

Коллоидный журнал, 2019, T. 81, № 6, стр. 782-787
Электрокинетические измерения при высоких концентрациях электролитов с многозарядными катионами
В. Д. Соболев 1, *, И. П. Сергеева 1, К. Г. Саббатовский 1
1 Институт физической химии и электрохимии им. Фрумкина РАН
119071 Москва, Ленинский проспект, 31, Россия
* E-mail: vsobolev@phiche.ac.ru
Поступила в редакцию 20.06.2019
После доработки 27.06.2019
Принята к публикации 01.07.2019
Аннотация
Методом капиллярной электрокинетики впервые проведены измерения тока течения в растворах солей с двух- и трехзарядными катионами при высоких концентрациях электролита. Показано, что ток течения пропорционален величине приложенного давления. При концентрации электролита 1 М диффузный слой полностью отсутствует. Наличие тока течения в этом случае свидетельствует о том, что гидродинамически неподвижных слоев у твердой, гладкой поверхности не существует.
ВВЕДЕНИЕ
Состояние твердой поверхности в контакте с растворами электролитов определяется пространственным распределением ионов вблизи нее. Такое распределение зарядов традиционно называют двойным электрическим слоем (ДЭС), хотя часто оно имеет более сложное строение, чем просто два слоя ионов. Структура ДЭС обсуждается уже более века, поскольку он обеспечивает коллоидные взаимодействия и во многом определяет физические и химические свойства поверхности.
В соответствии с современными представлениями о структуре ДЭС заряду твердой поверхности σ0 соответствует электрический потенциал ψo. Затем идет слой Гельмгольца, в котором различают внутреннюю часть, свободную от заряда, или слой Штерна, на границе которого располагаются гидратированные противоионы. Диффузная часть ДЭС, или слой Гуи, располагается за внешней плоскостью Гельмгольца, которой соответствуют заряд σd и потенциал ψd [1]. В работе [2] показано, что подобная структура ДЭС возникает при добавлении неэлектростатического гидратационного отталкивания к электростатическому кулоновскому потенциалу. Результатом электростатического притяжения и гидратационного отталкивания противоионов и поверхности является образование слоя, подобного слою Штерна, отделяющего диффузный слой от поверхности. Однако невозможность прямого измерения поверхностного потенциала в дисперсных системах не позволяет количественно определить структуру межфазного электрического слоя.
Тангенциальное движение жидкости вдоль заряженной поверхности приводит к возникновению электрокинетических явлений. Теория электрокинетических явлений была разработана в тесной связи с теорией ДЭС и электростатических поверхностных сил [1, 3–5]. Различают явления, связанные с движением твердой фазы относительно жидкости (электрофорез, электроосмос) под действием внешнего электрического поля, и потенциал течения, возникающий при движении жидкости под действием перепада давления. При этом считается, что течение имеет место вдоль так называемой плоскости скольжения, отделяющей застойную зону от области течения. Потенциал, соответствующий этой плоскости, называют электрокинетическим, или дзета-потенциалом. Величина дзета-потенциала, измеряемая в экспериментах, является характеристикой электрического состояния поверхности и широко применяется для оценки устойчивости электростатически стабилизированных коллоидов и объяснения их реологических свойств и процессов седиментации.
Однако вопрос о соотношении ζ-потенциала (и связанной с ним плотности электрокинетического заряда σel) с потенциалом на границе жидкость/твердое тело ψo (и связанной с ним поверхностной плотностью заряда σ0) полностью не решен. Рассчитанное из величины ζ-потенциала значение σel всегда меньше σ0. Это различие объясняют тем, что часть противозаряда включена в слой, который является гидродинамически неподвижным. Происхождение такого застойного слоя, его толщина и свойства до сих пор являются предметом дискуссии.
Другим важным межфазным электрокинетическим параметром является поверхностная проводимость, которую обычно относят к электропроводности тонкого слоя жидкости вблизи границы фаз, т.е. в застойной зоне. Избыток заряда в слое может двигаться под действием электрического поля, направленного тангенциально поверхности. Изучению поверхностной проводимости посвящено значительное число теоретических [1, 5–7] и экспериментальных работ, которые проводились на различных объектах [8–16]. Во всех работах отмечается, что влияние поверхностной проводимости необходимо учитывать в случае растворов с низкими значениями концентрации электролитов. В ряде работ установлено, что подвижность ионов в застойной зоне незначительно отличается от объемной [6, 8, 17] и понижена в присутствии полимера [16]. В результате этих исследований был сделан вывод о том, что плотный слой ведет себя так, как будто растворитель в нем неподвижен, в то время как подвижность ионов в нем сохраняется [17]. Моделирование с помощью молекулярной динамики показало, что между поверхностью и слоем ионов находится слой чистого растворителя [17]. В работе [18] методом рентгеновской электронной спектроскопии на жидких микроструях измеряли потенциал поверхности наночастиц диоксида кремния в растворах электролитов. На основании зависимости величины потенциала от радиуса гидратированного катиона был сделан вывод, что толщина слоя Штерна соответствует единичному слою молекул воды плюс радиус гидратированного катиона.
Несмотря на значительное число исследований, вопрос о физической природе плоскости скольжения и толщине застойной зоны нельзя считать полностью решенным.
Ранее в нашей лаборатории были проведены измерения потенциала и тока течения при высоких концентрациях хлорида калия, когда толщина диффузного слоя сравнима с толщиной слоя Гельмгольца [19]. Было выявлено наличие тока течения, на основании чего был сделан вывод об отсутствии гидродинамически неподвижных слоев у твердой гладкой поверхности.
Целью данной работы является выяснение влияния заряда катиона на течение в плотной части ДЭС.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы и методы
Растворы BaCl2 и LaCl3 с концентрацией 1 М готовили по навеске из реактивов BaCl2 · 2H2O и LaCl3 · 7H2O марки “х. ч.”. Растворы меньших концентраций получали последовательным разбавлением
Эксперименты проводили на установке, сконструированной в нашей лаборатории [20]. Метод основан на измерении потенциала и тока течения в тонком кварцевом капилляре радиусом 5–10 мкм. В контакте с водными растворами поверхность плавленого кварца заряжена отрицательно, плотность поверхностного заряда в нейтральной среде равна 1–2 мкКл/см2. Измерения проводили в капиллярах, для которых κr > 100 (κ – обратный дебаевский радиус, r – радиус капилляра). Для небольших концентраций раствора, вплоть до 10–2 М, измерение ζ-потенциала проводилось в режиме измерения потенциала течения. В этом случае ζ-потенциал рассчитывался по уравнению Гельмгольца–Смолуховского
В растворах с концентрациями, большими чем 0.01 М, проводились измерения тока течения при ступенчатом изменении давления: сначала при нулевом давлении, а затем при различных ненулевых его значениях. В этом случае ζ-потенциал рассчитывался по формуле
где l – длина капилляра, ΔI – изменение тока течения при изменении давления.Ток измеряли при помощи пикоамперметра А2-4 с погрешностью 0.5%. Общая погрешность измерения, включая погрешности измерений тока и давления, не превышала 2%.
Для экспериментов отбирали капилляры, для которых измеряемый потенциал в растворе KCl с концентрацией 10–4 М составлял 80–100 мВ и оставался неизменным при изменении давления, т.е. не зависел от скорости течения раствора через капилляр.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Метод капиллярной электрокинетики позволяет проводить измерения потенциала и тока течения при высоких перепадах давления, не выходя за пределы ламинарного режима течения. Поскольку ток течения прямо пропорционален величине давления, создается возможность измерения токов течения даже при низких значениях потенциала поверхности.
При высоких концентрациях электролитов измерения вели в режиме тока течения. Электрокинетический потенциал в этом случае рассчитывается по уравнению (2). Цикл измерений состоял из измерения в течение нескольких десятков секунд тока течения при выбранном давлении, затем давление уменьшалось до нуля и проводилось измерения при нулевом давлении, затем снова подавали давление и цикл повторялся несколько раз при разных давлениях. Время измерения одного значения тока составляло 0.6 с. Таким образом, при каждом давлении ток измерялся не менее 50–100 раз. Рассматривались только промежутки с постоянным давлением (ненулевым и нулевым), интервалы с падающим/возрастающим давлением исключались из рассмотрения [19].
Результаты измерений для раствора BaCl2 с концентрацией 0.1 М представлены на рис. 1. На рис. 1а показаны исходные результаты измерения тока течения при разном давлении, на основании которых были рассчитаны зависимости тока течения от приложенного давления (рис. 1б).
Рис. 1.
Зависимость тока течения 0.1 M раствора Ba-Cl2 от времени при разных значениях перепада давления (а) и от приложенного давления (ΔI/ΔP = 7.861 × × 10–4 нА/ат) (б).
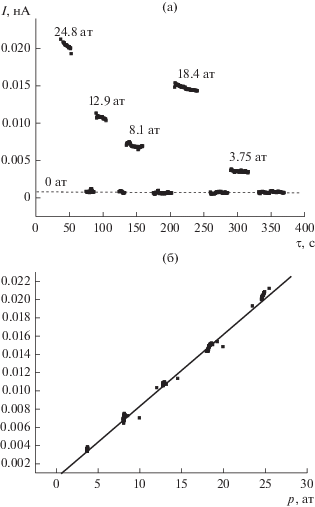
Массив полученных экспериментальных данных обрабатывали по стандартной методике методом наименьших квадратов. Как видно на рис. 1а, ток течения с достаточно хорошей точностью линейно зависит от давления, т.е. отношение ток/давление остается постоянным, что свидетельствует о постоянстве дзета-потенциала и, следовательно, о постоянстве положения плоскости скольжения. В этом случае угловой коэффициент B соответствующей линейной зависимости равен ΔI/ΔP. Для приведенного на рис. 1 примера ΔI/ΔP = 7.861 × 10–4 нА/ат.
На рис. 2 представлены зависимости тока течения от давления для раствора BaCl2 с концентрацией 1 М. Из приведенной зависимости следует, что при этой концентрации BaCl2 происходит перезарядка поверхности капилляра, и ток течения становится отрицательным. Зависимость тока течения от давления и в этом случае линейна; угловой коэффициент ΔI/ΔP = 0.00138 нА/ат.
Рис. 2.
Зависимость тока течения от приложенного давления для 0.1 M раствора BaCl2; ΔI/ΔP = = 0.00138 нА/ат.
На рис. 3 приведены аналогичные зависимости для растворов LaCl3 с концентрацией 0.1 М (рис. 3а) и 1 М (рис. 3б). Значения коэффициента B равны 0.00227 и 0.00296 соответственно.
Рис. 3.
Зависимости тока течения от приложенного давления для 0.1 М растворов LaCl3 (ΔI/ΔP = = 0.00227 нА/ат) (а) и LaCl3 (ΔI/ΔP = 0.00296 нА/ат) (б).

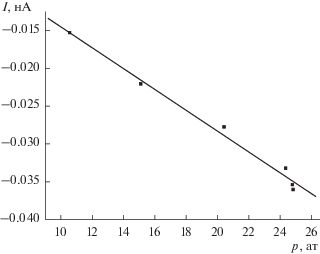
Наши измерения показали, что в растворе BaCl2 при концентрации 1 М происходит перезарядка поверхности капилляра. В растворах LaCl3 перезарядка наблюдается уже при концентрации 0.1 М. Для выяснения вопроса, при какой концентрации происходит перезарядка, нами были измерены зависимости ζ-потенциала от концентрации электролитов. Полученные зависимости показаны на рис. 4.

Из рисунка следует, что перезарядка в случае BaCl2 происходит при концентрации выше 0.1 М, а в случае LaCl3 уже при концентрации 10–3 М. Нами было установлено, что адсорбция BaCl2 на поверхности плавленого кварца обратима. После перезарядки поверхности при концентрации 1 М проводились измерения в растворе KCl с концентрацией 10–4 М. Значение ζ-потенциала полностью восстанавливалось. После измерения в растворах LaCl3 поверхность оставалась заряженной положительно даже после длительной фильтрации через капилляр 10–4 М раствора KCl под давлением 10 ат.
