

Коллоидный журнал, 2021, T. 83, № 1, стр. 3-34
Фотоиндуцированное изменение гидрофильности поверхности тонких пленок
А. В. Рудакова 1, *, А. В. Емелин 1, 2
1 Санкт-Петербургский государственный университет,
лаборатория “Фотоактивные нанокомпозитные материалы”
198504 Петергоф, Санкт-Петербург, ул. Ульяновская, 1, Россия
2 Санкт-Петербургский государственный университет, кафедра фотоники
198504 Петергоф, Санкт-Петербург, ул. Ульяновская, 1, Россия
* E-mail: aida.rudakova@spbu.ru
Поступила в редакцию 23.07.2020
После доработки 14.08.2020
Принята к публикации 20.08.2020
Аннотация
Выполнен обзор литературы по исследованию явления фотоиндуцированного изменения гидрофильности поверхности тонких пленок оксидов металлов, лежащего в основе способности фотоактивных материалов к самоочищению. Новизна работы состоит в обобщении и критическом анализе обширного материала по представленной тематике с момента открытия в 1997 г. эффекта фотоиндуцированной супергидрофильности диоксида титана до настоящего времени. Изложена суть явления, описаны предлагаемые механизмы эффекта, приведены примеры влияния различных факторов на кинетику и термодинамику данного фотопроцесса. Предложен механизм процесса, основанный на фотоактивации и фотодезактивации поверхностных центров, ответственных за последующую реструктуризацию адсорбированных молекул воды в полислойном гидроксильно-гидратном покрове на поверхности оксидных пленок, приводящую к изменению свободной поверхностной энергии и, как следствие, гидрофильности поверхности. В заключение рассмотрены возможные пути управления смачиваемостью поверхности фотоактивных материалов с помощью света, среди которых особо отмечен способ создания гетероструктурных покрытий.
1. ВВЕДЕНИЕ
В 1997–1998 гг. группа исследователей под руководством Фуджишимы и Хашимото с коллегами из японской компании ТОТО продемонстрировала возможность при помощи света изменять гидрофильность поверхности монокристаллов и тонких поликристаллических пленок диоксида титана [1, 2]. Явление заключалось в способности поверхности диоксида титана переходить из слабо гидрофильного состояния в супергидрофильное при воздействии на нее ультрафиолетового (УФ) излучения (рис. 1). Хранение образца с УФ-наведенной супергидрофильностью в темноте приводило к возвращению поверхности в исходное слабо гидрофильное состояние.
Рис. 1.
(a) Гидрофобная поверхность покрытия из диоксида титана (θ = 72 ± 1°) до воздействия УФ-света. (b) Супергидрофильная поверхность этого же покрытия (θ = 0 ± 1°) после УФ-облучения. (c) Взаимодействие гидрофобной поверхности (a) с парами воды. Образование тумана (маленькие капли воды) затрудняло просмотр текста на бумаге, помещенной за стеклом. (d) Создание УФ-облучением противотуманной поверхности, высокая гидрофильность которой предотвращает образование капель воды и делает текст хорошо видимым. Взято из работы [1].

Стоит заметить, что непосредственно термин ”фотоиндуцированная супергидрофильность”, обозначающий суть эффекта, не был применен в этих работах и впервые был использован в научной литературе позже, в 2000 г., тремя исследовательскими группами из Японии [3–7]. Этот термин стал общеупотребимым на сегодняшний день.
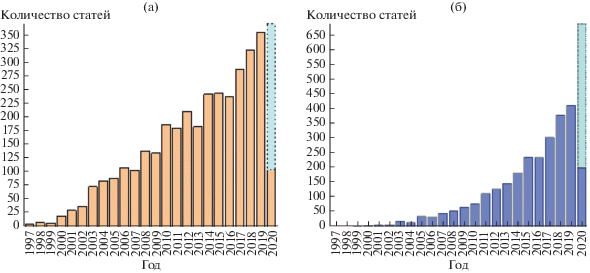
Начиная с момента открытия эффекта фотоиндуцированной супергидрофильности для поверхности диоксида титана, количество работ, посвященных исследованию фотоактивных материалов и покрытий, смачиваемость которых водой способна изменяться под действием света, неуклонно растет. На рис. 2а представлен график роста числа статей, опубликованных между 1997 и 2019 годами, в которых встречаются термины “hydrophilicity”, “wettability”, “photoinduced” и “light-controlled”. В последнее время эффект фотоидуцированной гидрофильности поверхности материалов и покрытий на их основе находит все большее применение в различных областях. Так, например, процесс разделения водно-масляных смесей можно реализовать на мембранах и сетках из фотоактивных материалов, обладающих фотоиндуцированной суперлиофобностью [8, 9].
Рис. 2.
Динамика роста числа публикаций за период с 1997 по 2020 г., процитированных в базе данных Scopus с использованием в качестве ключевых слов следующих терминов: (а) hydrophilicity, wettability, photoinduced и light-controlled, (б) hydrophilicity и self-cleaning. Для 2020 г. приведены данные за первые 4 месяца, и с учетом их динамики показано ожидаемое количество статей к концу года.
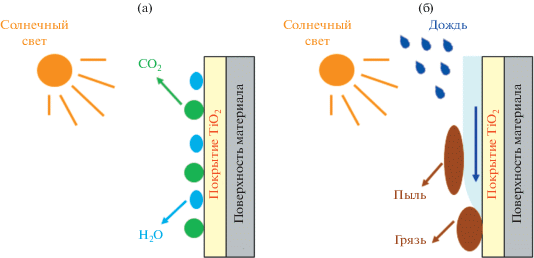
В пионерской работе [1], посвященной обнаруженному явлению фотоиндуцированной супергидрофильности диоксида титана, был сделан вывод о потенциально высокой практической значимости этого эффекта в сочетании с превосходными фотокаталитическими свойствами TiO2 для создания материалов, поверхности которых способны к самоочищению (self-cleaning surfaces). Эффект самоочищения фотокатализатора основывается на его высокой окислительной способности и явлении фотоиндуцированной супергидрофильности поверхности [10–13]. В некоторых случаях усиленная под действием света бактерицидная способность покрытий также рассматривается как неотъемлемая часть самоочищения поверхности материала. На рис. 3 схематически представлен механизм самоочищения поверхности фотоактивного материала. Под действием света фотокатализатор способен разлагать адсорбированные на поверхности загрязнения органического происхождения с образованием углекислого газа и воды. При этом самоочищающаяся поверхность переходит в супергидрофильное состояние, а воздействие воды (например, дождевой) очищает ее от остатков органических загрязнений, пыли и т.п., “смывая” их по поверхности слоя воды, сильно связанного с супергидрофильной поверхностью.
Рис. 3.
Иллюстрация способности фотоактивного материала к самоочищению поверхности, основанной на совместном проявлении фотокаталитической активности (а) и фотоиндуцированной супергидрофильности поверхности (б).
Прикладные исследования эффекта фотоиндуцированной супергидрофильности, начавшиеся сразу после его открытия, привели к созданию самоочищающихся и незапотевающих стекол, самоочищающихся материалов для внешней и внутренней отделки зданий, покрытий туннелей, автодорог и т.п. Материалами с самоочищающими свойствами покрыты стены и крыши различных известных строений по всему миру. На рис. 4 представлены примеры использования таких покрытий.
Рис. 4.
Примеры использования самоочищающихся покрытий. (а) Стекла заднего вида в автомобилях без (сверху) и с (внизу) самоочищающейся пленкой на основе диоксида титана. (б) Панорамное окно в музее фотокатализа в Академии наук и технологии Канагавы в Японии. На левой половине стекла нет, а на правой его половине есть самоочищающееся покрытие на основе диоксида титана. Самоочищающимися материалами покрыты такие известные сооружения как стадион “AT&T Stadium” (штат Техас, США) (в), Национальный центр исполнительских искусств (Пекин, Китай) (г), Сиднейский оперный театр (Сидней, Австралия).

Несмотря на широкое использование самоочищающихся покрытий, проводимые прикладные исследования до сих пор основаны, главным образом, на интуиции исследователей, так как вопрос о причинах и механизмах эффекта фотоиндуцированной супергидрофильности поверхности некоторых оксидов металлов остается открытым. На рис. 2б продемонстрирован ежегодный прирост публикаций, посвященных созданию и исследованию материалов, обладающих самоочищающимися свойствами. Судя по приведенным на нем данным, интерес к таким материалам постоянно увеличивается в связи с проблемами защиты окружающей среды и охраны здоровья людей.
В настоящем обзоре мы детально рассматрим индуцированные светом процессы на гладких поверхностях тонких пленок полупроводниковых оксидов, шероховатость которых не превышает ±10 нм. Фактор микрогеометрии поверхности оказывает большое влияние на смачиваемость, привнося особенности в механизм и кинетику фотоиндуцированного изменения гидрофильности поверхности, и требует отдельного рассмотрения [13–15].
2. ПОВЕРХНОСТНАЯ ЭНЕРГИЯ И УГОЛ СМАЧИВАНИЯ
Основной термодинамической характеристикой поверхности является ее свободная энергия, характеризующая избыточную энергию поверхности по сравнению с объемом. Иллюстративно формирование поверхности и “появление” поверхностной энергии представлено на рис. 5.

При этом при разделении объема вещества на две части на разрыв связей необходимо затратить работу (энергию) ${{W}_{{11}}},$ которая соответствует удвоеной, поскольку образуется две поверхности, свободной энергии (γ1) идеальной поверхности:
Очевидно, что источником поверхностной энергии является нарушение координации поверхностных атомов по сравнению с объемом вещества вследствие обрыва химических связей. Процессом, обратным образованию поверхностей, представленному на рис. 5, является их исчезновение в результате “слияния” двух объемов вещества, термодинамически выгодный процесс, сопровождающийся выделением энергии ${{W}_{{11}}}$ [16].Понятия поверхности и, соответственно, поверхностной энергии характерны для вещества в жидком и твердом состояниях, где роль межатомных и/или межмолекулярных взаимодействий в объеме является определяющей. Поскольку в результате появления поверхности общая энергия системы повышается, система будет стремиться к ее снижению. Для веществ в жидком состоянии такое снижение достигается за счет минимизации отношения поверхность/объем, т.е. жидкость будет стремиться преобрести форму сферы в отсутствие других действующих сил. Для твердого тела, структура которого задана сильными межатомными (межионными) связями, понижение поверхностной энергии, как правило, происходит за счет релаксации и реконструкции поверхности, т.е. ее перестройки и образования поверхностной структуры, отличной от структуры объема твердого тела [16]. Тем не менее, такая перестройка поверхности не приводит к полному исчезновению поверхностной энергии как атрибуту существования поверхности, а только частично снижает энергию системы.
Теоретически перестройку поверхности твердого тела также можно рассматривать как способ уменьшить отношение поверхность/объем. Для описания такого стремления вводят понятие силы поверхностного натяжения. Коэффициент пропорциональности, характеризующий силу (F), действующую на единицу длины поверхности (L), называется поверхностным натяжением ($\gamma $):
С энергетической точки зрения поверхностное натяжение – это коэффициент пропорциональности, связывающий изменение поверхностной энергии (G) с изменением площади поверхности (S):
Таким образом, поверхностное натяжение γ однозначно характеризует поверхностную энергию и силу поверхностного натяжения и стремление системы к снижению как поверхностной энергии, так и отношения объем/поверхность.
Способом снижения поверхностной энергии данного вещества является его взаимодействие с другим веществом. По сути дела, существование поверхностной энергии твердого тела является причиной таких явлений как адсобция, адгезия, смачиваемость, катализ и др. На рис. 6 представлена схема взаимодействия поверхностей двух веществ, каждая из которых характеризуется своей поверхностной энергией (поверхностным натяжением), отражающая процесс адгезии [16]. Энергия адгезии ${{W}_{{12}}},$ которая выделяется при таком взаимодействии, выражается как
здесь ${{\gamma }_{{12}}}$ – энергия межфазной поверхности (гетероперехода).
Очевидно, что взаимодействие двух фаз с понижением энергии всей системы происходит только при условии ${{\gamma }_{1}} + {{\gamma }_{2}} > {{\gamma }_{{12}}}.$ В противном случае взаимодействия не происходит. Выполнение данного условия является также движущей силой адгезии, смачивания, адсорбции молекул и катализа на поверхности твердого тела. В зависимости от значения энергии межфазной поверхности, ${{\gamma }_{{12}}},$ поверхности твердых тел можно разделить на два типа: высокоэнергетические и низкоэнергетические. Так, оксиды металлов, металлы, различные стекла и керамические материалы обладают высокой поверхностной энергией на воздухе, но являются низкоэнергетическими в воде. Уменьшение поверхностной энергии таких поверхностей на воздухе осуществляется за счет адсорбции молекул воды, углекислого газа, органических загрязнителей.
Одним из способов воздействия на данное соотношение является фотооблучение твердого тела, которое способно изменить поверхностную энергию твердого тела и инициировать такие процессы как фотостимулированная адсорбция и фотокатализ.
Изменение гидрофильного состояния поверхности твердого тела при облучении также связано с изменением его поверхностной энергии. Поскольку одним из компонентов системы является жидкость (например, вода), обладающая способностью к уменьшению поверхностной энергии при изменении формы, в этом случае следует рассматривать баланс поверхностных энергий на трех межфазных границах: твердое тело–газ, твердое тело–жидкость и жидкость–газ. Такой баланс определяется уравнением Юнга (5) [17, 18]
(5)
${{\gamma }_{{{\text{lv}}}}}\cos \theta = {{\gamma }_{{{\text{sv}}}}} - {{\gamma }_{{{\text{ls}}}}},$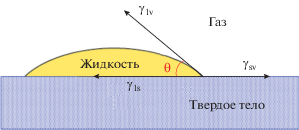
Из уравнения (5) следует, что значение угла смачивания задается соотношением
(6)
${\text{cos}}\theta = \frac{{{{\gamma }_{{{\text{sv}}}}} - {{\gamma }_{{{\text{ls}}}}}}}{{{{\gamma }_{{{\text{lv}}}}}}},$Для рассматриваемой трехкомпонентной системы твердое тело–жидкость–газ выражение для энергии адгезии (4) определяется уравнением Дюпре–Юнга (7) через угол смачивания θ:
(7)
${{W}_{{\text{a}}}} = {{\gamma }_{{{\text{lv}}}}} + {{\gamma }_{{{\text{sv}}}}} - {{\gamma }_{{{\text{ls}}}}} = {{\gamma }_{{{\text{lv}}}}}\left( {1 + \cos \theta } \right).$На практике при исследовании гидрофильности поверхности твердого тела для определения свободной поверхностной энергии основное внимание уделяют ее полярной (γps) и дисперсионной (γds) составляющим, измеряя углы смачивания одной и той же поверхности несколькими жидкостями с известными значениями этих составляющих (γp и γd); наиболее распространен метод двух жидкостей, основанный на подходе Оуэнса–Вендта [24]. В этом методе в качестве тестовых жидкостей обычно используют воду, как жидкость с высокой полярной составляющей (γp = 51.0 мДж/м2, γd = 21.8 мДж/м2), и йодистый метилен CH2I2, как жидкость с высокой дисперсионной составляющей (γd = 48.5 мДж/м2, γp = 2.3 мДж/м2). Полная свободная поверхностная энергия (γ tot) твердого тела находится как сумма найденных компонент γps и γds.
Таким образом, основным измеряемым макроскопическим параметром при исследовании гидрофильности/гидрофобности поверхности материала и ее изменения при облучении светом является угол смачивания (краевой угол смачивания, или контактный угол) θ водой или другой жидкостью. Измерения θ обычно проводят методами оптической тензиометрии [25]. Величина и направление изменения контактного угла θ в ходе облучения материала характеризуют способность его поверхности к фотоиндуцированному гидрофильному переходу. Значения θ < 10° (5°) характеризуют поверхность как супергидрофильную; при значениях θ > 120° поверхность считается супергидрофобной [13, 26].
Определяя изменение контактного угла как функцию времени облучения, получают кинетическую кривую фотоиндуцированного изменения θ для воды на поверхности фотоактивного материала. В этом случае за параметр, характеризующий эффективность процесса фотостимулированного перехода поверхности в иное, отличное от начального, состояние, принимают время достижения этого состояния или начальную скорость процесса.
Для количественного описания процесса фотоиндуцированного изменения гидрофильности поверхности были предприняты попытки использовать различные функции контактного угла. Например, в работах [27–30] было введено понятие скорости конверсии, представляющей собой скорость изменения обратной величины контактного угла. Очевидно, что применение этого параметра не имеет физического смысла, в отличие от скорости изменения контактного угла, которая пропорциональна скорости изменения поверхностной энергии. Опираясь на уравнение Юнга (5), легко продемонстрировать взаимосвязь изменений угла смачивания и поверхностной энергии твердого тела:
(8)
$\frac{{d{{\gamma }_{{{\text{sv}}}}}}}{{dt}} = - \frac{1}{{2{{\gamma }_{{{\text{lv}}}}}}}\left( {1 + \cos \theta } \right)\sin \theta \frac{{d\theta }}{{dt}},$В этом смысле использование cos θ в качестве параметра, связывающего скорость изменения измеряемого угла смачивания и скорость изменения поверхностной энергии, как это сделано в работе [32], более целесообразно, чем обратной величины этого угла.
Однако на сегодняшний день изменение угла смачивания остается наиболее удобным и часто используемым параметром при определении направления изменения поверхностной энергии твердого тела. Наряду с углом смачивания рекомендуется использовать данные по определению величины свободной поверхностной энергии и ее изменения в ходе облучения образца для более глубокого понимания природы межмолекулярных взаимодействий, имеющих место на поверхности во время фотостимулированного процесса.
При исследовании гидрофильности поверхности фотоактивных материалов основными до сих пор остаются две проблемы. Первая из них – это невоспроизводимость исходного состояния тонкой пленки (в частности, угол смачивания исходной пленки водой может варьироваться от 20° до 60°). Вторая проблема заключается в различиях в процедурах предварительной подготовки пленочных образцов и в условиях окружающей среды (температура, относительная влажность, чистота воздуха и т.д.) в ходе таких исследований [33–36]. В настоящий момент не существует единого подхода к получению исходного гидрофильного состояния поверхности, свободной от органических загрязнений, и проведению экспериментов по изучению фотоиндуцированного изменения гидрофильности поверхности тонких пленок. Это затрудняет систематические исследования влияния различных факторов на гидрофильность поверхности и делает практически невозможным сопоставление результатов, полученных в разных лабораториях. В работе [31] показано, что при отсутствии каких-либо органических загрязнений поверхности кинетика фотоиндуцированного уменьшения контактного угла не зависит от толщины нанопокрытий из диоксида титана. Одновременно предлагается методика формирования воспроизводимого начального состояния поверхности нанопокрытий, представленная на рис. 8, позволяющая получать углы смачивания с отклонением менее 10–15% от среднего значения для исследуемого набора покрытий.
Рис. 8.
Схема формирования воспроизводимого состояния поверхности, включающая термообработку, гидратацию, и последующую фотоактивацию гидратированного состояния [31].

Стоит отметить, что при использовании идентичной процедуры подготовки поверхностей покрытий различие в их фотоиндуцированном гидрофильном поведении может быть обусловлено другими многочисленными факторами, в том числе методом синтеза и типом подложки. Например, в работах [37, 38] продемонстрировано влияние подложки на параметры процесса фотостимулированного перехода поверхности поликристаллических покрытий из диоксида титана анатазной модификации, полученных методом золь–гель, в супергидрофильное состояние. Было обнаружено, что скорость такого перехода ниже для TiO2-покрытий на стеклянных подложках по сравнению с таковым на кварцевых подложках. Эффект объясняется присутствием в стекле ионов щелочных и щелочноземельных металлов, диффундирующих в слой фотоактивного материала в ходе термической обработки при синтезе покрытий. Для предотвращения такого влияния было предложено предварительно покрывать стеклянные подложки тонким слоем диоксида кремния.
3. ГИПОТЕЗЫ О МЕХАНИЗМАХ ФОТОИНДУЦИРОВАННОЙ СУПЕРГИДРОФИЛЬНОСТИ
Как отмечалось выше (смотри раздел 2), очевидной причиной проявления эффекта фотоиндуцированного изменения гидрофильности поверхности является изменение поверхностной энергии твердого тела при взаимодействии со светом. Вместе с тем, существует множество возможных причин изменения поверхностной энергии при каком-либо воздействии. Основным макроскопическим проявлением изменения гидрофильности поверхности является изменение ее смачиваемости водой (т.е. угла смачивания). Однако переход к причинам изменения поверхностной энергии на “атомарном” уровне и установление детального механизма эффекта фотоиндуцированного изменения гидрофильности, в том числе и фотоиндуцированной супергидрофильности, является сложной задачей, которая на сегодняшний день в полном объеме не решена и остается на уровне предлагаемых гипотез.
Анализ современного состояния фундаментальных исследований позволяет выделить три основные гипотезы о причинах, приводящих к эффекту фотоиндуцированной гидрофильности [39].
Согласно первой гипотезе этот эффект в случае оксидов металлов связан только с их фотокаталитической активностью в процессе разложения биографических или привнесенных извне органических загрязнений поверхности [30, 34, 40–44]. Так, Хендерсон с коллегами исследовали поверхность монокристаллического TiO2 с адсорбированным монослоем 3-метилацетата, проявляющую гидрофобность [40]. Авторы продемонстрировали, что УФ-облучение этой системы в присутствии кислорода приводит к разложению органического соединения на поверхности оксида и она становится гидрофильной. Аналогичные результаты для монокристалла диоксида титана представлены в работах [41–43]. Однако данные сравнительного исследования фотокаталитической активности ряда оксидов металлов в реакции разложения метиленового голубого и их способности к проявлению эффекта фотоиндуцированной супергидрофильности, представленные в работах [45, 46], не подтверждают рассматриваемую гипотезу. В этих работах продемонстрировано, что не для всех оксидов металлов, проявляющих фотокаталитическую активность при разложении адсорбированных молекул красителя, гидрофильность поверхности увеличивается под действием света, и только покрытия из некоторых оксидов, а именно TiO2, SnO2 и ZnO, проявляют эффект фотоиндуцированного перехода в супергидрофильное состояние.
Методом поверхностной рентгеновской дифракции авторами работы [47] обнаружено увеличение шероховатости поверхности (110) монокристалла TiO2 (рутил), свободной от органических загрязнений, вызванное УФ-освещением в присутствии влажного воздуха. Это наблюдение было объяснено образованием метастабильных наноразмерных гидрофильных областей на поверхности вследствие миграции на нее фотогенерированных электронно-дырочных пар, что совпадает с предположением, сделанным в пионерской работе [1], и свидетельствует о независимости обнаруженного эффекта от наличия поверхностных загрязнений органического происхождения.
В работе [10] получены экспериментальные результаты, показывающие, что высокая гидрофильность поверхности диоксида титана является результатом не столько ее чистоты, сколько изменения ее структуры и образования метастабильных областей. Представленные в работе рентгеновские фотоэлектронные спектры в области пика С1s для диоксида титана до и после УФ-облучения в течение 12 ч свидетельствуют, что супергидрофильное состояние поверхности может быть достигнуто даже при наличии углеродных загрязнений. (Обязательное присутствие таких загрязнений в виде карбоксилатных групп на поверхности монокристалла диоксида титана (110), находящегося на воздухе или в растворе, подтверждается результатами исследования [48]). Вместе с тем, экспериментальным путем показано, что зависимости между степенью достижения супергидрофильного состояния (значением контактного угла воды) и количеством углеродных загрязнений на поверхности не существует. Кроме того, в работе [10] отмечена высокая стабильность достигнутого при УФ-облучении супергидрофильного состояния поверхности TiO2, что подтверждается величиной энергии активации (62.4 кДж/моль) перехода поверхности из супергидрофильного в менее гидрофильное состояние, т.е. обратного процесса. Таким образом, сделан вывод, что удаление органических загрязнений за счет фотоокисления может сделать поверхность только умеренно гидрофильной в условиях окружающей среды, при этом контактный угол воды достигает значений 10°–20°, а супергидрофильность поверхности может быть достигнута благодаря образованию неких метастабильных состояний на поверхности под действием УФ-облучения.
К похожему выводу о наличии, по крайней мере, двух этапов в процессе фотоиндуцированного перехода поверхности диоксида титана в супергидрофильное состояние пришли авторы [49], изучая спектральную зависимость кинетических кривых фотоиндуцированного изменения контактного угла воды на поверхности пленок этого оксида. По виду кинетических зависимостей процесс был разделен ими на два этапа. При этом на первом этапе происходит фоторазложение органических загрязнений поверхности (θ воды уменьшается до 10°–20°). На втором этапе механизм изменений состояния поверхности (при этом θ воды становится меньше 10°–20°) не связан с фотокаталитической активностью TiO2, но, как показано, зависит от состава или структуры поверхности.
Вторая гипотеза относительно механизма фотоиндуцированного изменения гидрофильности поверхности, высказанная впервые в работе [50], связана с термическим действием света, вызывающим десорбцию внешних слабосвязанных слоев адсорбированной воды с поверхности, что повышает поверхностную энергию и, следовательно, гидрофильность поверхности твердого тела. При этом последующая реадсорбция молекул воды восстанавливает исходную структуру гидратного покрова, что понижает поверхностную энергию. Методом инфракрасной (ИК) фурье-спектроскопии диффузного отражения авторы работы [50] исследовали изменения в спектрах воды, адсорбированной на поверхности порошкообразного диоксида титана, до и после его УФ-облучения и продемонстрировали, что в результате облучения в течение 3 ч температура образца увеличивалась от комнатной (25°C) до 55°C. Однако в этой же работе было показано, что использование водяного фильтра, блокирующего ИК-излучение, являющееся источником тепловой энергии, значительно снижает рост температуры, до 5–6°C, даже при столь длительном облучении образца. Термоэффект такой величины можно объяснить трансформацией энергии, высвобождающейся при рекомбинации фотогенерированных носителей или при электрон-фононном взаимодействии, в тепловую энергию. Так, в работе [51] методом ИК-фурье-спектроскопии in situ продемонстрирована десорбция адсорбированных молекул воды с поверхности анатаза во время 10-секундного воздействия УФ-излучения в вакууме (ртутная лампа с водяным фильтром и светофильтром с максимумом пропускания на 365 нм, плотность излучения 90 мВт/см2), по величине сопоставимая с ее десорбцией при нагреве этого же образца до 90°C, и столь же быстрое восстановление (примерно за 50 с) широкой полосы поглощения OH-групп этой воды (с максимумом при 3000 см–1), что указывает на значительную роль электронного, а не термического, фактора в процессе фотостимулированного изменения гидрофильности поверхности.
Этот же вывод следует из результатов ряда других исследований. Например, в работе [52] представлены результаты фотоэлектрохимических исследований поверхности электрода, покрытого пленкой TiO2, демонстрирующие зависимости вольтамперных характеристик электрода от наличия облучения и его интенсивности, а также угла смачивания от величины потенциала, приложенного к такому образцу. Вывод о необходимости электронного фотовозбуждения в процессе фотостимулированного перехода поверхности диоксида титана в супергидрофильное состояние подтверждается также зависимостями скорости перехода в это состояние и угла смачивания от спектрального состава и интенсивности действующего света, полученными в работах [49] и [31]. Оказалось, что только облучение светом с энергией фотонов больше ширины запрещенной зоны дает положительный результат, а спектральные зависимости изменений поверхностной энергии и краевого угла [31] подобны спектральным зависимостям квантовых выходов фотоадсорбции некоторых газов на диоксиде титана [53, 54], а также фоторазложения фенолов [55]. При этом в спектральных зависимостях присутствуют особенности, совпадающие с энергией первых непрямого и прямого межзонного переходов в диоксиде титана. Спектральная зависимость фотоиндуцированного гидрофильного перехода для поверхности ZnO, полученная в работе [56], также свидетельствует о важной роли электронного фотовозбуждения твердого тела. Сходство между спектральными зависимостями эффективности фотоиндуцированного гидрофильного перехода и отношениeм поверхностных концентраций электронов и дырок, оцененных на основе соотношения между квантовыми выходами фотостимулированной адсорбции кислорода и метана [54], указывает на то, что возможной причиной изменения гидрофильности является перераспределение поверхностного заряда, вызванное действием света на твердое тело. Таким образом, представленные экспериментальные данные прямо указывают на то, что процесс фотоиндуцированного изменения гидрофильности поверхности твердого тела непосредственно связан с его электронным фотовозбуждением.
Согласно третьей гипотезе [2, 6, 27, 52, 57–60], эффект фотоиндуцированной супергидрофильности объясняется электронным фотовозбуждением твердого тела, приводящим к генерации свободных электронов и дырок. Однако полагается, что следствием этого является не фотокаталитическая “очистка” поверхности от органических загрязнений, как предлагается в первой гипотезе [40–43], а захват фотогенерированных носителей поверхностными центрами с последующей реорганизацией поверхности и изменением структуры гидратного покрова. На настоящий день существуют несколько механизмов с участием фотоносителей, причем все они касаются процесса именно на поверхности диоксида титана. Рассмотрим основные предлагаемые механизмы.
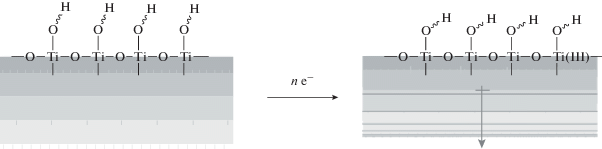
Первое объяснение механизма фотоиндуцированной супергидрофильности поверхности диоксида титана было предложено Фуджишимой с коллегами после обнаружения этого эффекта [2, 6, 57]. По результатам исследования монокристаллических поверхностей диоксида титана, представленных разными кристаллографическими плоскостями – (110), (100) и (001) – было предположено, что при УФ-облучении на поверхности образуются анионные вакансии в результате захвата дырок и разрядки анионов кислорода, а ионы ${\text{T}}{{{\text{i}}}^{{4 + }}}$ восстановливаются до ${\text{T}}{{{\text{i}}}^{{3 + }}}$ при захвате электронов, что создает новые центры для диссоциативной адсорбции молекул воды. Схематически эти процессы можно представить как
В результате такой адсорбции на фотоиндуцированных дефектных участках поверхности образуются новые гидроксильные группы. Считалось, что в предложенном “восстановительном” механизме электронные центры играют определяющую роль при формировании новых поверхностных дефектов – центров адсорбции воды, а молекулы воды “залечивают” образующиеся на поверхности под действием света кислородные вакансии [60].
В дальнейшем эти же исследователи показали, что приложение положительного потенциала к TiO2-электроду заметно ускоряет достижение супергидрофильности его поверхности. Это предполагает значимую роль фотогенерированных дырок в механизме фотоиндуцированной супергидрофильности диоксида титана [27, 52, 58–60]11. На основании полученных результатов был предложен другой, так называемый “окислительный”, механизм, в соответствии с которым происходит фотоокисление поверхностного мостикового кислорода с возникновением кислородной вакансии. Дальнейшее взаимодействие молекул воды с такой вакансией приводит к образованию гидроксильных групп, ответственных за увеличение гидрофильности поверхности, согласно следующей схеме:

Существует несколько модификаций этого механизма, однако во всех случаях ключевая роль отводится образующимся кислородным вакансиям. Одновременно фотогенерированные электроны захватываются поверхностными ионами Ti4+, но необязательно связанными с этими кислородными вакансиями.
Умозрительное предположение об образовании и вовлечении кислородных вакансий в процесс гидрофилизации поверхности является слабым местом предложенного механизма, так как идентифицировать образование таких вакансий в условиях окружающей среды практически невозможно – они активно взаимодействуют с кислородом и водой атмосферы еще до попытки их регистрации. Более того, экспериментальные результаты некоторых работ опровергают возможность образования кислородных вакансий на поверхности монокристалла диоксида титана под действием УФ-света в условиях высокого вакуума, т.е. до взаимодействия с молекулами окружающей среды [40, 41].
Для описания процесса фотостимулированной трансформации реальной поверхности с адсорбированной водой в супергидрофильное состояние предложенные ранее механизмы были модифицированы [52]. Одна из таких модификаций представлена на приведенной ниже схеме.

Как видно из этой схемы, в случае гидратированной поверхности существование кислородных вакансий невозможно, однако в ходе процесса фотоактивации происходит значительная перестройка гидратно-гидроксильного покрова, в том числе увеличение числа гидроксильных групп, связанных непосредственно с атомами поверхности. Именно этот механизм послужил отправной точкой для расширенной гипотезы, рассматривающей процессы в полислое воды, адсорбированной на поверхности твердого тела (смотрите раздел 4).
В последнее время высказывалось мнение о том, что изменение гидрофильности поверхности связано с одновременным действием фотоносителей заряда обоих знаков (электронов и дырок), в том числе, на поверхности диоксида титана. Так, в работе [60] авторы предложили модель комбинированного механизма, согласно которому могут протекать как восстановительные (с участием фотоэлектронов), так и окислительные (с участием фотодырок) поверхностные процессы, приводящие к увеличению гидрофильности поверхности диоксида титана (рис. 9).
Рис. 9.
Модель комбинированного окислительно-восстановительного механизма фотоиндуцированного гидрофильного перехода. (а) В отсутствие кислорода протоны молекул воды восстанавливаются с образованием межузельных атомов водорода. (б) В присутствии кислорода его молекулы восстанавливаются до супероксида или перекиси водорода. В обоих случаях фотодырки взаимодействуют с молекулами воды, что приводит к образованию разнообразных гидрофильных фрагменты. Взято из работы [60].
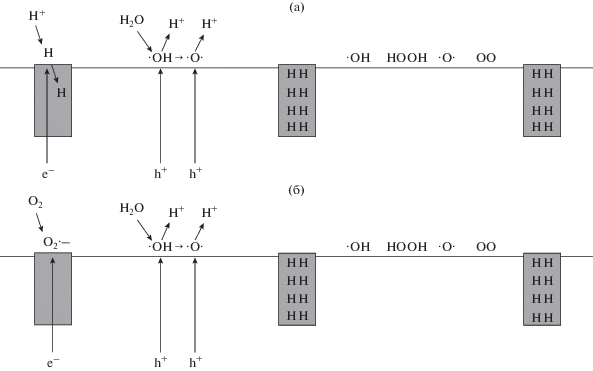
В ходе процесса, представленного на рис. 9, предполагается образование различных радикалов. Идентификация таких нестабильных соединений является достаточно трудной задачей и требует использования таких методов как, например, спектроскопия электронного парамагнитного резонанса. Однако Хоффманн с коллегами, используя ИК-фурье-спектроскопию диффузного отражения, смогли обнаружить образование двух типов гидроксильных радикалов на гидратированной поверхности поликристаллического диоксида титана после УФ-облучения: новые полосы поглощения при 3716 и 3683 см–1 были отнесены соответственно к валентным колебаниям поверхностных групп ${\text{Ti}}_{{\text{s}}}^{{3 + }}{\text{O}}{\kern 1pt} - {\kern 1pt} {{{\text{H}}}^{\centerdot }}$ и ${{{\text{O}}}_{{\text{s}}}}{\kern 1pt} - {\kern 1pt} {{{\text{H}}}^{\centerdot }}$ [61, 62]. Из исследований в области фотокатализа известно, что, действительно, эффективный восстановительный процесс с участием поверхностных электронов способствует повышению эффективности процессов с участием дырок, в том числе образованию новых поверхностных центров при захвате дырок на адсорбированных молекулах воды.
В заключение описания электронного механизма в рамках третьей гипотезы хотелось бы отметить, что до сих пор не существует единого доказанного представления об электронных процессах, происходящих на поверхности. Вместе с тем, достоверно продемонстрирована роль дефектности поверхности и наличия как электронных, так и дырочных центров, также отмечена важная роль кислорода [27, 42, 44, 50, 60] и степени гидратации поверхности [30, 50, 57, 60, 63] в процессе фотоиндуцированного гидрофильного перехода.
В литературе встречаются также исследования, в которых экспериментальные результаты и зависимости объясняют с помощью комбинации сразу двух или трех гипотез, описанных выше [49, 60, 64], или выявляются специфические факторы, влияющие на изменение гидрофильности поверхности и присущие конкретным образцам пленок. Так, например, в работах [65, 66] упоминается влияние на скорость процесса фотоиндуцированного гидрофильного перехода такого фактора как остаточное напряжение пленки, зависящее от природы подложки вследствие различия в коэффициентах теплового расширения материалов покрытия и подложки. Обнаружено [65], что растягивающее напряжение, приложенное к пленке диоксида титана, усиливает эффективность фотоиндуцированного перехода ее поверхности в супергидрофильное состояние, в то время как сжимающее напряжение подавляет этот эффект. Представленный пример демонстрирует возможность разнонаправленного изменения поверхностной энергии твердого тела и соответственно разнонаправленного изменения адгезии молекул одного и того же вещества к его поверхности. Очевидно, что изменение поверхностной энергии может достигаться с помощью различных видов воздействия на материал, например, механического, химического или физического. В свою очередь, достигнутое изменение поверхностной энергии может приводить к изменению гидрофильности поверхности. Однако следует помнить, что, несмотря на схожесть конечных результатов изменения поверхностной энергии и адгезионных свойств поверхности, причины и механизмы таких изменений могут быть разными.
В работе [46] продемонстрирован эффект фотоиндуцированного изменения гидрофильности поверхности различных оксидов металлов. Однако данные о механизме этого процесса на поверхности оксидов металлов, отличных от диоксида титана, и факторах, влияющих на этот переход, практически отсутствуют. Таким образом, следует признать, что на сегодняшний день единого общепризнанного понимания причин и механизма явления фотоиндуцированной супергидрофильности поверхности диоксида титана и, тем более, фотоиндуцированного изменения гидрофильности тонких пленок других оксидов металлов, не существует. В связи с этим фундаментальные исследования в этой области остаются актуальными по сей день.
4. МОДЕЛЬ ПОЛИСЛОЙНОЙ СТРУКТУРЫ АДСОРБИРОВАННОЙ ВОДЫ
Как следует из рис. 3, демонстрирующего способность фотоактивных покрытий к самоочищению, чтобы реализовать “очистку” поверхности покрытия за счет явления фотоиндуцированной супергидрофильности, поверхность должна быть покрыта слоем воды. При этом свет переводит поверхность в супергидрофильное состояние, которое может сохраняться на протяжении длительного времени, например покрытие из диоксида титана может оставаться супергидрофильным в течение 24–48 ч. Таким образом, очевидно, что изменение смачиваемости поверхности под действием света неразрывно связано с наличием слоя воды, адсорбированного на этой поверхности.
Результаты ИК-спектроскопических исследований, проведенных Анпо с коллегами [50, 67–69], свидетельствуют о существовании полислоя воды на поверхностях твердых тел. Авторы предлагают структурную модель адсорбированных кластеров воды, молекулы в которой различаются числом водородных связей, образованных ими с молекулами-соседями. В такой полислойной структуре внешний слой представлен координационно-ненасыщенными молекулами воды, т.е. частично свободными от водородных связей подобно кластерам молекул H2O в газовой газе. В то же время молекулы воды в объеме этого полислоя жестко связаны друг с другом H-связями, причем такое связывание отличается большей силой взаимодействия, чем H-связывание в объеме жидкой воды [68]. Было предположено, что эта особенность объясняется способностью поверхностных катионов титана координировать адсорбированные молекулы воды, что оказывает влияние на структуру последующих, формирующихся на этих молекулах воды, гидратных слоев. Возможность молекулярной адсорбции воды на атомах титана на поверхности диоксида титана также продемонстрирована совместным экспериментальным и теоретическим исследованием адсорбции воды на стехиометрической и восстановленной поверхности (011) TiO2 рутильной модификации при разной температуре [70], а также расчетами колебательных частот для молекул воды на кластере ${\text{T}}{{{\text{i}}}_{8}}{{{\text{O}}}_{{16}}}$ [71].
Наличие различных типов адсорбированной воды на поверхности диоксида титана подтверждается данными, полученными методами термопрограммируемой десорбции, спектроскопии ядерного магнитного резонанса (ЯМР) и ИК-спектроскопии [72, 73]. На основании выводов, сделанных из экспериментальных данных о строении адсорбированного слоя воды, предложена модель полислоя воды на поверхности оксидов металлов, представленная на рис. 10. Видно, что весь слой адсорбированной воды формально можно разделить на три подслоя: внешний – на границе с газовой атмосферой, внутренний – на границе с твердым телом и “буферный” (трансляционный), расположенный между внешним и внутренним слоями. Предложенная модель облегчает понимание процессов, происходящих при воздействии различных факторов на систему и приводящих к изменению гидрофильности поверхности материалов.
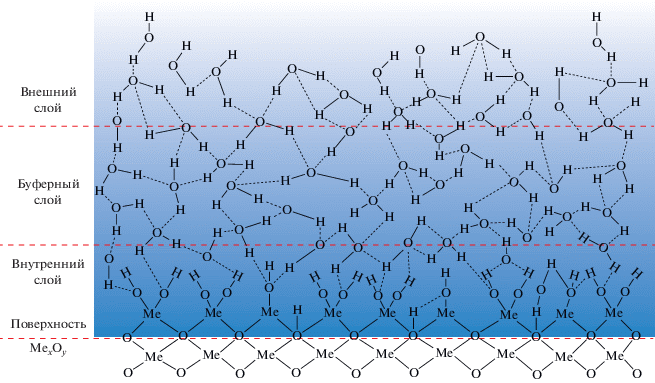
Упрощенная модель начальной стадии перестройки гидроксильно-гидратного покрова была представлена Хоффманном [62]. В предложенном механизме локализация электрона на поверхностном центре типа Ti4+ приводит к образованию состояния Ti3+. В результате изменяется электростатическое взаимодействие точечного заряда и диполей соседних поверхностных гидроксильных групп. В том числе, изменяется угол наклона гидроксильных групп по отношению к поверхности, т.е. происходит локальная реструктуризация гидроксильного слоя на поверхности TiO2 (рис. 11).
Рис. 11.
Изменение структуры гидроксильного слоя при локализации электрона на поверхностном катионе ${\text{T}}{{{\text{i}}}^{{4 + }}}$. Взято из работы [62].
Из предложенной модели с очевидностью следует, что при локализации на поверхностных состояниях достаточного числа носителей заряда можно достичь значительного возмущения и изменения структуры внутреннего гидроксильного слоя. Естественно, что такое возмущение отразится на величине поверхностной энергии на границе фаз твердое тело–вода. Это, в свою очередь, может привести к возмущению и перестройке последующих слоев гидроксильно-гидратного полислоя и к повышению поверхностной энергии в целом.
Формирование на поверхности полислоя адсорбированной воды и его реструктуризация при УФ-облучении подтверждаются экспериментальными данными, полученными с помощью различных методов. Так, интересные результаты продемонстрированы в работе [74], где методами спектроскопии генерации суммарной частоты лазерного излучения и пьезокварцевого микровзвешивания показано, что гидратный покров поверхности тонкой пленки диоксида титана, состоящий из 14 монослоев, увеличивался до 21 монослоя после УФ-облучения образца в отсутствие кислорода. То есть, воздействие облучения на поверхность диоксида титана способствует улучшению адгезии воды в результате фотовозбуждения твердого тела. При этом отмечено, что становится различимой структура полислоя адсорбированной воды, состоящая из слоя воды, непосредственно связанного с поверхностью, слоя воды, схожего по структуре с жидкой водой, и слоя воды на границе с воздухом. Предлагаемая авторами структура такого полислоя воды однозначно соответствует модели, представленной на рис. 10.
Экспериментальные наблюдения [74] согласуются с результатами, полученными методами ЯМР-спектроскопии и усиленной поверхностью инфракрасной абсорбционной спектроскопии [73, 75, 76]. Продемонстрировано увеличение количества слабо связанной воды на поверхности TiO2, т.е. образующей внешний слой полислойной структуры (рис. 10), после УФ-облучения независимо от присутствия кислорода. При этом отмечается, что количество воды, дополнительно адсорбированной в результате фотоактивации поверхности, заметно снижается в присутствии кислорода, что указывет на его существенную роль в явлении фотоиндуцированной гидрофильности.
Приведенные выше заключения подтверждаются результатами изучения тонких пленок диоксида титана с помощью атомно-силовой микроскопии и фотоэлектронной спектроскопии. Обнаружено, что после 90-минутного воздействия УФ-излучения толщина слоя воды на поверхности пленок TiO2 увеличивается на 30 нм [77]. Изучение спектров пропускания этих пленок показало [77, 78], что после УФ-облучения происходит сдвиг интерференционных максимумов в коротковолновую область, что указывает на уменьшение коэффициента преломления пленки TiO2 с полислойным гидратным покровом.
Следует отметить, что до сих пор неясно, как конкретно происходит реконструкция гидратно-гидроксильного покрова на поверхности материала при его облучении, так как данные, представленные разными авторами, зачастую противоречат друг другу. Вместе с тем, очевидно, что именно этот процесс отвечает за фотостимулированное изменение поверхностной энергии и гидрофильности поверхности.
Результаты, полученные методами ИК-спектроскопи и спектроскопии в УФ/видимой/ближней ИК областях [69], показывают, что при УФ-облучении порошка диоксида титана в вакууме десорбируется значительная доля слабо связанной воды, но ее количество восстанавливается при напуске в систему кислорода. Вместе с тем, такой значительной фотодесорбции воды с поверхности гидратированного образца не наблюдалось в присутствии кислорода даже при облучении в течение 3 ч, что обусловлено, предположительно, более сильным связыванием иона титана ${\text{T}}{{{\text{i}}}^{{4 + }}}$ с адсорбируемой молекулой воды. Такое поведение подтверждается и расчетами: частота деформационных колебаний молекулы, адсорбированной на ионе титана, имеет довольно низкое значение, 1560 см–1, по сравнению с таковым для водородно-связанных молекул воды в гидратно-гидроксильном слое (≈1620–1645 см–1) [71].
Изучение зависимостей гидрофильного поведения пленок ${\text{Ti}}{{{\text{O}}}_{2}}$ и ${\text{ZnO\;}}$ от температуры прогрева и поверхностной кислотности также демонстрирует наличие полислойного гидратного покрова на поверхности оксидов и важности участия отдельных его слоев как в формировании исходного состояния поверхности, так и в процессе фотоиндуцированного изменения гидрофильности [31, 79].
Таким образом, изменение смачиваемости поверхности обеспечивается коллективным поведением молекул воды в гидратно-гидроксильном слое, что подтверждает “трансляционную” схему изменений, происходящих в полислое воды на поверхности, рассмотренную выше. К такому же выводу пришли авторы работы [80], исследуя фотоиндуцированную смачиваемость покрытий из нитрида галлия. Определено, что один атом примесного кислорода посредством сети водородных связей способен влиять на состояние 1 × 105 молекул поверхностной воды. В работе [31] также проведена оценка количества поверхностных центров, ответственных за фотоиндуцированный переход диоксида титана в супергидрофильное состояние. Оказалось, что они составляют лишь небольшую долю от общего числа поверхностных центров, равную приблизительно 10–3–10–4.
В связи с изложенными фактами, очевидно, что в описанной выше гипотезе, предполагающей участие фотоносителей в процессе фотоиндуцированного изменения гидрофильности поверхности оксидов, требуется коррекция механизма для учета существования полислоя воды на поверхности оксидов металлов и его возможной реструктуризации как следствия изменения электронного состояния поверхностных центров под воздействием света.
Согласно термодинамическому подходу, переход поверхности из одного гидрофильного состояния в другое состояние, отличное от начального, как под действием света, так и при других воздействиях, связан с повышением/понижением величины свободной поверхностной энергии. В свою очередь, изменение поверхностной энергии связано с изменением типа и энергии взаимодействия адсорбированных молекул воды с поверхностью и между собой и, в частности, с изменением ее составляющих – полярной и/или дисперсионной. Это приводит к увеличению/уменьшению сродства поверхности пленки к молекулам воды или, иначе говоря, к улучшению/ухудшению адгезии воды к поверхности пленки.
5. ФАКТОРЫ, ВЛИЯЮЩИЕ НА ПРОЦЕСС ФОТОИНДУЦИРОВАННОГО ИЗМЕНЕНИЯ ГИДРОФИЛЬНОСТИ ПОВЕРХНОСТИ ТВЕРДОГО ТЕЛА
Опираясь на предложенную модель гидратно-гидроксильного полислоя (рис. 10), факторы, определяющие исходное гидрофильное состояние поверхности и влияющие на процесс фотостимулированного изменения ее смачиваемости, можно формально разделить на две группы: внешние и внутренние. Внешние факторы влияют на перестройку структуры адсорбционного полислоя воды со стороны его границы с газовой фазой (со стороны внешнего слоя), а внутренние – со стороны границы полислоя с твердым телом (со стороны внутреннего слоя). Тип фактора определяет направление передачи результата его воздействия через трансляционный слой от внутреннего слоя к внешнему слою или наоборот.
Внешние факторы приводят к изменению свободной поверхностной энергии пленки за счет изменения состава и структуры гидроксильно-гидратного полислоя воды на ее поверхности. В качестве примера внешнего фактора можно назвать кислотность поверхности. Так, предварительная выдержка покрытий из оксидов титана и цинка в водных растворах с разным значением pH приводит к изменению начального краевого угла воды на их поверхности, что говорит о различиях в начальном состоянии гидроксильного покрова на поверхности одного и того же материала (рис. 12) [31, 79]. В результате изменения кислотности поверхности происходит перестройка молекул и ионов, образующих гидратно-гидроксильный полислой, и, как следствие, изменяется величина свободной поверхностной энергии. В дальнейшем, как видно на рис. 13, это приводит и к изменению вида кинетики фотоиндуцированного изменения гидрофильности поверхности.

Рис. 13.
Кинетика фотоиндуцированного изменения краевого угла воды на поверхности тонких пленок ${\text{Ti}}{{{\text{O}}}_{2}}$ (а) и ${\text{ZnO}}$ (б) после их предварительной выдержки в воде с разным значением pH: 1 – 3.0, 2 – 5.7, 3 – 10.0. Облучение пленок проводилось светом ксеноновой лампы мощностью 150 Вт [31, 79].
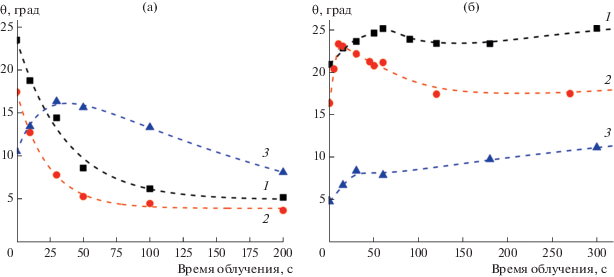
Эти примеры демонстрируют зависимость механизма фотоиндуцированного изменения гидрофильности от строения гидратного слоя воды на одной и той же поверхности, причем разную для различных высокоэнергетических поверхностей оксидов. В табл. 1 представлены значения свободной поверхностной энергии, а также ее дисперсионной и полярной составляющих для покрытий из оксидов титана и цинка, подвергнутых различным манипуляциям. Можно отметить, что дисперсионная и полярная составляющие поверхностной энергии представленных оксидов в разной мере “откликаются” на воздействие различных факторов, согласно схеме на рис. 8. Это означает, что различаются и механизмы перестройки гидратно-гидроксильных слоев на их поверхностях, и различно фотоиндуцированное гидрофильное поведение, о чем убедительно свидельствуют кривые 2 на рис. 13.
Таблица 1.
Значения свободной поверхностной энергии (γtot) и ее дисперсионной (γd) и полярной (γp) составляющих для покрытий из ${\text{Ti}}{{{\text{O}}}_{2}}$ и ${\text{ZnO}}$ после их термообработки при 500°C, выдержки в ультрачистой воде с рН 5.7 и УФ-облучения в течение 15 мин
| Покрытие | ${\text{Ti}}{{{\text{O}}}_{2}}$ | ${\text{ZnO}}$ | ||||
|---|---|---|---|---|---|---|
| Значения γ tot, γd и γp, мДж/м2 | γtot | γd | γp | γtot | γd | γp |
| Прогрев при 500°C | 80.1 | 48.5 | 31.6 | 75.9 | 38.6 | 37.3 |
| Выдержка в H2O | 74.9 | 47.6 | 27.3 | 74.5 | 40.4 | 34.1 |
| УФ-облучение | 80.4 | 48.8 | 31.6 | 73.0 | 42.5 | 30.5 |
Влияние температуры предварительной обработки поверхности на изменение контактного угла воды и свободной поверхностной энергии исследовали для покрытий из диоксида титана в различных состояниях – после термоактивации, после гидратации, после фотоактивации гидратированного состояния, согласно схеме на рис. 8 [31]. Термообработка изменяет степень гидратирования/гидроксилирования поверхности, а, следовательно, поверхностную энергию. Как следует из представленных на рис. 14 данных, максимальное изменение контактного угла воды и поверхностной энергии наблюдается в температурном диапазоне 175–300°C. Такое поведение поверхности объяснено удалением различных форм предварительно адсорбированных молекул воды, энергия десорбции которых различается. Наличие разных форм адсорбированной воды на поверхности диоксида титана подтверждается данными термодесорбционного исследования [27]. Так, термодесорбция слабо связанной воды при температуре 175–200°C (предположительно из внешнего слоя гидратного покрытия) приводит к уменьшению гидрофильности поверхности (угол смачивания возрастает, поверхностная энергия уменьшается). В то же время удаление более прочно связанных молекул воды при температуре 200–300°C способствует переходу поверхности в супергидрофильное состояние (угол смачиания уменьшается до 5°, поверхностная энергия увеличивается до значений, превосходящих поверхностную энергию чистой воды при 20°C (72.8 мДж/м2)).
Рис. 14.
Зависимость краевого угла воды (a) и поверхностной энергии (б) от температуры для трех образцов пленки ${\text{Ti}}{{{\text{O}}}_{2}}{\text{:}}$ 1 – предварительно прогретый при 500°C в течение 10 ч, 2 – дополнительно выдержанный в ультрачистой воде и высушенный в потоке воздуха, 3 – подвергнутый затем УФ-облучению [31].
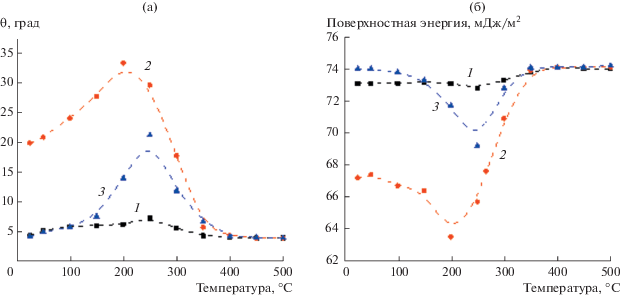
Таким образом, с точки зрения модели гидратного слоя можно сделать вывод, что наличие различных форм адсорбированной на поверхности воды согласно структуре полислоя (рис. 10) определяет величину свободной энергии поверхности, характеризующейся в каждом случае определенным гидрофильным состоянием.
В качестве следующего примера воздействия внешнего фактора рассмотрим влияние состава окружающей атмосферы. В работе [30] исследовали влияние влажности окружающей среды на фотоиндуцированный гидрофильный переход поверхности, что, несомненно, влияет на степень ее гидратации, т.е. на толщину полислоя адсорбированной воды.
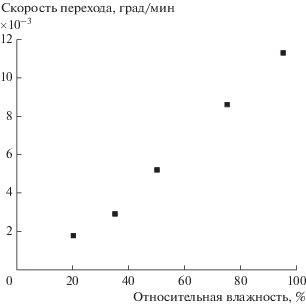
Рисунок 15 демонстрирует зависимость скорости фотоиндуцированного перехода поверхности пленки ${\text{Ti}}{{{\text{O}}}_{2}}$ в супергидрофильное состояние от относительной влажности воздуха. Видно, что эта зависимость представляет собой линейную функцию и чем выше влажность окружающего воздуха, тем больше скорость перехода. Эти данные хорошо согласуются с результатами исследования степени гидратации поверхности TiO2 [63, 81].
Рис. 15.
Зависимость скорости фотоиндуцированного перехода поверхности пленки ${\text{Ti}}{{{\text{O}}}_{2}}$ в супергидрофильное состояние от относительной влажности воздуха при 25°C. Взято из [30].
Многие исследования доказывают существование зависимости времени достижения супергидрофильного состояния поверхности диоксида титана от состава окружающей атмосферы, например присутствия/отсутствия кислорода [27, 42, 44, 50, 60, 76]. Авторами отмечается, что так называемый процесс старения достигнутого фотоиндуцированного супергидрофильного состояния, т.е. обратный переход супергидрофильной поверхности в менее гидрофильное состояние в темноте, также зависит от состава газовой атмосферы. В качестве примера рис. 16 демонстрирует изменение угла смачивания водой супергидрофильной поверхности тонкой пленки TiO2 при хранении ее в темноте в газах с различной электронодонорной способностью, а именно, в кислороде (электроноакцепторе) и в водороде (электронодоноре).
Рис. 16.
Зависимость фотоиндуцированного супергидрофильного состояния поверхности тонких пленок ${\text{Ti}}{{{\text{O}}}_{2}}$ от времени хранения в атмосфере кислорода (1) и водорода (2).
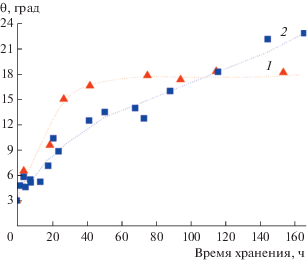
Очевидно, что газы по-разному влияют на “разрушение” супергидрофильного состояния поверхности, наведенного УФ-облучением. Поверхность диоксида титана переходит из супергидрофильного в менее гидрофильное состояние (θ = 15°–17°) в течение первых двух дней постепенно в водороде (рис. 16, кривая 2) и резко – в кислороде (рис. 16, кривая 1). Однако после этого характер влияния этих газов становится различным: гидрофильное состояние стабилизируется и практически не меняется в присутствии кислорода, в то время как в водороде угол смачивания продолжает монотонно увеличиваться с той же скоростью, что и в первые 48 ч.
Существует мнение, что молекулы кислорода способны частично вытеснять адсорбированные молекулы воды из внутреннего слоя (рис. 10), тем самым изменяя структуру адсорбционного полислоя, однако далее гидрофильное состояние поверхности стабилизируется благодаря уменьшению поверхностной электрон-дырочной рекомбинации из-за захвата электронов адсорбированными молекулами кислорода. В атмосфере электронодонорного газа водорода происходит рекомбинация захваченных на поверхностных центрах фотоэлектронов и фотодырок, усиленная “поставкой” электронов от молекул водорода при взаимодействии с фотоактивированной поверхностью твердого тела. Эти наблюдения позволяют сделать вывод, что именно наличие на поверхности дырок в значительной степени определяет эффективность фотостимулированного формирования супергидрофильного состояния гидратированной поверхности диоксида титана.
Внутренние факторы влияют на состояние активных центров поверхности, ответственных за фотоиндуцированный гидрофильный переход, что впоследствии приводит к изменениям в гидратно-гидроксильном слое на поверхности оксидной пленки и, следовательно, к изменению свободной энергии поверхности и ее гидрофильности. К этим факторам относятся интенсивность и спектральный состав действующего света, дефектность поверхности или структура активных поверхностных центров.
На рис. 17 и 18 представлены экспериментальные зависимости начальной скорости изменения краевого угла воды на поверхности покрытий из TiO2 и ZnO от интенсивности и длины волны действующего света, полученные в работах [31, 56].
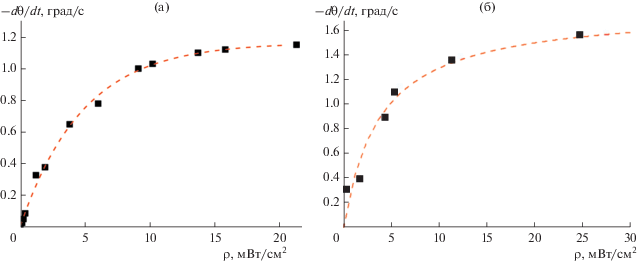
В работах [27, 81, 82] также получены зависимости начальной скорости фотоиндуцированного изменения угла смачиваемости водой поверхности диоксида титана от интенсивности действующего света, представленные в логарифмических координатах lg(dθ/dt)–lg(ρ), где ρ – интенсивность падающего света (мВт/см2). При построении в координатах dθ/dt–ρ эти зависимости имеют экспоненциальный вид, аналогичный кривым на рис. 17. При этом все полученные зависимости хорошо аппроксимируются уравнением
где a и b – постоянные, не зависящие от интенсивности света.Отмечено, что такой тип зависимости характерен для гетерогенных фотохимических и фотокаталитических реакций и может быть интерпретирован аналогичным образом с точки зрения фотостимулированной активации и дезактивации поверхностных центров, благодаря которым происходит фотоиндуцированный гидрофильный переход [53, 55, 83–85]. Из уравнения (9) следует, что при достаточно больших значениях интенсивности действующего света скорость процесса перестает зависеть от интенсивности.
Существование спектральной зависимости угла смачивания или начальной скорости его изменения в процессе облучения (так называемого спектра действия), подтверждает факт того, что именно электронное фотовозбуждение твердого тела является начальной стадией исследуемых фотопроцессов. Влияние спектрального состава действующего света на параметры рассматриваемого процесса легко продемонстрировать, изменяя энергию фотонов путем перехода из области собственного поглощения (межзонное фотовозбуждение) твердого тела в область несобственного поглощения (область поглощения дефектов) [31, 49, 56, 86–89].
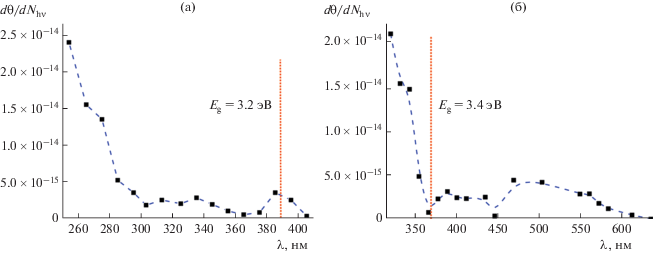
При условии линейной зависимости начальной скорости процесса от интенсивности света были получены спектральные зависимости начальной скорости фотоиндуцированного изменения угла смачивания для поверхностей тонких пленок (покрытий) ${\text{Ti}}{{{\text{O}}}_{2}}$ и ${\text{ZnO,}}$ представленные на рис. 18а и 18б соответственно [31, 56].
Как видно на рис. 18а, в спектре действия для покрытия из TiO2 имеются хорошо выраженные особенности (максимумы) около 385 и 335 нм, которые коррелируют с энергиями первого непрямого (3.2 эВ) и первого прямого (≈3.7 эВ) межзонных электронных переходов в диоксиде титана анатазной модификации. При этом в области несобственного поглощения (λ > 385 нм, E < 3.2 эВ) изменения гидрофильного перехода не наблюдается. Для покрытия из оксида цинка (рис. 18б) эффективность фотоиндуцированной гидрофильной конверсии поверхности достаточно высока в области собственного поглощения ${\text{ZnO}}$ (энергии фотонов ниже 3.37 эВ). В то же время в области несобственного поглощения ZnO наблюдаются две широкие спектральные особенности вблизи 487 нм (2.55 эВ) и 400 нм (3.10 эВ), связанные с возбуждением собственных дефектных состояний в ZnO. Примечательно, что представленные спектральные зависимости качественно соответствуют спектральным зависимостям квантовых выходов и фотонной эффективности фотостимулированных реакций на поверхности этих оксидов [54, 90, 91]. Этот факт подтверждает, что первичные электронные процессы при фотовозбуждении твердого тела, ответственные за фотокаталитическую активность и за фотоиндуцированный гидрофильный переход, идентичны и связаны с фотогенерацией свободных носителей заряда.
Дефектность поверхности определяет природу и концентрацию поверхностных активных центров, тем самым задавая характер и силу взаимодействия этих центров с адсорбированными молекулами во внутреннем гидроксильном слое (рис. 10) и структуру самого гидратно-гидроксильного слоя на поверхности материала. Таким образом, изменяя элементный состав поверхности можно влиять на параметры рассматриваемого поверхностного процесса. Состав и дефектность поверхности материала можно изменить разными способами. Среди них, например, варьирование методов и/или условий синтеза [56, 58, 79, 92, 93], использование различных кристаллических плоскостей [66, 93–98], создание остаточных напряжений в пленках при их формировании на разных подложках [65, 66], формирование границы между слоями с разной дефектностью [63, 99], допирование материала [28, 29, 49, 88, 89, 100–103]. В перечисленных работах продемонстрировано, как, варьируя перечисленные внутренние факторы, можно не только изменить начальное значение угла смачивания, но и повлиять на скорость и направление его изменения при фотовозбуждении фотоактивных материалов.
6. КИНЕТИКА И МЕХАНИЗМ ФОТОИНДУЦИРОВАННОГО ГИДРОФИЛЬНОГО ПЕРЕХОДА ПОВЕРХНОСТИ ТОНКИХ ПЛЕНОК ФОТОАКТИВНЫХ МАТЕРИАЛОВ
С учетом полученных экспериментальных зависимостей изменения смачиваемости поверхности фотоактивного материала как функции времени УФ-облучения были предприняты попытки создания теоретических основ для анализа и описания исследуемого процесса, а также влияния на его кинетику различных факторов. Рассмотрим некоторые из них, которые укладываются в рамки третьей гипотезы механизма фотоиндуцированного изменения гидрофильности поверхности в результате фотовозбуждения твердого тела.
Первый теоретический подход к анализу фотоиндуцированного гидрофильного перехода (применительно к поверхности диоксида титана) был предложен японскими учеными Секи и Тачия в рамках представления о существовании гидрофильных/гидрофобных участков на поверхности и увеличения доли гидрофильных участков при УФ-облучении [32]. В качестве экспериментального параметра для описания кинетики процесса авторы этого подхода использовали косинус угла смачивания (cos θ).
При облучении поверхность диоксида титана становится более гидрофильной (угол смачивания θ уменьшается, cos θ увеличивается), что, согласно уравнению Юнга (5), означает рост величины $\left( {{{\gamma }_{{{\text{sv}}}}}{\text{\;}} - {{\gamma }_{{{\text{ls}}}}}} \right).$ Для установления взаимосвязи между этими межфазными энергиями и долей гидрофильных участков (с) на поверхности вводится следующая линейная функция:
(10)
${{\gamma }_{{{\text{sv}}}}} - {{\gamma }_{{{\text{ls}}}}} = {{\gamma }_{1}}c + {{\gamma }_{2}}.$(11)
$f \equiv \cos \theta = {\text{\;}}\frac{{{{\gamma }_{{{\text{sv}}}}} - {{\gamma }_{{{\text{ls}}}}}}}{{{{\gamma }_{{{\text{lv}}}}}}} = {{f}_{1}}c + {{f}_{2}},$Как отмечалось ранее, поверхность, предварительно переведенная УФ-излучением в гидрофильное состояние при выдержке в темноте со временем снова становится гидрофобной. Если переход поверхности в гидрофильное состояние и обратный процесс охарактеризовать соответственно с помощью единых констант скорости, ${{k}_{{\text{f}}}}$ и ${{k}_{{\text{b}}}},$ то кинетику изменения доли гидрофильных участков можно представить как
Таким образом, подстановка выражения (12) в уравнение (11) дает выражение (13) для зависимости косинуса угла θ на поверхности диоксида титана от времени УФ-облучения t:(13)
$\begin{gathered} f\left( t \right) = {{f}_{1}}\left[ {\frac{{{{k}_{{\text{f}}}}}}{{{{k}_{{\text{f}}}}~ + ~{{k}_{{\text{b}}}}}} - \left( {\frac{{{{k}_{{\text{f}}}}}}{{{{k}_{{\text{f}}}}~ + {\text{\;}}{{k}_{{\text{b}}}}}} - c\left( 0 \right)} \right) \times } \right. \\ \left. {^{{^{{^{{^{{^{{}}}}}}}}}} \times \,\,{\text{exp}}\left\{ { - \left( {{{k}_{{\text{f}}}} + {{k}_{{\text{b}}}}} \right)t} \right\}} \right] + {{f}_{2}}. \\ \end{gathered} $(15)
$f\left( {t \to \infty } \right) = \frac{{{{f}_{1}}{{k}_{{\text{f}}}}}}{{{{k}_{{\text{f}}}}~ + ~{{k}_{{\text{b}}}}}} + {{f}_{2}}.$(17)
$\frac{{d\left[ {\text{M}} \right]}}{{dt}} = g\rho - {{k}_{{\text{h}}}}\left[ {\text{M}} \right] - {{k}_{{{\text{e}} - {\text{h}}}}}\left[ {\text{M}} \right]\left[ {{{{\text{M}}}_{{\text{e}}}}} \right],$(18)
$\frac{{d\left[ {{{{\text{M}}}_{{\text{e}}}}} \right]}}{{dt}} = g\rho - {{k}_{{\text{e}}}}\left[ {{{{\text{M}}}_{{\text{e}}}}} \right] - {{k}_{{{\text{e}} - {\text{h}}}}}\left[ {\text{M}} \right]\left[ {{{{\text{M}}}_{{\text{e}}}}} \right],$В результате математических преобразований с учетом равенства ${{k}_{{\text{h}}}}\left[ {\text{M}} \right] = {{k}_{{\text{e}}}}\left[ {{{{\text{M}}}_{{\text{e}}}}} \right]~$в работе [32] получено следующее выражение для концентрации состояний $\left[ {\text{M}} \right]$:
(19)
$\left[ {\text{M}} \right] = \frac{{{{k}_{{\text{e}}}}}}{{2{{k}_{{{\text{e}} - {\text{h}}}}}}}\left[ {{{{\left( {1 + \frac{{4{{k}_{{{\text{e}} - {\text{h}}}}}g\rho }}{{{{k}_{{\text{e}}}}{{k}_{{\text{h}}}}}}} \right)}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}} - 1} \right].$(21)
$\left[ {\text{M}} \right]\sim \sqrt {\frac{{{{k}_{{\text{e}}}}g\rho }}{{{{k}_{{\text{h}}}}{{k}_{{{\text{e}} - {\text{h}}}}}}}} .$Введение некоего параметра c, характеризующего долю гидрофильных и гидрофобных участков на поверхности, позволило описать кинетику фотоиндуцированного изменения контактного угла воды (его косинуса) с помощью экспоненциальной функции. С точки зрения термодинамики дополнительной информации об изменении состояния поверхности в процессе облучения и “старения” ее фотоиндуцированного супергидрофильного состояния, а именно об изменении свободной поверхностной энергии и ее дисперсионной и полярной компонент, в предлагаемом подходе получить невозможно. Несмотря на это, в рамках этого подхода впервые была сделана попытка найти связь скорости исследуемого процесса с концентрацией неких поверхностных центров, ответственных за протекание фотоиндуцированного гидрофильного перехода, т.е. найти функцию, связывающую изменение макропараметра, угла смачивания θ, с молекулярными характеристиками поверхности – концентрацией поверхностных центров, концентрацией фотоносителей. Однако в предложенном подходе не представлено конкретной модели механизма, введенные константы скорости неких промежуточных процессов описаны нечетко, что затрудняет восприятие кинетических уравнений в целом. Две части подхода не связаны между собой.
Второй подход к описанию кинетики и механизма фотоиндуцированного изменения гидрофильности поверхности, предложенный авторами настоящего обзора, основывается на том, что первой стадией этого процесса является электронное фотовозбуждение. Для описания экспериментальной зависимости начальной скорости фотоиндуцированного изменения угла смачивания и поверхностной энергии для диоксида титана было предложено уравнение (9) [31]. При этом известно [53, 55, 83–85, 104, 105], что такой тип зависимости характерен для различных гетерогенных фотокаталитических реакций, и для ее интерпретации можно использовать механизм, включающий стадии фотостимулированной активации и дезактивации поверхностных центров. В соответствии с этой предпосылкой, предложен механизм фотостимулированного перехода поверхности диоксида титана в супергидрофильное состояние, определяющую роль в котором играют фотодырки. Механизм был представлен следующим образом [31, 106]:
где ki – константы скорости соответствующих стадий. Здесь стадия (22.1) описывает процесс фотоактивации поверхностных активных центров (S), ответственных в дальнейшем за фотоиндуцированный переход поверхности в новое гидрофильное состояние (H) (стадия (22.3)), связанный с перестройкой гидроксильно-гидратного полислоя. В то же время стадии (22.2) и (22.4) представляют собой процессы фотостимулированной и термической дезактивации возбужденного состояния поверхностных активных центров (S+) и перестроенной структуры гидроксильно-гидратного полислоя (Н) в окружении возбужденных состояний поверхностных активных центров, отвечающих за более высокую гидрофильность. Для поверхности ${\text{Ti}}{{{\text{O}}}_{2}}$ доля дефектных центров, ответственных за изменение поверхностной энергии, а, следовательно, и за гидрофильный переход при УФ-облучении, оценивается в среднем равной 10–4–10–3 от общего числа поверхностных цетров [31].Константа скорости k4 стадии нефотостимулированной дезактивации (22.4) довольно мала по сравнению с константами скоростей других трех стадий, особенно, при значениях интенсивности света ρ выше 4 мВт/см2 (рис. 17а). Следовательно, решение системы кинетических уравнений, соответствующих механизму (22), с учетом того что ${{k}_{4}} \ll {{k}_{1}},{{k}_{2}},{{k}_{3}}$ приводит к следующему выражению для начальной скорости рассматриваемого фотоиндуцированного процесса:
(23)
$\frac{{dH}}{{dt}} = \frac{{{{k}_{1}}{{k}_{2}}{{S}_{0}}{{n}_{1}}}}{{{{k}_{2}}{{n}_{2}} + {{k}_{3}}}},$Из уравнения (23), учитывая, что концентрации фотоносителей, n1 и n2, прямо пропорциональны интенсивности действующего света $\rho $ ($n = g\tau = \alpha \rho \tau ,$ где $g$ – скорость генерации носителей, $\alpha $ – коэффициент поглощения, $\tau $ – время жизни носителей) [104, 105], можно определить зависимость начальной скорости процесса от ρ. При малых значениях ρ (${{k}_{2}}\alpha \rho \tau \ll {{k}_{3}}$) скорость процесса линейно зависит от интенсивности света, а при больших ρ (${{k}_{2}}\alpha \rho \tau \gg {{k}_{3}}$) скорость перестает зависеть от интенсивности, что и наблюдается в эксперименте (рис. 17).
Заметим, что представленные подходы к описанию фотостимулированного процесса рассматриваются только применительно к поверхности диоксида титана с соответствующим предположением, что фотодырки играют ключевую роль. Однако в общем случае это предположение может быть неверным, так как любой из фотоносителей (или их соотношение) может играть определяющую роль в рассматриваемом фотоиндуцированном процессе, и направление изменения гидрофильности тонкой пленки определенного оксида металла может отличаться от такового для пленки из диоксида титана [45, 46, 58, 79, 93, 107]. Следует также отметить ограниченность механизма (22), заключающуюся в возможности описания только однонаправленных фотоиндуцированных переходов гидрофильности для поверхности фотоактивного материала. Вместе с тем, известны случаи разнонаправленного фотоиндуцированного гидрофильного поведения поверхности одного и того же материала. Так, представленные в работе [106] экспериментальные данные для слоистых покрытий из оксидов титана и цинка демонстрируют способность поверхности одних и тех же материалов переходить под действием облучения как в более, так и в менее гидрофильное состояние.
В связи со сказанным выше механизм (22) был нами пересмотрен. Чтобы объяснить возможность изменения гидрофильности поверхности в обоих направлениях – в сторону более высокой и в сторону более низкой гидрофильности – под действием света одного и того же спектрального диапазона, начальное гидрофильное состояние поверхности стали характеризовать не нулевым значением H, а неким начальным количеством гидрофильных участков H0. При этом было предположено, что исходные гидрофильные участки могут быть дезактивированы путем захвата фотогенерированных носителей заряда определенного знака. В результате этих поправок модифицированный механизм был представлен следующим образом:
где ki – константы скорости соответствующих стадий, S и S* – это основное и возбужденное состояния поверхностных активных центров, ответственных за фотостимулированный гидрофильный переход.С учетом начальных условий (S0 и H0) решение набора кинетических уравнений (24.1)–(24.5) представляет собой выражение
(25)
$\Delta H\left( t \right) = H\left( t \right) - {{H}_{0}} = \left( {\frac{A}{B}{{S}_{0}} - {{H}_{0}}} \right)\left( {1 - {{e}^{{ - \,\,\frac{C}{B}t}}}} \right),$Анализ выражения (25) для скорости фотоиндуцированного изменения гидрофильности поверхности дает возможность определить факторы, влияющие на направление и величину изменения смачиваемости исследуемых фотоактивных материалов. Так, направление изменения гидрофильности поверхности $\Delta H\left( t \right) = H\left( t \right) - {{H}_{0}}$ будет положительным (т.е. поверхность становится более гидрофильной, краевой угол в процессе облучения уменьшается), ΔH > 0, если выполняется неравенство (A/B)S0 > H0. Это может быть реализовано при выполнении следующих условий: (а) H0 → 0, т.е. исходно поверхность гидрофобна или имеет низкую гидрофильность (начальный краевой угол большой), или (б) A $ \gg $ B, т.е. n1$ \gg $ n2, в том числе для поверхностей, являющихся исходно супергидрофильными, когда значение H0 высоко. Практически, последнее условие означает, что состояние супергидрофильности при фотовозбуждении сохраняется.
Противоположное направление изменения гидрофильности поверхности, т.е. когда поверхность становится менее гидрофильной/более гидрофобной, ΔH < 0, может быть реализовано при выполнении неравенства (A/B)S0 < H0. В свою очередь, это возможно, если значение H0 изначально достаточно высоко, например поверхность супергидрофильна, и A $ \ll $ B (n1$ \ll $ n2), т.е. дезактивация поверхностных гидрофильных центров (стадия (24.2)) происходит быстрее, чем их генерация (стадия (24.1)).
Таким образом, модифицированный механизм, представленный уравнениями (24.1)–(24.5), включает в себя основные стадии, приводящие к изменению электронного состояния активных центров поверхности, что, в свою очередь, определяет процессы реструктуризации гидратно-гидроксильного слоя. Несмотря на определенную упрощенность стадий фотопроцесса, предложенный механизм отражает все установленные на сегодняшний день экспериментально термодинамические и кинетические зависимости фотостимулированного изменения гидрофильности поверхности твердого тела.
7. ВОЗМОЖНЫЕ СПОСОБЫ УПРАВЛЕНИЯ ПРОЦЕССОМ ИЗМЕНЕНИЯ СМАЧИВАЕМОСТИ ПОВЕРХНОСТИ С ПОМОЩЬЮ СВЕТА
Создание материалов с заданной смачиваемостью поверхности является одной из важных задач современного материаловедения, науки о новых функциональных материалах для различных отраслей промышленности и человеческой жизнедеятельности. Как известно, такой параметр материала как морфология поверхности в значительной степени определяет его гидрофильность/ гидрофобность [13, 108]. Так, в последнее время стали очень популярными биомиметические материалы, смачиваемость которых обеспечивается специально созданной микро(нано)структурой их поверхности [14, 15]. Большинство из таких иерархических микро/наноструктур, реализованных по принципам живой природы, обладают супергидрофильностью или супергидрофобностью, что актуально при формировании материалов с высокой механической стабильностью и долговечностью, обладающих при этом самоочищающимися, противотуманными и антиотражающими свойствами.
Однако в настоящем обзоре предметом рассмотрения являются гладкие поверхности фотоактивных материалов. Опираясь на модель полислоя адсорбированной воды и механистические представления о фотовозбуждении твердого тела как неотъемлемой стадии процесса, можно выделить два основных подхода к управлению фотоиндуцированным изменением гидрофильности поверхности материала.
Первый подход заключается в изменении поверхностной энергии фотоактивного материала в исходном состоянии (до воздействия на него света), что впоследствии оказывает существенное влияние на термодинамические и кинетические параметры поверхностных фотопроцессов. Данный подход можно реализовать различными путями, причем в результате свободная поверхностная энергия твердого тела может как понизиться, так и повыситься. Некоторые из этих путей мы уже упоминали выше, когда рассматривали факторы, влияющие на рассматриваемый фотопроцесс. Например, на скорость гидрофильного перехода в случае монокристаллических диоксида титана, оксида цинка и титаната стронция можно повлиять, используя их различные кристаллографические плоскости, различающиеся химическим составом и структурой поверхности [66, 93–98].
Изменить свободную поверхностную энергию можно, химически модифицируя поверхность, например, за счет изменения состава гидратного слоя (изменение кислотности поверхности, обработка перекисью водорода) [31, 79, 109] или адсорбции тех или иных молекул [13, 31, 49, 110, 111]. Возможно использование и физического (например, обработка ультразвуком) [112, 113] или механического (измельчение частиц, шлифование поверхности, создание напряжений) [65, 66, 112, 114] воздействия. Несмотря на то, что механизмы таких воздействий различны, все они приводят к изменению поверхностной энергии, что, как показывают экспериментальные результаты, в дальнейшем оказывает влияние на фотоиндуцированное поведение гидрофильности поверхности. Стоит также отметить, что при использовании всех перечисленных выше методов фундаментальные характеристики материала, его физические и химические свойства остаются прежними, а изменяются только поверхностные свойства.
В отличие от методов модификации поверхности, химическая модификация самого материала, например, с помощью допирования или со-допирования металлами или неметаллами, изменяет не только химический состав поверхности, влияющий на тип поверхностных гидроксильных групп и адсорбированных молекул воды, но и его электронные свойства [13, 28, 29, 49, 80, 88, 89, 100–102, 115].
Показательные результаты по влиянию допирования материала на гидрофильность его поверхности представлены в работе [80]. Авторы исследовали смачиваемость покрытий из нитрида галлия n- и p-типов, нанесенных на сапфировую подложку. Было обнаружено, что УФ-облучение в течение 30 мин способствует гидрофилизации только поверхности n-GaN (допированного кремнием): угол смачивания водой уменьшился с 61.3° до 8.5°. Гидрофильность поверхности p-GaN (допированного магнием) при облучении существенно не изменялась: начальный угол смачивания водой был равен 48.5°, а после облучения составил 46.4°.
Всестороннее изучение свойств покрытий показало, что за фотоиндуцированное изменение гидрофильности поверхности нитрида галлия ответственны кислородные вакансии, образующиеся при облучении благодаря присутствию примесных атомов кислорода. По сравнению с GaN p-типа, GaN n-типа содержит заметно большее количество примесей кислорода и характеризуется изгибом зон вверх вблизи поверхности, что способствует ее обогащению фотодырками, которые необходимы для образования анионных вакансий. На рис. 19 представлены предлагаемые авторами схемы, иллюстрирующие электронные процессы вблизи поверхности при возбуждении нитрида галлия n- и p-типов и механизм фотоиндуцированного перехода поверхности n-GaN в гидрофильное состояние.
Рис. 19.
(а) Схема изгиба зон вблизи поверхности ${\text{GaN}}$ n- и p-типов. (б) Схема фотоиндуцированного гидрофильного перехода поверхности ${\text{GaN}}$ n-типа. Взято из работы [80].

На сегодняшний день определено, что механизм фотоиндуцированного гидрофильного перехода зависит от природы самого материала и типов существующих на поверхности активных центров. Однако из имеющихся в литературе экспериментальных результатов не ясно, какой тип фотоносителей, электроны или дырки, или их определенное соотношение на поверхности определяет кинетику процессов активации и дезактивации поверхностных центров, ответственных за фотоиндуцированное изменение гидрофильности и, соответственно, за направление фотоиндуцированного процесса. Что общего и чем различаются механизмы этих процессов на поверхности различных фотоактивных материалов? Этот вопрос до сих пор остается открытым.
Второй способ управления смачиваемостью поверхности с помощью света был сформулирован на основе предложенного нами механизма фотоиндуцированной конверсии гидрофильности поверхности фотоактивных материалов, описанного стадиями (24.1)–(24.5), и анализа кинетического выражения (25). Этот способ объединяет методы, с помощью которых можно изменить соотношение концентраций фотоносителей противоположных знаков n1/n2 (выражение (25)), определяющих изменение электронного состояния активных поверхностных центров в процессе облучения. Фотоиндуцированное изменение состояний поверхностных активных центров, ответственных за реструктуризацию гидратно-гидроксильного слоя на поверхности, изменяет величину свободной поверхностной энергии, и, как следствие, степень гидрофильности поверхности данного материала.
В результате химической модификации материала допированием одним или несколькими металлами/неметаллами, как уже было упомянуто, изменяется не только химический состав поверхности, но и электронные свойства материала благодаря созданию дефектных уровней определенного типа в запрещенной зоне или стабилизации собственных дефектов, что влияет на скорость электрон-дырочной рекомбинации и доминирующий тип носителей заряда [13, 88, 100, 103, 115–117]. В этом случае целесообразно исследовать фотоиндуцированные поверхностные процессы и электрофизические свойства материала в зависимости от концентрации допанта. Так, в работе [115] исследовали влияние допирования на электропроводимость и фотоиндуцированную гидрофильность пленок диоксида титана, полученных магнетронным распылением. Обнаружено, что допирование ионами ниобия приводит к значительному увеличению темнового тока и фототока, в то же время гидрофильность поверхности при облучении пленок УФ-светом в течение 30 ч значительно повышалась по сравнению с недопированным образцом (рис. 20, кривые 1 и 2). Вместе с тем, слишком высокая степень допирования приводит как к падению значений фототока, так и к уменьшению эффекта фотоиндуцированной гидрофилизации (рис. 20, кривая 3).
Рис. 20.
Влияние допирования ниобием на свойства пленок диоксида титана, полученных магнетронным распылением при 250 Вт: 1 – исходная пленка TiO2, 2, 3 – пленки TiO2 допированные Nb. (а) Зависимость фототока от мощности УФ-излучения. (б) Кинетика фотоиндуцированного изменения краевого угла воды на поверхности этих пленок (мощностm УФ-излучения – 1.0 мВт/см2). Взято из работы [115].

Полученные результаты подтверждают, что изменения гидрофильных и электрофизических свойств пленок ${\text{Ti}}{{{\text{O}}}_{2}}$ в зависимости от степени допирования взаимосвязаны. Это объяснено образованием энергетических уровней примесных ионов ниобия в запрещенной зоне диоксида титана, способных выступать в роли ловушек носителей заряда и изменять положение уровня Ферми. Практическая значимость работы заключается в определении концентрации допанта (ниобия), необходимой для получения пленок TiO2 с более высокой гидрофильностью, чем пленки чистого диоксида титана, нанесенные методом магнетронного распыления.
Использование допирования металлами и неметаллами оксидов металлов считается одним из способов улучшения свойств самоочищающихся покрытий за счет сенсибилизации материалов к видимому свету и повышения фотокаталитической активности в окислительных реакциях [11].
Исследование изменений гидрофильности электрода из фотоактивного материала, к которому приложен электрический потенциал, под действием облучения также зарекомендовало себя как метод, дающий ценную информацию о влиянии носителя заряда того или иного типа на смачиваемость поверхности и, тем самым, о механизме этого фотопроцесса [52, 113]. Этот способ был использован при изучении механизма фотоиндуцированной супергидрофильности поверхности электрода из диоксида титана [52]. Изменения гидрофильности поверхности пленочных TiO2-электродов при облучении УФ-светом в водном растворе были изучены в условиях управления прилагаемым потенциалом. При приложении к освещаемому электроду потенциала, равного потенциалу плоской зоны (Efb ≈ –0.8 В), изменений в гидрофильности его поверхности не наблюдалось (рис. 21).
Рис. 21.
Кинетические зависимости фотоиндуцированной гидрофилизации поверхности TiO2-электрода при разном приложенном потенциале: (а) –0.8, (б) –0.5, (в) –0.2, (г) +0.2, (д) +0.5, (е) +0.8 В. Взято из работы [52].
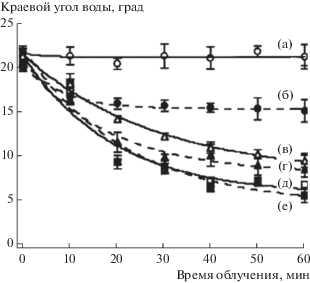
При анодных потенциалах, т.е. бóльших, чем потенциал плоской зоны, наблюдалась фотоиндуцированная гидрофилизация поверхности, и ее скорость возрастала с увеличением потенциала. Таким образом, кинетические параметры процесса для электрода из одного и того же материала можно успешно варьировать, изменяя значение прилагаемого потенциала.
Варьирование интенсивности действующего света является эффективным способом изменения концентрации фотогенерированных электронов и дырок и, следовательно, изменения скорости фотоиндуцированного процесса, в то время как изменение его спектрального состава приводит, в первую очередь, к изменению соотношения концентраций носителей обоих типов. Таким образом, можно изменить направление рассматриваемого поверхностного фотопроцесса. Способ варьирования спектрального состава действующего света продемонстрировал свою действенность в отношении изменения кинетических параметров фотоиндуцированного изменения гидрофильности поверхности оксидов титана, цинка и циркония [31, 49, 56, 79, 86–89]. Зависимости скорости исследуемых поверхностных процессов от интенсивности и спектрального состава излучения представлены и обсуждены для тонких пленок диоксида титана и оксида цинка выше (рис. 17 и 18) [31, 56, 79].
Влияние спектрального состава света на фотоиндуцированное изменение гидрофильности поверхности диоксида циркония былo исследованo с использованием измерений контактного угла воды на поверхности пленки ZrO2, а также методами рентгеновской фотоэлектронной спектроскопии и спектроскопии люминесценции [86, 87]. Было обнаружено, что облучение покрытий из ${\text{Zr}}{{{\text{O}}}_{2}}$ в области собственного поглощения оксида (светом с длиной волны меньше 254 нм) приводит к переходу поверхности пленки ${\text{Zr}}{{{\text{O}}}_{2}}$ в супергидрофильное состояние (угол смачивания водой падает до значений меньших 5°). В то же время облучение светом с длиной волны больше 254 нм, поглощаемым электронными дефектами в диоксиде циркония, переводит поверхность в более гидрофобное состояние. Кинетические кривые фотоиндуцированного изменения угла смачивания водой поверхности ${\text{Zr}}{{{\text{O}}}_{2}}$ для обоих случаев представлены на рис. 22а. Такое поведение было объяснено разницей в концентрации фотоэлектронов и фотодырок, которые участвуют в фотоиндуцированных поверхностных процессах (рис. 22б). По результатам исследования [86] сделан вывод, что именно поверхностные центры, захватывающие дырки, такие как поверхностные гидроксильные группы и адсорбированные молекулы воды, играют решающую роль в фотоиндуцированном переходе поверхности диоксида циркония в супергидрофильное состояние.
Рис. 22.
(а) Кинетика фотоиндуцированного изменения угла смачивания водой поверхности ZrO2 при ее облучения светом различного спектрального состава: 1 – λ < 300 нм, 2 – λ > 300 нм. Поверхность Zr${{{\text{O}}}_{2}}$ предварительно выдерживали в воде с pH 5.5. (б) Схемы электронных процессов, протекающих при фотовозбуждении ZrO2 в различных областях его поглощения – собственного (λ < 300 нм) и несобственного (λ > 300 нм). Взято из [87].

Обнаружена возможность обратимого и многократного изменения гидрофильности поверхности диоксида циркония при изменении спектрального состава действующего света (рис. 23) [87].
Рис. 23.
Обратимое переключение между супергидрофильным (θ < 10°) и менее гидрофильным (θ > 25°) состояниями поверхности ZrO2 путем изменения спектрального состава действующего света: hν1 соответствует свету с λ < 300 нм, hν2 – свету с λ > 300 нм. Взято из [87].

Этот эффект интересен с точки зрения практического применения, особенно, при использовании покрытий, нанесенных на обе стороны различных подложек. В случае кварцевой подложки облучение светом с λ < 300 нм приводит к супергидрофильности покрытий из ${\text{Zr}}{{{\text{O}}}_{2}}$ на обеих сторонах подложки, а в случае стеклянных подложек – только покрытия со стороны непосредственного действия света, а покрытие на обратной (необлучаемой) стороне становится менее гидрофильным (так называемый эффект дождевых капель). Эффект продемонстрирован на рис. 24.
Рис. 24.
Иллюстрация фотоиндуцированного гидрофильного поведения двусторонних покрытий ${\text{Zr}}{{{\text{O}}}_{2}}$ на стеклянной (слева) и кварцевой (справа) подложках. Белая стрелка означает прохождение полного солнечного света, красная стрелка – только видимой части солнечного света. Взято из [87].

Недавние исследования [106] показали, что создание слоистых гетероструктурных покрытий из полупроводниковых материалов, образующих гетеропереход II типа, можно эффективно использовать для управления смачиваемостью поверхности под действием света. Особенность гетеропереходов II типа, или так называемых ступенчатых гетеропереходов, заключается в том, что вследствие локализации электронов и дырок по разные стороны его границы происходит разделение носителей заряда [118, 119]. Таким образом, в результате формирования гетероперехода типа II внешняя поверхность таких композитных покрытий может быть обогащена одним определенным типом носителей заряда, электронами или дырками, в зависимости от структуры гетероперехода и природы образующих его полупроводниковых материалов. Отметим, что подход, основанный на формировании гетеропереходов, широко применяется при проектировании фотоэлектрохимических и фотоэлектрических систем для повышения эффективности их работы [13, 120, 121]. Однако в литературе встречаются работы, в которых сочетание различных полупроводниковых материалов при создании самоочищающихся покрытий используют и как способ улучшения способности материалов к самоощищению [11, 13, 98, 122–127]. Следует отметить, что речь идет, в первую очередь, о покрытиях на основе диоксида титана и о повышении фотокаталитической активности материалов в отношении окислительных процессов благодаря эффективному разделению зарядов и уменьшению электрон-дырочной рекомбинации на поверхности. Например, Лиу и Миаучи исследовали смачиваемость пленки с сэндвич-структурой ${{{{{\text{W}}{{{\text{O}}}_{3}}} \mathord{\left/ {\vphantom {{{\text{W}}{{{\text{O}}}_{3}}} {{\text{ITO}}}}} \right. \kern-0em} {{\text{ITO}}}}} \mathord{\left/ {\vphantom {{{{{\text{W}}{{{\text{O}}}_{3}}} \mathord{\left/ {\vphantom {{{\text{W}}{{{\text{O}}}_{3}}} {{\text{ITO}}}}} \right. \kern-0em} {{\text{ITO}}}}} {{\text{CaF}}{{{\text{e}}}_{2}}{{{\text{O}}}_{4}}}}} \right. \kern-0em} {{\text{CaF}}{{{\text{e}}}_{2}}{{{\text{O}}}_{4}}}}$ и установили, что ее поверхность становится супергидрофильной при облучении видимым светом, что было приписано эффективному разделению зарядов в поверхностном слое ${\text{W}}{{{\text{O}}}_{3}}$ (рис. 25) [122]. Авторами был сделан вывод, что стратегия объединения полупроводниковых материалов открывает новые возможности для разработки супергидрофильных материалов.
Рис. 25.
(а) Схема электронных процессов, протекающих в структуре ${{{{{\text{W}}{{{\text{O}}}_{3}}} \mathord{\left/ {\vphantom {{{\text{W}}{{{\text{O}}}_{3}}} {{\text{ITO}}}}} \right. \kern-0em} {{\text{ITO}}}}} \mathord{\left/ {\vphantom {{{{{\text{W}}{{{\text{O}}}_{3}}} \mathord{\left/ {\vphantom {{{\text{W}}{{{\text{O}}}_{3}}} {{\text{ITO}}}}} \right. \kern-0em} {{\text{ITO}}}}} {{\text{CaF}}{{{\text{e}}}_{2}}{{{\text{O}}}_{4}}\left( {{\text{CFO}}} \right)}}} \right. \kern-0em} {{\text{CaF}}{{{\text{e}}}_{2}}{{{\text{O}}}_{4}}\left( {{\text{CFO}}} \right)}}.$ (б) Фотоиндуцированное изменение краевого угла воды на поверхности пленок ${\text{CFO,}}$ ${\text{W}}{{{\text{O}}}_{3}},$ ${{{\text{W}}{{{\text{O}}}_{3}}} \mathord{\left/ {\vphantom {{{\text{W}}{{{\text{O}}}_{3}}} {{\text{CFO}}}}} \right. \kern-0em} {{\text{CFO}}}}$ и ${{{{{\text{W}}{{{\text{O}}}_{3}}} \mathord{\left/ {\vphantom {{{\text{W}}{{{\text{O}}}_{3}}} {{\text{ITO}}}}} \right. \kern-0em} {{\text{ITO}}}}} \mathord{\left/ {\vphantom {{{{{\text{W}}{{{\text{O}}}_{3}}} \mathord{\left/ {\vphantom {{{\text{W}}{{{\text{O}}}_{3}}} {{\text{ITO}}}}} \right. \kern-0em} {{\text{ITO}}}}} {{\text{CFO}}}}} \right. \kern-0em} {{\text{CFO}}}}$ при облучении видимым светом. Взято из работы [122].
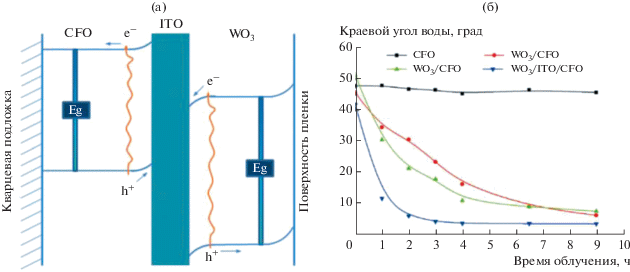
Подход, основанный на создании гетероперехода II типа путем объединения полупроводниковых оксидов, был впервые целенаправленно использован для механистического изучения фотоиндуцированных изменений гидрофильности поверхности тонких пленок оксидов металлов в работе [106]. Было продемонстрировано, что состав и структура сэндвичевых покрытий ${\text{Ti}}{{{\text{O}}}_{2}}{\kern 1pt} --{\kern 1pt} {\text{ZnO}}$ влияют на процесс фотоиндуцированного изменения гидрофильности поверхностей диоксида титана и цинка. На рис. 26а представлены кинетические кривые для случаев фотоиндуцированного изменения смачиваемости поверхности индивидуальных пленок TiO2 и ZnO и пленок слоистого строения ${{{\text{Ti}}{{{\text{O}}}_{2}}} \mathord{\left/ {\vphantom {{{\text{Ti}}{{{\text{O}}}_{2}}} {{\text{ZnO}}}}} \right. \kern-0em} {{\text{ZnO}}}}$ и ${{{\text{ZnO}}} \mathord{\left/ {\vphantom {{{\text{ZnO}}} {{\text{Ti}}{{{\text{O}}}_{2}}}}} \right. \kern-0em} {{\text{Ti}}{{{\text{O}}}_{2}}}}.{\text{\;}}$ Они наглядно демонстрируют, что кинетические параметры фотоиндуцированного изменения гидрофильности поверхности могут быть значительно изменены за счет создания гетероперехода между компонентами покрытия. Особенно заметно изменялось поведение поверхности пленки ${\text{ZnO}}$ при “подкладывании” под нее слоя диоксида титана: фотовозбуждение системы приводило к переходу поверхности ZnO из менее гидрофильного состояния (θ ≈ 20°) (рис. 26а, кривая 2) в высокогидрофильное (θ < 6°) (рис. 26а, кривая 4).
Рис. 26.
(а) Кинетика фотоиндуцированного изменения краевого угла воды θ на поверхности тонких пленок TiO2 (1), (2), TiO2/ZnO (3), ZnO/TiO2 (4). Погрешность измерения θ составляла ±1.0°. (б) Схема фотоиндуцированного переноса заряда для гетероперехода ${{{\text{Ti}}{{{\text{O}}}_{2}}} \mathord{\left/ {\vphantom {{{\text{Ti}}{{{\text{O}}}_{2}}} {{\text{ZnO}}}}} \right. \kern-0em} {{\text{ZnO}}}}{\text{.}}$ Взято из [106].
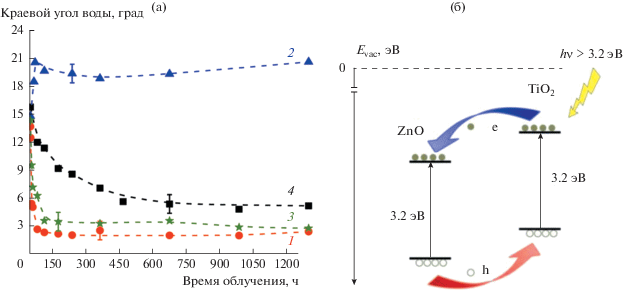
Согласно схеме фотоиндуцированного переноса заряда на гетеропереходе ${\text{Ti}}{{{\text{O}}}_{2}}{\kern 1pt} - {\kern 1pt} {\text{ZnO}}$ (рис. 26б) и экспериментальным данным по измерению работы выхода, полученным в работе [106], сделано предположение, что обогащение поверхности ${\text{ZnO}}$ фотоэлектронами, захваченными поверхностными электронными центрами, приводит к увеличению гидрофильности поверхности оксида цинка. В то же время для поверхности ${\text{Ti}}{{{\text{O}}}_{2}}$ ключевым фактором для супергидрофильного перехода, индуцированного УФ-излучением, является не концентрация определенного типа фотоносителей, а соотношение концентраций фотоносителей обоих типов (n1/n2), участвующих в активации и дезактивации гидрофильных участков поверхности (механизм (24) и выражение (25)).
Таким образом, предложенный подход, основанный на формировании гетероструктурных покрытий II типа, представляется многообещающим и продуктивным как для фундаментальных исследований фотоиндуцированной поверхностной гидрофильности, так и для создания самоочищающихся покрытий с контролируемой смачиваемостью. Интересным продолжением этих исследований может являться использование селективного фотовозбуждения отдельных компонентов гетероструктурной системы, что возможно реализовать при комбинации широкозонного полупроводникового материала с узкозонным полупроводником, например, комбинация оксидов титана или цинка с оксидами одно- и двухвалентной меди, сульфидом кадмия, оксидом вольфрама(VI) и другими. Эффективность разделения зарядов в таких системах уже продемонстрирована повышенной фотокаталитической и фотоэлектрохимической эффективностью в различных реакциях. [13, 120, 121, 128–130].
8. ЗАКЛЮЧЕНИЕ
Проведенный анализ работ по исследованию фотоиндуцированного изменения гидрофильности поверхности тонких пленок оксидов металлов обобщает информацию, накопленную за период с момента открытия эффекта фотоиндуцированной супергидрофильности поверхности диоксида титана до настоящего времени. Очевидно, что количество прикладных работ, в которых предлагается все большее число разнообразных способов использования обнаруженного эффекта и разработки самоочищающихся материалов, значительно превышает число фундаментальных исследований данного фотостимулированного процесса. Это обусловленно, прежде всего, проблемами, связанными с установлением экспериментально и теоретически обоснованных связей между “микроскопическими” стадиями фотовозбуждения и локализации возбуждения на поверхности, влиянием возбужденного состояния поверхности на перестройку адсорбированного слоя, с одной стороны, и такими макроскопическими параметрами как поверхностная энергия и связанный с ней угол смачивания водой, с другой стороны.
На настоящий момент для фотоактивных материалов, поверхность которых свободна от органических загрязнений, гипотеза об электронном механизме с участием фотогенерированных носителей является наиболее адекватно объясняющей экспериментальные результаты по влиянию различных факторов на термодинамику и кинетику процесса фотоиндуцированной супергидрофильности. Электронные процессы на поверхности твердого тела при его фотовозбуждении способны изменить силу и тип взаимодействий активных поверхностных центров с адсорбированными молекулами воды, находящимися в непосредственном контакте с поверхностью. Дальнейшая реструктуризация гидратно-гидроксильного покрова в полислое воды приводит к изменению величины свободной поверхностной энергии и, как следствие, макроскопического свойства поверхности твердого тела – ее смачиваемости.
На параметры, характеризующие процесс фотоиндуцированного изменения гидрофильности поверхности, такие как начальная скорость изменения краевого угла и направление процесса и предельно достижимое гидрофильное состояние, наряду с химической и электронной природой фотоактивного материала существенное влияние оказывает и ряд других факторов. Среди них – кислотность поверхности, температура обработки, состав окружающей газовой атмосферы, интенсивность и спектральный состав действующего света и другие. Воздействие этих факторов обусловлено их влиянием либо на значение поверхностной энергии исходного покрытия, либо на изменение соотношения концентраций фотогенерированных носителей, участвующих в первичных электронных процессах при фотовозбуждении материала.
Достигнутое к настоящему времени понимание поверхностных процессов, происходящих на микроуровне (молекулярные процессы) и макроуровне (адгезия воды к поверхности твердого тела), позволило предложить разнообразные пути управления смачиваемостью поверхности фотоактивных материалов с помощью света. Одним из наиболее эффективных, на наш взгляд, является способ комбинирования фотоактивных материалов, например, двух полупроводников, образующих гетеропереход II типа, или полупроводника и металла. Такой подход представляется многообещающим и при механистических исследованиях процесса фотоиндуцированной гидрофильности, а именно для выяснения деталей механизмов микро- и макропроцессов на поверхности и определения взаимосвязи между ними.
На сегодняшний день диоксид титана остается основным объектом фундаментальных и прикладных исследований процесса фотоиндуцированной супергидрофильности. Однако использование в дальнейших исследованиях композитных покрытий позволит расширить круг самоочищающихся материалов и повысить эффективность их действия.
Список литературы
Wang R., Hashimoto K., Fujishima A., Chikuni M. et al. // Nature. 1997. V. 388. P. 431.
Wang R., Hashimoto K., Fujishima A. et al. // Adv. Mater. 1998. V. 10. P. 135.
Tadanaga K., Morinaga J., Matsuda A., Minami T. // Chem. Mater. 2000. V. 12. P. 590.
Tadanaga K., Morinaga J., Minami T. // J. Sol-Gel Sci. Technol. 2000. V. 19. P. 211.
Fujishima A., Rao T.N., Tryk D.A. // Electrochim. Acta. 2000. V. 45. P. 4683.
Fujishima A., Rao T.N., Tryk D.A. // J. Photochem. Photobiol. C. 2000. V. 1. P. 1.
Hattori A., Kawahara T., Uemoto T. et al. // J. Colloid Interface Sci. 2000. V. 232. P. 410.
Gao S.J., Shi Z., Zhang W.B. et al. // ACS Nano. 2014. V. 8. P. 6344.
Sawai Y., Nishimoto S., Kameshima Y. et al. // Langmuir. 2013. V. 29. P. 6784.
Hashimoto K., Irie H., Fujishima A. // Jpn. J. Appl. Phys. 2005. V. 44. P. 8269.
Zhang L., Dillert R., Bahnemann D., Vormoo M. // Energy Environ. Sci. 2012. V. 5. P. 7491.
Midtdal K., Jelle B.P. // Sol. Energy Mater. Sol. Cells. 2013. V. 109. P. 126.
Banerjee S., Dionysiou D.D., Pillai S.C. // Appl. Catal. B. 2015. V. 176−177. P. 396.
Bhushan B., Jung Y.C. // Prog. Mater. Sci. 2011. V. 56. P. 1.
Gao X., Guo Z. // J. Bionic Eng. 2017. V. 14. P. 401.
Щукин Е.Д., Перцов А.В., Амелина Е.А. Коллоидная химия: учебник для бакалавров. М.: Издательство Юрайт, 2014.
Young T. // Philos. Trans. Royal Soc. 1805. V. 95. P. 65.
Hondros E.D. // J. Mater. Sci. 2005. V. 40. P. 2119.
Della Volpe C., Maniglio D., Brugnara M. et al. // J. Colloid Interface Sci. 2004. V. 271. P. 434.
Fowkes F.M. // J. Phys. Chem. 1962. V. 66. P. 382.
Good R.J., van Oss C.J. // Modern Approaches to Wettability / Ed. by Schrader M.E., Loeb G.I. Boston: Springer, 1992. P. 27.
van Oss C.J. // Colloids Surf. A. 1993. V. 78. P. 1.
Lee L.-H. // Langmuir. 1996. V. 12. P. 1681.
Żenkiewicz M. // Polym. Test. 2007. V. 26. P. 14.
https://www.biolinscientific.com/measurements/surface-free-energy.
Law K.-Y. // J. Phys. Chem. Lett. 2014. V. 5. P. 686.
Sakai N., Fujishima A., Watanabe T., Hashimoto K. // J. Phys. Chem. B. 2003. V. 107. P. 1028.
Irie H., Washizuka S., Yoshino N., Hashimoto K. // Chem. Commun. 2003. P. 1298.
Irie H., Washizuka S., Hashimoto K. // Thin Solid Films. 2006. V. 510. P. 21.
Simonsen M.E., Li Z., Søgaard E.G. // Appl. Surf. Sci. 2009. V. 255. P. 8054.
Emeline A.V., Rudakova A.V., Sakai M. et al. // J. Phys. Chem. C. 2013. V. 117. P. 12086.
Seki K., Tachiya M. // J. Phys. Chem. B. 2004. V. 108. P. 4806.
Lee F.K., Andreatta G., Benattar J.-J. // Appl. Phys. Lett. 2007. V. 90. 181928.
Mills A., Crow M. // Int. J. Photoenergy. 2008. V. 2008. 470670.
Foran P.S., Boxall C., Denison K.R. // Langmuir. 2012. V. 28. P. 17647.
Kawasaki S., Holmström E., Takahashi R. et al. // J. Phys. Chem. C. 2017. V. 121. P. 2268.
Watanabe T., Fukayama S., Miyauchi M. et al. // J. Sol-Gel Sci. Technol. 2000. V. 19. P. 71.
Yu J., Zhao X. // Mater. Res. Bull. 2000. V. 35. P. 1293.
Рудакова А.В., Опаричева У.Г., Гришина А.Е. и др. // Фундаментальные исследования. 2013. С. 1959.
White J.M., Szanyi J., Henderson M.A. // J. Phys. Chem. B. 2003. V. 107. P. 9029.
Mezhenny S., Maksymovych P., Thompson T.L. et al. // Chem. Phys. Lett. 2003. V. 369. P. 152.
Zubkov T., Stahl D., Thompson T.L. et al. // J. Phys. Chem. B. 2005. V. 109. P. 15454.
Thompson T.L., Yates J.T. // Chem. Rev. 2006. V. 106. P. 4428.
Ohtsu N., Masahashi N., Mizukoshi Y., Wagatsuma K. // Langmuir. 2009. V. 25. P. 11586.
Miyauchi M., Nakajima A., Fujishima A. et al. // Chem. Mater. 2000. V. 12. P. 3.
Miyauchi M., Nakajima A., Watanabe T., Hashimoto K. // Chem. Mater. 2002. V. 14. P. 2812.
Hennessy D.C., Pierce M., Chang K.-C. et al. // Electrochim. Acta. 2008. V. 53. P. 6173.
Balajka J., Hines M.A., De Benedetti W.J.I. et al. // Science. 2018. V. 361. P. 786.
Yan X., Abe R., Ohno T. et al. // Thin Solid Films. 2008. V. 516. P. 5872.
Takeuchi M., Sakamoto K., Marta G. et al. // J. Phys. Chem. B. 2005. V. 109. P. 15422.
Bulanin K.M., Bahnemann D.W., Rudakova A.V. // Rev. Sci. Instrum. 2019. V. 90. 105113.
Sakai N., Fujishima A., Watanabe T., Hashimoto K. // J. Phys. Chem. B. 2001. V. 105. P. 3023.
Emeline A.V., Rudakova A.V., Ryabchuk V.K., Serpone N. // J. Phys. Chem. B. 1998. V. 102. P. 10906.
Emeline A.V., Kuzmin G.N., Purevdorj D. et al. // J. Phys. Chem. B. 2000. V. 104. P. 2989.
Emeline A.V., Ryabchuk V.K., Serpone N. // J. Photochem Photobiol. A. 2000. V. 133. P. 89.
Rudakova A.V., Oparicheva U.G., Grishina A.E. et al. // J. Phys. Chem. C. 2015. V. 119. P. 9824.
Wang R., Sakai N., Fujishima A. et al. // J. Phys. Chem. B. 1999. V. 103. P. 2188.
Sun R.-D., Nakajima A., Fujishima A. et al. // J. Phys. Chem. B. 2001. V. 105. P. 1984.
Fujishima A., Zhang X. // C. R. Chim. 2006. V. 9. P. 750.
Fujishima A., Zhang X., Tryk D.A. // Surf. Sci. Rep. 2008. V. 63. P. 515.
Szczepankiewicz S.H., Colussi A.J., Hoffmann M.R. // J. Phys. Chem. B. 2000. V. 104. P. 9842.
Szczepankiewicz S.H., Moss J.A., Hoffmann M.R. // J. Phys. Chem. B. 2002. V. 106. P. 7654.
Spagnol V., Cachet H., Baroux B., Sutter E. // J. Phys. Chem. C. 2009. V. 113. P. 3793.
Schneider J., Matsuoka M., Takeuchi M. et al. // Chem. Rev. 2014. V. 114. P. 9919.
hibata T., Irie H., Hashimoto K. // J. Phys. Chem. B. 2003. V. 107. P. 10 696
Kamisaka H., Yamashita K. // Proc. SPIE. 2006. V. 6340. 634 004.
Takeuchi M., Martra G., Coluccia S., Anpo M. // J. Phys. Chem. B. 2005. V. 109. P. 7387.
Takeuchi M., Bertinetti L., Martra G. et al. // Appl. Catal. A. 2006. V. 307. P. 13.
Takeuchi M., Martra G., Coluccia S., Anpo M. // J. Phys. Chem. C. 2007. V. 111. P. 9811.
Di Valentin C., Tilocca A., Selloni A. et al. // J. Am. Chem. Soc. 2005. V. 127. P. 9895.
Kevorkyants R., Rudakova A.V., Chizhov Y.V., Bu-lanin K.M. // Chem. Phys. Lett. 2016. V. 662. P. 97.
Soria J., Sanz J., Sobrados I. et al. // J. Phys. Chem. C. 2007. V. 111. P. 10590.
Nosaka A.Y., Kojima E., Fujiwara T. et al. // J. Phys. Chem. B. 2003. V. 107. P. 12042.
Uosaki K., Yano T., Nihonyanagi S. // J. Phys. Chem. B. 2004. V. 108. P. 19086.
Nosaka A.Y., Fujiwara T., Yagi H. et al. // Chem. Lett. 2002. V. 3. P. 420.
Nakamura R., Ueda K., Sato S. // Langmuir. 2001. V. 17. P. 2298.
Liu B., Wen L., Zhao X. // Appl. Surf. Sci. 2008. V. 255. P. 2752.
Emeline A.V., Rudakova A.V., Oparicheva U.G. // Proc. of 4th Int. Conf. on Semiconductor Photochemistry (SP4). Prague, 2013. P. KL6-44.
Rudakova A.V., Oparicheva U.G., Grishina A.E. et al. // J. Colloid Interface Sci. 2016. V. 466. P. 452.
Jia R., Zhao D., Zhang L. et al. // J. Phys. Chem. C. 2018. V. 122. P. 24818.
Honjo Y., Furuya M., Takamasa T., Okamoto K. // Proc. 16th Int. Conf. on Nuclear Engineering (ICONE16). Orlando, 2008. P. 353.
Sakai N., Fukuda K., Shibata T. et al. // J. Phys. Chem. B. 2006. V. 110. P. 6198.
Egerton T.A., King C.J. // JOCCA. 1979. V. 62. P. 386.
Okamoto K., Yamamoto Y., Tanaka H., Itaya A. // Bull. Chem. Soc. Jpn. 1985. V. 58. P. 2023.
D’Oliveira J.-C., Al-Sayyed G., Pichat P. // Environ. Sci. Technol. 1990. V. 24. P. 990.
Rudakova A.V., Maevskaya M.V., Emeline A.V., Bahnemann D.W. // Sci. Rep. 2016. V. 6. 34285.
Rudakova A.V., Emeline A.V., Bulanin K.M. et al. // J. Photochem. Photobiol. A. 2018. V. 367. P. 397.
Zeng R.-J., Pang Q., Lin Z.-H. et al. // Chinese J. Inorg. Chem. 2003. V. 19. P. 678.
Nishijima K., Naiton H., Tsubota T., Ohno T. // J. Ceram. Soc. Jpn. 2007. V. 115. P. 310.
Emeline A.V., Salinaro A., Serpone N. // J. Phys. Chem. B. 2000. V. 104. P. 11202.
Emeline A.V., Smirnova L.G., Kuzmin G.N. et al. // J. Photochem. Photobiol. A. 2002. V. 148. P. 97.
Yamagishi M., Kuriki S., Song P.K., Shigesato Y. // Thin Solid Films. 2003. V. 442. P. 227.
Miyauchi M., Shimai A., Tsuru Y. // J. Phys. Chem. B. 2005. V. 109. P. 13 307.
Katsumata K., Okayasu Y., Matsushita N., Okada K. // Chem. Lett. 2013. V. 42. P. 618.
Watanabe T., Nakajima A., Wang R. et al. // Thin Solid Films. 1999. V. 351. P. 260.
Nakajima A., Koizumi S., Watanabe T., Hashimoto K. // J. Photochem. Photobiol. A. 2001. V. 146. P. 129.
Song P.K., Yamagishi M., Odaka H., Shigesato Y. // Jpn. J. Appl. Phys. 2003. V. 42. P. L1529.
Katayama M., Miyakawa T., Ohsawa T. et al. // Jpn. J. Appl. Phys. 2007. V. 46. P. L562.
Zhuang J., Weng S., Dai W. et al. // J. Phys. Chem. C. 2012. V. 116. P. 25 354.
Yu J., Zhou M., Yu H. et al. // Mater. Chem. Phys. 2006. V. 95. P. 193.
Kaleji B.K., Hosseinabadi N. // J. Sol-Gel Sci. Technol. 2014. V. 69. P. 412.
Dai W.-X., Wang X.-X., Fu X.-Z. et al. // Acta Phys-Chim. Sin. 2005. V. 21. P. 1274.
Talinungsang, Purkayastha D.D., Krishna M.G. // Appl. Surf. Sci. 2018. V. 447. P. 724.
Рябчук В.К., Емелин А.В. Нанофотоника гетерогенных систем. Учебное пособие. СПб.: СПбГУ, 2007.
Serpone N. // Catalysts. 2018. V. 8. P. 553.
Rudakova A.V., Emeline A.V., Bahnemann D.W. // J. Phys. Chem. C. 2019. V. 123. P. 8884.
Shtarev D.S., Shtareva A.V., Kevorkyants R. et al. // J. Mater. Chem. C. 2020. V. 8. P. 3509.
Lee H.Y., Park Y.H., Ko K.H. // Langmuir. 2000. V. 16. P. 7289.
Tseng Y.-H., Kuo C.-S., Huang C.-H. et al. // Micro Nano Lett. 2010. V. 5. P. 81.
Tang J., Quan H., Ye J. // Chem. Mater. 2007. V. 19. P. 116.
Lahann J., Mitragotri S., Tran T.-N. et al. // Science. 2003. V. 299. P. 371.
Sakai N., Wang R., Fujishima A. et al. // Langmuir. 1998. V. 14. P. 5918.
Gras S.L., Mahmud T., Rosengarten G. et al. // ChemPhysChem. 2007. V. 8. P. 2036.
Kamei M., Mitsuhashi T. // Surf. Sci. 2000. V. 463. P. L609.
Nakamura M., Aoki T., Hatanaka Y. // Vacuum. 2000. V. 59. P. 506.
Purkayastha D.D., Brahma R., Krishna M.G., Madhurima V. // Bull. Mater. Sci. 2015. V. 38. P. 203.
Liu Z., Wang Y., Peng X. et al. // Sci. Technol. Adv. Mater. 2012. V. 13. 025001.
Esaki L. // IEEE J. Quantum Electron. 1986. V. QE-22. P. 1611.
Rana F. Lecture Course on Semiconductor Optoelectronics / Cornell University. https://courses.cit.cornell.edu/ece533/Lectures/handout2.pdf.
Wang Z., Wu W., Xu Q. et al. // Nano Energy. 2017. V. 38. P. 518.
vanov S.A., Piryatinski A., Nanda J. et al. // J. Am. Chem. Soc. 2007. V. 129. P. 11708
Liu Z., Miyauchi M. // Chem. Commun. 2009. P. 2002.
Balachandran S., Prakash N., Thirumalai K. et al. // Ind. Eng. Chem. Res. 2014. V. 53. P. 8346.
Hwang Y.K., Patil K.R., Kim H.-K. et al. // Bull. Korean Chem. Soc. 2005. V. 26. P. 1515.
Qureshi U., Manning T.D., Parkin I.P. // J. Mater. Chem. 2004. V. 14. P. 1190.
Kim H.-M., Kim D., Kim B. // Surf. Coat. Technol. 2015. V. 271. P. 18.
Miyauchi M., Nakajima A., Hashimoto K., Watanabe T. // Adv. Mater. 2000. V. 12. P. 1923.
Serpone N., Maruthamuthu P., Pichat P. et al. // J. Photochem. Photobiol. A. 1995. V. 85. P. 247.
Low J., Yu J., Jaroniec M. et al. // Adv. Mater. 2017. 1601694.
Serpone N., Emeline A.V. // J. Phys. Chem. Lett. 2012. V. 3. P. 673.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал