакций. Особенно это относится к гетерогенным катализаторам, для которых в настоящее время отсутствуют адекватные теоретические модели. “Информатика катализа” тесно связано с “хемоинформатикой” и “информатикой материалов”. Ее отличие заключается в том, что катализ представляет собой динамический процесс, на который влияют структура катализатора и химическая природа каталитически активных центров. Для дальнейших исследований необходимо связать теоретические модели на основе данных и сложные реальные системы с учетом динамики каталитического процесса.
Прямое же использование опубликованных данных для методов МО не представляется целесообразным, поскольку может привести к обнаружению только узкого диапазона точно настроенных вариаций ранее изученных катализаторов. Более логичным будет применять такие наборы данных в качестве обучающих для моделей МО. В этом смысле экспериментальные данные должны быть получены при одинаковых или сопоставимых условиях реакции и обобщены в единую базу данных. Ее появление может ускорить использование моделей машинного обучения в области катализа. Кроме того, сочетание науки о данных с теоретическими и экспериментальными подходами способно привести к углубленному пониманию существующих каталитических процессов.
В итоге интеграция методов МО с имеющимися химическими и физическими моделями в перспективе может объединить разные аспекты каталитического процесса от молекулярного до реакторного уровня и значительно ускорить процесс внедрение новых технологий и решений в химическую индустрию.
Список литературы
Крупнов А.А., Погосбекян М.Ю. // Кинетика и катализ. 2019. Т. 60. № 2. С. 181.
Дюсембаева А.А., Вершинин В.И. // Кинетика и катализ. 2019. Т. 60. № 1. С. 129.
Landrum G.A., Penzotti J.E., Putta S. // Meas. Sci. Technol. 2005. V. 16. P. 270.
Erdem Günay M., Yıldırım R. // Catal. Rev. Sci. Eng. 2021. V. 63. P. 120.
Jollife I.T., Cadima J. // Philosophical Transactions of the Royal Society. A: Mathematical, Physical and Engineering Sciences. 2016. V. 374. P. 1.
Wei J., Cao S. // International Conference on Intelligent Computing and Control Systems (ICCS), IEEE, Secunderabad, India, 27–28 June 2019. P. 85.
Landrum G. A., Penzotti J. E., Putta S. // Meas. Sci. Technol. 2004. V. 16. № 1. P. 270.
Dai Q., Zhang C., Wu H. // Int. J. Database Theory and Application. 2016. V. 9. P. 1.
Somvanshi M., Chavan P. // International conference on computing communication control and automation (ICCUBEA), IEEE, Pune, Maharashtra, India, 12–13 August 2016. P. 1.
Jinnouchi R., Asahi R. // J. Phys. Chem. Lett. 2017. V. 8. P. 4279.
Saikia P., Baruah R.D., Singh S.K., Chaudhuri P.K. // Comput. Geosci. 2020. V. 135. P. 1.
Li H., Zhang Z., Liu Z. // Catalysts. 2017. V. 7. P. 1.
Yano J., Yachandra V.K. // Photosynth. Res. 2009. V. 102. P. 241.
Cibin G., Gianolio D., Parry S.A., Schoonjans T., Moore O., Draper R., Miller L.A., Thoma A., Doswell C.L., Graham A. // Radiat. Phys. Chem. 2020. V. 175. P. 1.
Mathew K., Zheng C., Winston D., Chen C., Dozier A., Rehr J.J., Ong S.P., Persson K.A. // Scientific Data. 2018. V. 5. P. 1.
Ravel B., Newville M. // J. Synchrotron Radiat. 2005. V. 12. P. 537.
Zheng C., Mathew K., Chen C., Chen Y., Tang H., Dozier A., Kas J.J., Vila F.D., Rehr J.J., Piper L.F.J., Persson K.A., Ong, S. P. // Comput. Mater. 2018. V. 4. P. 1.
Timoshenko J., Jeon H.S., Sinev I., Haase F.T., Herzog A., Cuenya B.R. // Chem. Sci. 2020. V. 11. P. 3727.
Timoshenko J. Lu D., Lin Y., Frenkel A.I. // J. Phys. Chem. Lett. 2017. V. 8. P. 5091.
Miyazato I., Takahashi L., Takahashi K. // Mol. Syst. Des. Eng. 2019. V. 4. P. 1014.
Weckhuysen B.M. // Phys. Chem. Chem. Phys. 2003. V. 5. P. 4351.
Serhan M., Sprowls M., Jackemeyer D., Long M., Perez I.D., Maret W., Forzani, E. // AIChE Annual Meeting, Conference Proceedings, 2019.
Penner-Hahn J.E. / In: Comprehensive Coordination Chemistry. II. Eds. Mccleverty J.A., Meyer T.J. Amsterdam–Oxford–New York–San Diego: Elsevier–Pergamon Press, 2003. V. 2. P. 159.
Wu L.B., Wu L.H., Yang W.M., Frenkel A.I. // Catal. Sci. Technol. 2014. V. 4. P. 2512.
Huth F., Schnell M., Wittborn J., Ocelic N., Hillenbrand R. // Nature Mater. 2011. V. 10. P. 352.
Ellis D.I., Broadhurst D., Clarke S.J., Goodacre R. // Analyst. 2005. V. 130. P. 1648.
Howley T., Madden M.G., O’Connell M.-L., Ryder A.G. // International Conference on Innovative Techniques and Applications of Artificial Intelligence. Cambridge, United Kingdom. 12–14 December 2005. P. 209.
Zou T., Dou Y., Mi H., Zou J., Ren Y. // Analyt. Biochem. 2006. V. 355. P. 1.
Kale M.J., Christopher P. // ACS Catal. 2016. V. 6. P. 5599.
Lansford J.L., Vlachos D.G. // Nature Commun. 2020. V. 11. P. 1.
Davies J.C., Nielsen R.M., Thomsen L.B., Chorkendorff I., Logadottir A., Lodziana Z., Besenbacher F. // Fuel Cells. 2004. V. 4. P. 309.
Steininger H., Lehwald S., Ibach H. // Surf. Sci. 1982. V. 123. P. 264.
Lansford J.L., Vlachos D.G. // ACS Nano. 2020. V. 14. P. 17295.
Agarwal N., Sondhi A., Chopra K., Singh G. / In: Tiwari S., Trivedi M., Mishra K., Misra A., Kumar K., Suryani, E. (eds) Smart Innovations in Communication and Computational Sciences. Advances in Intelligent Systems and Computing. Springer, Singapore, 2021. V. 1168. P. 145.
Chanussot L., Das A., Heras-Domingo J., Goyal S., Ho C., Lavril T., Palizhati A., Parikh D., Riviere M., Shuaibi M., Tran K., Ulissi Z., Yoon J., Zitnick C.L. // 2020 Virtual AIChE Annual Meeting.2020.
Дополнительные материалы отсутствуют.
Инструменты
Кинетика и катализ