При расчете ζ-потенциала по формуле Смолуховского помимо отношения ΔI/ΔP необходимо знать величины диэлектрической константы и вязкости при высоких концентрациях электролитов. Зависимость диэлектрической проницаемости от концентрации соли изучалась во многих работах как теоретически [21–23], так и экспериментально [24–26]. Теоретические расчеты и данные экспериментов свидетельствуют об уменьшении диэлектрической константы с ростом концентрации соли (диэлектрический декремент). В работе [24] обнаружена также зависимость диэлектрической константы от размера катиона в ряду солей щелочных металлов. Измерения в растворах NaCl [26] показали, что при малых концентрациях, вплоть до 10–1 диэлектрическая константа изменяется незначительно. Причиной электрического декремента является тот факт, что локальное электрическое поле вокруг каждого иона больше, чем внешнее поле, и ориентирует диполи воды в его окрестности. Это создает гидратную оболочку, которая окружает ионы. В результате уменьшается воздействие внешнего поля на молекулы воды и, следовательно, понижается диэлектрическая проницаемость [22].
Для оценки значений ζ-потенциала нами были использованы величины диэлектрической проницаемости, полученные в работе [25]. При концентрации 1 М для раствора KCl ε = 68.5, для BaCl2 ε = 64, для LaCl3 ε0 = 64.
Экспериментальному измерению вязкости водных растворов электролитов посвящен ряд работ [27–30]. Показано, что в растворах галогенидов Na и K наблюдается незначительное изменение вязкости вплоть до концентрации 2 M [27–29]. Вязкость 0.1 М растворов BaCl2 и LaCl3 равна 1.040 [30] и 1.068 сПз [29], а их 1 М растворов – 1.233 [30] и 1.745 сПз [29] соответственно.
Расчеты по уравнению (2) дали следующие значения ζ-потенциала:
1) 0.1 М KCl – ζ = –21.8 мВ, 1 М KCl – ζ = –6.8 мВ [19],
2) 0.1 М BaCl2 – ζ = –3.82 мВ, 1 М BaCl2 – ζ = 9.94 мВ,
3) 0.1 М LaCl3 – ζ = 11.33 мВ, 1 М LaCl3 – ζ = 23.43 мВ.
К сожалению, исходя из полученных данных, трудно выявить закономерность относительно влияния заряда катиона на ток течения в плотном слое. Перезарядка поверхности кварца в 1 М растворе BaCl2 и в 0.1 М и 1 М растворах LaCl3 приводит к тому, что ДЭС формируется вблизи поверхностей, заряд которых имеет разные знаки. KCl является нейтральным электролитом и не адсорбируется на поверхности кварца. В этом случае может происходить уменьшение заряда вследствие понижения степени диссоциации силанольных групп в растворе с более высокой ионной силой и, как следствие, уменьшение величины измеряемого потенциала. В растворах BaCl2 и LaCl3 положительный заряд поверхности увеличивается с ростом их концентрации, и мы наблюдаем увеличение ζ-потенциала.
Основной вывод, который можно сделать на основе наших экспериментов, состоит в том, что гидродинамически неподвижные слои вблизи гладкой поверхности отсутствуют. Ранее мы сделали такой же вывод исходя из результатов измерений тока течения в растворах хлорида калия [19]. При концентрации раствора KCl 1 М толщина диффузного слоя сравнима с размером молекулы воды (0.3 нм), т.е. диффузный слой практически отсутствует.
Толщина диффузного слоя рассчитывается по уравнению
где F0 – число Фарадея, R0 – универсальная газовая постоянная, Т – температура, I – ионная сила раствора, $I = \frac{1}{2}\sum\nolimits_1^n {{{c}_{i}}z_{i}^{2}} .$ Для водных растворов ${\delta }\sim \frac{{3{\text{\;}} \times {\text{\;}}{{{10}}^{{ - 10}}}}}{{\sqrt I }}$ [м].В соответствии с уравнением (3) толщина диффузного слоя уменьшается с ростом ионной силы раствора. Ионная сила растворов BaCl2 в 3 раза, а растворов LaCl3 в 6 раз больше, чем растворов KCl той же концентрации. Следовательно, в этих случаях диффузный слой отсутствует полностью. Таким образом, наличие измеренного нами тока течения может быть связано только с течением в плотной части ДЭС.
ЗАКЛЮЧЕНИЕ
Впервые проведены измерения тока течения при высоких концентрациях растворов солей с двух- и трехзарядными катионами. Показано, что ток течения пропорционален величине приложенного давления. При концентрации электролитов 1 М толщина диффузного слоя меньше размера молекулы воды (0.3 нм), т.е. диффузный слой полностью отсутствует. Наличие тока течения в этом случае говорит о том, что гидродинамически неподвижных слоев у твердой гладкой поверхности не существует. Результаты нашей работы свидетельствуют о том, что современные представления о строении ДЭС требуют дальнейшего уточнения.
Список литературы
Delgado A.V., González-Caballero F., Hunter R.J., Koopal L.K., Lyklema J. // J. Colloid Interface Sci. 2007. V. 309. P. 194.
Brown M.A., Bossa G.V., May S. // Langmuir. 2015. V. 31. P. 11477.
Духин С.С. Электропроводность и электрокинетические свойства дисперсных систем. Киев: Наукова думка, 1975.
Hunter R.J. Foundations of Colloid Science. Oxford: Oxford University Press, 2001. Ch. 8.
Lyklema J. Fundamentals of Interfaces and Colloid Science V. II. N.Y.: Academic Press, 1995. Chs. 3, 4.
Lyklema J. // J. Phys. Condensed Matter. 2001. V. 13. P. 5027.
Erickson D., Li D., Werner C. // J. Colloid Interface Sci. 2000. V. 232. P. 186.
Kijlstra J., van Leewan H.P., Lyklema J. // Langmuir. 1993. V.9. P. 1625.
Lobbus M., van Leewan H.P., Lyklema J. // J. Colloid Interface Sci. 2000. V. 229. P. 174.
Leroy P., Devau N., Revil A., Bizi M. // J. Colloid Interface Sci. 2013. V. 410. P. 81.
Leroy P., Tournassat C., Bernard O., Devau N., Azaroual M. // J. Colloid Interface Sci. 2015. V. 451. P. 21.
Li S., Leroy P., Heberlink F., Devau N., Jougnot D., Chiaberg C. // J. Colloid Interface Sci. 2015. V. 468. P. 262.
Jimenez M.L., Arroyo F.J., Carrique F., Delgado A.V. // J. Colloid Interface Sci. 2007. V. 316. P. 836.
Barchini R., van Leewan H.P., Lyklema J. // Langmuir. 2000. V. 16. P. 8338.
van der Wal A., Minor M., Norde W., Zehnder A.J.B., Lyklema J. // Langmuir. 1997. V. 13. P. 165.
Minor M., van Leewan H.P., Lyklema J. // Langmuir. 1999. V. 15. P. 6677.
Lyklema J., Rovillard S., de Coninck J. // Langmuir. 1998. V. 14. P. 5659.
Brown M.A., Abbas Z., Kleibert A., Green R.G., Goel A., May S., Squires T.M. // Phys. Rev. X. 2016. V. 6. 011007.
Сергеева И.П., Соболев В.Д. // Коллоид. журн. 2018. Т. 80. С. 91.
Churaev N.V., Sergeeva I.P., Sobolev V.D., Derjaguin B.V. // J. Colloid Interface Sci. 1981. V. 84. P. 451.
Persson R.A.X. // J. Chem. Chem. Phys. 2017. V. 19. P. 1982.
Levy A., Andelman D., Orland H. // Phys. Rev. Lett. 2012. V. 108. 227801.
Gavish N., Promislov K. // Phys. Rev. E. 2016. V. 94. 012611.
Wei Y.-Z., Chiang P., Sridhar S. // J. Chem. Phys. 1992. V. 96. P. 4569.
Hasted J.B., Ritson D.M., Colle C.H. // J. Chem. Phys. 1948. V. 16. P. 1.
Norteman K., Hilland J., Kaatze U. // J. Chem. Phys. 1997. V. 101. P. 6864.
Takaeshi S., Koteaki H. // Bull. Chem. Soc. Jpn. 1961. V. 34. P. 1260.
Goldsack D.E., Franchetto R. // Can. J. Chem. 1977. V. 55. P. 1062.
Liu Y.-S., Hu Y.F., Hao Q.-C., Zhang X.-M., Liu Z.-C., Li J.-G. // J. Chem. Eng. Data. 2009. V. 54. P. 739.
Азизов Н.Д., Ахундов Т. С. // Журн. физ. химии. 1997. Т. 71. С. 1955.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал