

Коллоидный журнал, 2021, T. 83, № 2, стр. 187-196
Термодинамическое исследование солюбилизации краунзамещенного фталоцианината магния в водных растворах катионных ПАВ
Т. Г. Мовчан 1, *, А. И. Русанов 1, 2, Е. В. Плотникова 1
1 Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
119071 Москва, Ленинский просп., 31, Россия
2 Менделеевский центр, Санкт-Петербургский государственный университет
199034 Санкт-Петербург, Университетская наб., 7/9, Россия
* E-mail: movchan_tamara@mail.ru
Поступила в редакцию 20.10.2020
После доработки 26.10.2020
Принята к публикации 30.10.2020
Аннотация
Проведено спектрофотометрическое исследование солюбилизации краунзамещенного фталоцианината магния(I) в водных растворах бромидов тетрадецилтриметиламмония(II), гексадецилтриметиламмония(III) и гексадецилтрифенилфосфония. Опыты проводились с насыщенными растворами I в условиях термодинамического равновесия раствора с осадком I. На основе экспериментальных данных определены термодинамические характеристики солюбилизации: солюбилизационная емкость мицелл, коэффициент распределения солюбилизата между мицеллами и окружающим раствором, а также стандартное сродство солюбилизации I. Показано, что, несмотря на все различия в структуре ПАВ, стандартная работа внесения молекулы I в мицеллы всех трех ПАВ почти одинакова. В расчете на одну молекулу I в мицеллах II и III найденные значения солюбилизационной емкости приводят к аномально большим числам агрегации. Среди объяснений этого явления – возможность возникновения бимодального распределения мицелл, в котором мицеллы с солюбилизатом сосуществуют с “пустыми” мицеллами, благодаря чему среднее число молекул солюбилизата в мицелле может оказаться меньше единицы.
ВВЕДЕНИЕ
Фталоцианины и их производные применяются в самых разных областях, включая молекулярную электронику, химию и медицину. Среди функционально замещенных фталоцианинов одним из перспективных для широкого использования является сравнительно новый и еще мало изученный окта[(4'-бензо-15-краун-5)окси]фталоцианинат магния (I) [1–6]. По целому ряду причин (например, для проявления хромофорных свойств молекул) желательно иметь его в мономерной форме. В [1–3] было показано, что I находится преимущественно в мономерной форме в дихлорметане, которой на спектрах поглощения отвечает резкий пик с длиной волны 690 нм и сопутствующий малый максимум при длине волны 616 нм. Для коэффициента экстинкции ε в дихлорметане приводятся значения (в единицах 105 М–1 см–1): 1.64 [1] и 2.09 [3]. Характерные полосы мономеров I при длине волны 686 нм наблюдались в спектрах поглощения I, растворенного в водных мицеллярных средах анионных ПАВ: додецилсульфата натрия, ДСН (ε = 1.7 × × 105 М–1 см–1) [1, 2], дезоксихолата натрия [2] и додецилбензолсульфоната натрия (ε = 1.89 × × 105 М–1 см–1) [3]. Однако в растворах бромида гексадецилтриметиламмония концентрации 8.6 мМ в работах [1, 3] фиксировали преимущественно агрегированное состояние I. Согласно работе [4], в спектрах поглощения широкая полоса поглощения с максимумом в области 634 нм отвечает димерам.
Эффект мономеризации I в мицеллярных средах анионных ПАВ достигается с помощью солюбилизации и поэтому мог бы проявиться и в растворах катионных ПАВ. Впервые удалось обнаружить его для I в присутствии бромида гексадецилтрифенилфосфония (IV на рис. 1) и его гомологов [5, 6]. Переход от димеров I к мономерам в процессе солюбилизации под влиянием ПАВ более детально был изучен на примере ДСН [7]. Установлено, что мономеризация I начинается при концентрациях гораздо ниже критической концентрации мицеллообразования (ККМ) ДСН, а при еще меньших концентрациях специфическое действие ДСН повышает растворимость димеров I. Найдена растворимость I в чистой воде, которая составила 7.38 мкМ. В [7] были определены термодинамические характеристики солюбилизации: солюбилизационная емкость мицелл, коэффициент распределения солюбилизата между мицеллами и окружающим раствором, а также стандартное сродство солюбилизации I.

Данная работа продолжает начатое в [7] исследование солюбилизации краунзамещенного фталоцианината магния в водных растворах ПАВ, но теперь уже на примере катионных ПАВ: бромидов тетрадецилтриметиламмония (II), гексадецилтриметиламмония (III) и гексадецилтрифенилфосфония (IV).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы
Вещество I представляет собой довольно сложное комплексное соединение металла с плоской молекулярной структурой макроцикла и подвижными (благодаря связи через кислородный мостик) бензо-краун-группами (рис. 1). Синтез и очистка I, произведенные в Институте физиологически активных веществ РАН совместно с Институтом физической химии и электрохимии им. А.Н. Фрумкина РАН, описаны в работе [1]. К нам препарат поступил в виде кристаллического порошка, что свидетельствует о его достаточно высокой чистоте.
Вещества II, III и IV – препараты фирмы “Acros Organics” с содержанием основного вещества 99.8% использовались без дальнейшей очистки. Вода бралась в виде тридистиллята с удельной электропроводностью не выше 4 × 10–4 См/м при 27°С.
Методика измерений и результаты
Главной задачей эксперимента было определение равновесной концентрации I в мицеллярных растворах II, III и IV. Для этого использовался спектрофотометрический метод, подразумевающий измерение оптической плотности A раствора для характерной длины волны. Как уже отмечалось ранее [1–6], I может находиться в растворах в агрегированном (в виде димеров) и мономерном состояниях. Для нас особый интерес представляли мономеры. Мономерам I отвечает резкий пик в видимой части спектра при длине волны 690 нм в дихлорметане [1–3], 686 нм – в водных мицеллярных растворах ДСН и додецилбензолсульфоната натрия [1–3], 683 нм – в мицеллярных средах IV [5, 6], а димерам – широкая полоса с максимумом при длине волны 634 нм [4]. Для определения характерной длины волны I в растворах II и III (наша первая методическая задача) были измерены электронные спектры поглощения при разной концентрации ПАВ и заданной концентрации I. Здесь и в дальнейшем условимся обозначать концентрацию солюбилизатора (ПАВ) как ${{с}_{1}},$ а концентрацию солюбилизата как с2.
Приведенные на рис. 2а спектры поглощения I в растворах II (с концентрацией с1 ≤ 1.36 мМ) имеют широкую полосу с максимумом при длине волны 634 нм, что характерно для димеров I [4]. Увеличение концентрации II сначала повышает растворимость I в виде димеров, т.е. первичная стадия солюбилизации происходит на уровне димеров. При дальнейшем повышении содержания II ситуация меняется: при той же концентрации I (с2 = 1.1 мкМ) при с1 = 3 мМ наряду с присутствием максимума димеров заметен пик мономеров I (кривая 1 на рис. 2б), высота которого резко возрастает уже при с1 = 4 мМ (кривая 2 на рис. 2б). Заметим, что появление полосы мономера происходит в области концентраций ниже ККМ II, составляющей около 3.8 мМ [8, 9]. Подобный эффект наблюдался в растворах додецилбензолсульфоната натрия [3] и ДСН [7]. На рис. 2в видно, что при всех значениях ${{с}_{2}}$ мономерам I в растворах II соответствует хорошо выраженный пик при λ = 683 нм.
Рис. 2.
Электронные спектры поглощения I при с2 = 1.1 мкМ в водных растворах II: (а) в домицеллярной области при с1 = 0.16 (1) и 1.36 мМ (2), (б) в домицеллярной при с1 = 3 мМ (1) и мицеллярной при с1 = = 4 мМ (2) областях, (в) в мицеллярной области при с1 = 4 (1) и 12 мМ (2, 3) и с2 = 1.1 (1), 2 (2) и 4 мкМ (3). l = 1 см.

На рис. 3 и 4 приведены электронные спектры поглощения I в растворах III и IV [5]. При низкой (с1 = 0.1 мМ) концентрации III, как и в случае II, это – полосa димеров (кривaя 1 на рис. 3). Но в растворе IV в спектре поглощения I четко просматривается полоса мономера (кривая 1 на рис. 4) при с1 = 0.037 мМ, т.е. значительно ниже ККМ IV (0.197 мМ [9], 0.17 мМ [10, 11] при 30°C и 0.14 мМ [12] при 25°C). В мицеллярных средах III (ККМ при 30°C около 1мМ [8, 9, 13]) и IV измеренные спектры демонстрируют высокоинтенсивные полосы, характерные для мономера I (кривые 2 на рис. 3 и 4) с положением максимума при λ = 683 нм. Таким образом, была решена первая методическая задача – нахождение длины волны, соответствующей поглощению мономера в растворах катионных ПАВ II, III и IV.
Рис. 3.
Электронные спектры поглощения I (с2 = = 1.1 мкМ) в диапазоне длин волн 500–850 нм в водных растворах III: в домицеллярной области при с1 = 0.1 мМ (1) и в мицеллярной – при с1 = 5 мМ (2). Длина оптического пути 1 см.

Рис. 4.
Электронные спектры поглощения I (в диапазоне длин волн 550–700 нм) в водных растворах IV при c2 = 1.1 мкМ: в домицеллярной области ПАВ при c1 = 0.037 мМ (1) и в мицеллярной области при c1 = = 0.4 мМ (2) [5]. l = 1 см.

Связь оптической плотности (экстинкции) A с концентрацией поглощающего свет вещества в растворе c дается законом Ламберта–Бера
где l – длина пути светового луча. Из (1) видно, что для определения концентрации по оптической плотности необходимо знать коэффициент экстинкции ε. Эта важная характеристика вещества должна быть найдена из независимого эксперимента, что составляло вторую методическую задачу данной работы. В литературе имеются значения ε в единицах 105 М–1 см–1, полученные для I в дихлорметане (1.64 [1], 2.09 [2, 3] (при длине волны λ = 690 нм) и 4.43 [7], в диметилсульфоксиде (2.9) [7]). Однако, как указывалось в [7], разнобой в этих данных указывает на нецелесообразность использования органических растворителей для калибровки I.Если коэффициент экстинкции вещества зависит от природы среды, возникает естественное желание найти его при использовании такой среды, в которой и будет проводиться эксперимент. В работе [7] для нахождения коэффициента экстинкции I как константы закона Ламберта–Бера была построена калибровочная кривая при разном его содержании в мицеллярном растворе ДСН (задание концентрации ПАВ определяет среду), показывающая (своей прямолинейностью), что этот закон действительно выполняется. Известно, что после прохождения ККМ свойства мицеллярной системы меняются незначительно. В соответствии с требованиями термодинамики химический потенциал ПАВ продолжает возрастать с концентрацией, но настолько медленно, что ситуация напоминает фазовый переход, когда химический потенциал не изменяется совсем. Очень слабо меняется и средняя активность ионного ПАВ, а потому можно надеяться, что калибровка, проведенная при некоторой концентрации ПАВ выше ККМ, работает в довольно широком интервале концентраций мицеллярного раствора.
В соответствии со сказанным выше, оптическую плотность полосы поглощения I в растворах II, III и IV измеряли при постоянной концентрации ПАВ, значение которой (естественно, выше ККМ) выбирали из условия полного растворения взятого количества солюбилизата. На рис. 5 показана калибровочная кривая для I в растворах II при с1 = 12 мМ, III при с1 = 10 мМ и IV при с1 = 1 мМ. Видно, что экспериментальные зависимости A(c2) при λ = 683 нм в интервале ${{с}_{2}}$ = 1–15 мкМ хорошо (с квадратом смешанной корреляции около 0.995) отображается прямыми линиями, наклон которых однозначно определяет значение молярного коэффициента экстинкции. Результаты калибровки суммированы в табл. 1. Эти значения и использовались для определения концентрации мономеров I во всех наших опытах при λ = 683 нм.
Рис. 5.
Калибровочные зависимости оптической плотности I в водных растворах II при c1 = 12 мМ (1), III при c1 = 10 мМ (3) и IV при c1 = 1 мМ (2).

Таблица 1.
Значения молярного коэффициента экстинции ε для I в разных средах
| Среда | с1, мМ | λ, нм | ε, 105 М–1 см–1 |
|---|---|---|---|
| Водный раствор II | 0 | 634 | 0.435 |
| 1 | |||
| 12 | 683 | 2.13 | |
| Водный раствор III | 0.1 | 634 | 0.57 |
| 0.4 | 0.93 | ||
| 10 | 683 | 2.64 | |
| Водный раствор IV | 0 | 634 | 0.435 |
| 1 | 683 | 2.35 |
Что касается домицеллярного раствора ПАВ, то для него калибровку в отношении солюбилизата нужно проводить для каждой концентрации ПАВ раздельно. Результатом такой кропотливой работы должна быть функция $\varepsilon ({{с}_{1}}),$ описывающая зависимость коэффициента экстинкции от концентрации ПАВ в диапазоне от чистой воды до ККМ. В данной работе были рассчитаны “точечные” значения коэффициентов экстинкции димеров I по заданным концентрациям (с2) и измеренным значениям оптической плотности (А) в максимуме полосы димера (λ = 634 нм). Так, в растворе II при с2 = 1.1 мкМ для с1 = 1 мМ получено значение A = 0.04785. Отсюда по формуле (1) ε = 0.435 × 105 М–1 см–1. В случае III аналогичные расчеты при с2 = 1.13 мкМ, с1 = 0.1 мМ и A = 0.0644 дают значение ε = 0.57 × 105 М–1 см–1, а при с2 = = 0.21 мкМ, с1 = 0.4 мМ и A = 0.0195 имеем ε = = 0.93 × 105 М–1 см–1 (табл. 1).
После проведения калибровки можно было начать эксперимент по исследованию солюбилизации. Его процедура была той же, что и в [7]. В насыщенный водный раствор I добавляли навеску сухого ПАВ (II, III или IV) и выдерживали его в закрытой емкости при комнатной температуре в защищенном от света месте в течение двух-трех суток (до установления равновесия), после чего измеряли оптическую плотность раствора. На рис. 6 в качестве примера приведены спектры поглощения I в растворе III, измеренные через полчаса и через сутки после добавления ПАВ в водный насыщенный раствор I. Следует отметить не только количественное изменение оптической плотности раствора, произошедшее за сутки, но и существенное изменение самого спектра: переход от широкой полосы с двумя слабо выраженными максимумами к интенсивной полосе с максимумом при 683 нм, характерной для мономера. Далее величина оптической плотности при 683 нм (кривая 2 на рис. 6) практически не изменялась.
Рис. 6.
Электронные спектры поглощения I в его насыщенном водном растворе после введения 1.9 мМ III: через 30 мин (1) и через сутки (2). l = 1 см.

С помощью коэффициента экстинкции рассчитывали концентрацию солюбилизата. Процедура повторялась несколько раз, в результате чего строилась изотерма солюбилизации. Интервал задаваемых концентраций ПАВ составлял 0.2–10 ККМ (при указанном во введении определении ККМ через константу закона действия масс сама ККМ становится единицей концентрации [14–16]).
Измерения проводились с помощью однолучевого сканирующего спектрофотометра UV-2800 (UNICO, США) в диапазоне длин волн 190−900 нм с использованием стандартных кварцевых кювет с длиной l оптического пути 1 мм и 1 см. Для обсуждения выбран интервал 500–850 нм (Q-диапазон), в котором проявляются полосы поглощения димеров и мономеров I. Нужно подчеркнуть, что все измерения относились к насыщенным растворам, в условиях, когда раствор все время находился в равновесии с твердой фазой I на дне сосуда. Это значит, что все наши исследования проводились в условиях постоянства химического потенциала I.
На рис. 7 приведены спектры поглощения I в его насыщенных водных растворах без ПАВ (1) и при концентрации III с1 = 0.4 мМ, соответствующей домицеллярной области (2). Поскольку в присутствии ПАВ оптическая плотность раствора I существенно больше, чем в чистой воде, то можно говорить об увеличении растворимости вещества I даже при концентрации ПАВ меньше ККМ. В то же время в спектре еще отсутствует характерный пик мономеров I. Это означает, что сначала присутствие III в воде повышает растворимость I в виде димеров, т.е. первичная стадия солюбилизации происходит на уровне димеров. При увеличении содержания III до с1 = 1.9 мМ растворимость I в воде повышается за счет роста концентрации мономеров (кривая 2 на рис. 6).
Рис. 7.
Электронные спектры поглощения I (в диапазоне длин волн 500–850 нм) в его насыщенных водных растворах без ПАВ (1) и в домицеллярной области III при c1 = 0.4 мМ (2). l = 1 см.

На рис. 8, 9 приведены экспериментальные данные об оптической плотности (A), полученные для насыщенных растворов I в присутствии II, III и IV с концентрациями в интервале 0.2–10 ККМ. При этом данные, представленные отдельными точками, относятся к димерам I, а кривыми и точками – к его мономерам.
Рис. 8.
Зависимость оптической плотности (А) насыщенных водных растворов I от концентрации c1 II (квадраты) и III (треугольники) при значениях λ 683 и 634 нм (соответствующие символы вне кривых II и III).

Рис. 9.
Зависимость оптической плотности (A) насыщенных водных растворов I от концентрации IV (c1) при значениях λ 683 (1) и 634 нм (2).
Из значений оптической плотности и коэффициента экстинкции (из табл. 1) вычислялась концентрация I в растворах исследуемых ПАВ. Полученные величины приведены в табл. 2. Ее первые две (для II и III) и первая одна (для IV) численные строки отвечают концентрациям ПАВ ниже ККМ. В этой области расчет велся с использованием значений коэффициента экстинкции (4.35–9.3) × × 104 М–1 см–1, приведенных в табл. 1 для димеров. Такие значения с гарантией действуют в первой строке для раствора III в табл. 2, но полученное во второй строке явно завышенное значение с2 = 11.8 мкМ (при с1 = 0.4 мМ) свидетельствует о том, что в растворе уже появляются мономеры. Ввиду того, что коэффициент экстинкции для мономеров примерно в пять раз больше, чем для димеров, даже незначительное присутствие мономеров при с1 = 0.4 мМ может при расчете снизить концентрацию с2 = 11.8 мкМ до разумных значений. Фактически при с1 = 0.4 мМ и выше в растворе III присутствует смесь димеров и мономеров, и коэффициент экстинкции должен иметь эффективное значение между значениями (0.5 и 0.93) × 105 М–1 см–1, указанными в табл. 1. Это значение нам не известно, а потому для III (с1 = = 1.9 мМ) в третьей строке табл. 2 расчет ${{с}_{2}}$ не проводился.
Таблица 2.
Оптическая плотность (А) и концентрация солюбилизата I (c2, мкМ) как функция концентрации II, III и IV (c1, мМ) в водном растворе при заданном коэффициенте экстинкции (ε, 105 М–1 см–1)
| Среда | Параметры | |||
|---|---|---|---|---|
| с1 | А | ε | с2 | |
| Вода | 0 | 0.321 | 0.435 | 7.38 |
| Водный раствор II | 1.4 | 0.33 | 7.59 | |
| 3.0 | 0.50 | – | – | |
| 4.0 | 0.80 | 2.13 | – | |
| 6.0 | 1.52 | 7.10 | ||
| 8.0 | 2.50 | 11.70 | ||
| 10.0 | 3.44 | 16.20 | ||
| 12.4 | 4.75 | 22.30 | ||
| 15.0 | 6.20 | 29.10 | ||
| Водный раствор III | 0.1 | 0.425 | 0.57 | 7.46 |
| 0.4 | 1.1 | 0.93 | 11.8 | |
| 1.9 | 1.75 | 2.64 | – | |
| 3.75 | 2.63 | 9.96 | ||
| 5.0 | 3.61 | 13.67 | ||
| 7.0 | 5.3 | 20.1 | ||
| 10.0 | 8.12 | 30.76 | ||
| Водный раствор IV | 0.037 | 0.330 | 0.435 | 7.59 |
| 0.20 | 1.698 | 2.35 | 7.23 | |
| 0.40 | 3.16 | 13.45 | ||
| 0.50 | 4.534 | 19.29 | ||
| 0.70 | 6.344 | 27.00 | ||
| 1.00 | 9.485 | 40.36 | ||
Как было отмечено выше, при заданной концентрации I в растворах II при с1 = 3 мМ наряду с димерами присутствуют мономеры (рис. 2б). Подобная ситуация наблюдается и в насыщенных растворах I при с1 = 3 мМ. Расчет ${{с}_{2}}$ не проводился из-за отсутствия данных о коэффициенте экстинкции I в смеси димеров с мономерами при с1 = = 3 и 4 мМ. Начиная же с четвертой строки табл. 2, а для IV – с третьей, расчет относится к концентрациям ПАВ выше ККМ, где с гарантией работают значения ε для мономеров, которые присутствуют в растворе с большим преобладанием.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
В ряде работ изучалась зависимость оптической плотности водных растворов ПАВ от их концентрации при заданной брутто-концентрации I [1–6]. При этом, очевидно, в процессе прибавления ПАВ идет солюбилизация и мономеризация I до тех пор, пока все количество I не перейдет в мономерную форму. Мы же работали в условиях постоянства химического потенциала солюбилизата, а осадок на дне сосуда играл роль резервуара вещества. По мере поступления ПАВ и солюбилизации свободного I в растворе, химический потенциал I уменьшался, что вызывало поток новой порции I из осадка в раствор для восстановления прежнего значения химического потенциала. Таким образом, у нас поступление ПАВ в раствор всегда сопровождалось переходом в жидкую фазу новой порции I. Это создавало стационарные условия для продолжения такого процесса как угодно долго, а зависимость A или ${{с}_{2}}$ от ${{с}_{1}}$ была всегда возрастающей (напомним, что номером 1 мы обозначили солюбилизатор, а номером 2 – солюбилизат).
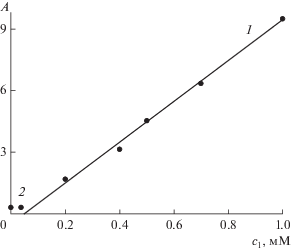
Поскольку расчет разных участков солюбилизационной кривой ${{с}_{2}}({{с}_{1}})$ требует разных и порой неизвестных нам значений коэффициента экстинкции I, мы не можем изобразить эту кривую сплошной линией. На рис. 10 показана солюбилизационная кривая ${{с}_{2}}({{с}_{1}})$ в мицеллярной области II, III и IV. Для последних четырех (в случае III) или пяти (в случае II и IV) значений ${{с}_{2}}({{с}_{1}})$ в табл. 2 точки хорошо (с квадратом смешанной корреляции 0.998) ложатся на прямые линии, для которых аппроксимация дает следующие уравнения (обе концентрации в мМ):
Из уравнений (2)–(4) следует, что пересечение этих прямых с осью абсцисс происходит соответственно при 3.25, 0.88 и 0.075 мМ, и это были бы ККМ II, III и IV, определенные классическим методом солюбилизации (причем весьма правдоподобные: немного меньше известных значений 3.8 мМ [8, 9], 1 мМ [8, 9, 13] и 0.17 мМ [10, 11] для II, III и IV соответственно). Ведь было установлено [9, 17] и подтверждено теоретически [18], что солюбилизат всегда понижает ККМ ПАВ.
Следуя стандартной процедуре, мы должны положить наклоны каждой солюбилизационной прямой (т.е. коэффициенты при ${{с}_{1}}$ в уравнениях (2)–(4)) равными солюбилизационным емкостям s мицелл II, III и IV. Каждая из них является отношением чисел агрегации солюбилизата z и солюбилизатора n:
В данном случае мы заранее знали, что в каждую мицеллу II, III и IV входит лишь одна молекула I (в этом суть явления мономеризации). Положив z = 1, находим значения n 417, 303 и 23 для II, III и IV соответственно.Здесь уже есть, что обсудить. ПАВ II и III отличаются друг от друга только длиной углеводородной цепи: у III она больше на две группы CH2. Как известно из физикохимии ПАВ, чем длиннее неполярный хвост молекулы ПАВ, тем больше его число агрегации в прямых мицеллах. В согласии с этим правилом, из опыта имеем n ≈ 67 для II [19] и n ≈ 90 для III [20]. У нас же получилось, что число агрегации II (417), наоборот, гораздо больше числа агрегации III (303). Как это понимать? Дело в том, что указанное правило относится к обычным прямым мицеллам без солюбилизата, когда радиус углеводородного ядра мицеллы приравнивается длине растянутой углеводородной цепи. Чем больше радиус, тем больше объем углеводородного ядра, а значит, тем больше и число агрегации. У нас же роль ядра мицеллы ПАВ играет посторонняя молекула I, причем размер ядра один и тот же для обоих ПАВ II и III. Очевидно, чем крупнее молекулы ПАВ, тем меньше их может поместиться на поверхности ядра. Таким образом, мы приходим к прямо противоположному правилу: для мицелл разных ПАВ с одним и тем же солюбилизационным ядром, чем крупнее молекулы ПАВ, тем меньше их число агрегации. Что касается IV, то длина углеводородного радикала у этого ПАВ та же, что и у III, но полярная головная часть значительно объемнее. Видимо, это заметно увеличивает посадочную площадь молекулы IV (по сравнению с III) и приводит к числу агрегации 23, близкому к числу агрегации для обычных мицелл IV: n ≈ 15 [12] и 18 [9]. Однако для таких малых чисел агрегации как 1 для I и 23 для IV вряд ли годятся общие рассуждения. Нужно просто смотреть с химической точки зрения, какие агрегативные структуры здесь возможны.
Проблема правдоподобности большого числа агрегации для мицеллы с солюбилизатом I обсуждалась в [7] с попыткой оценить число посадочных мест для углеводородных радикалов. Хотя расчеты касались лишь самого макроцикла молекулы I и дали величину в три раза меньше (110 против 309 для ДСН), но все же это была величина того же порядка. Трудно сказать, сколько еще ионов ДСН могут повиснуть гроздьями на периферийных краунзамещенных функциональных группах молекулы I. В пользу сильного возрастания числа агрегации говорят, например, данные работы [21] по солюбилизации мономеров замещенного фталоцианината цинка (структура I с заменой Mg на Zn при R = SO3H на рис. 1). При использовании в качестве солюбилизаторов II и III методом динамического рассеяния света было установлено, что при солюбилизации диаметр мицелл возрастает от 2.69 до 255 и 295 нм соответственно, т.е. примерно в 100 раз!
Как было отмечено в [7], нельзя исключать и того, что число агрегации ПАВ в мицелле с солюбилизатом I является завышенным по той простой причине, что мы вместо какого-то распределения агрегатов по числам агрегации рассматриваем только один вид мицелл (одномодальное распределение). Если, например, в реальной мицеллярной системе мицеллы с солюбилизатом I сосуществуют с “пустыми” мицеллами (бимодальное распределение), то на языке одномодального распределения это означает, что среднее число молекул солюбилизата в мицелле (число z в формуле (5)) может быть дробным. Тогда и число агрегации n для II и III будет меньше. Числа же 417 и 303 приобретают другой смысл – это среднее соотношение чисел молекул ПАВ и I в системе.
Перейдем теперь к рассмотрению общих термодинамических характеристик солюбилизации – коэффициента распределения солюбилизата и стандартного сродства солюбилизации. Коэффициент распределения (в общем случае любого вещества i) между мицеллами (область внутри мицеллы обозначается символом фазы α) и раствором (фаза β) определяется из опыта как
где ${{x}_{i}}$ – мольная доля солюбилизата, индекс x показывает, что коэффициент распределения выражен в формализме мольных долей. Сформулированная недавно [18] термодинамическая теория солюбилизации с участием реальных мицелл связывает коэффициент распределения со стандартным сродством солюбилизации ${{A}_{{i\operatorname{s} }}}$ соотношением(7)
$kT\ln {{K}_{x}} = \mu _{{i\operatorname{s} }}^{\beta } - \mu _{{i\operatorname{s} }}^{\alpha } \equiv {{A}_{{i\operatorname{s} }}},$Полученные нами экспериментальные данные позволяют рассчитать величины (6) и (7) для II, III и IV. Начнем с оценки мольных долей и сначала – средней мольной доли I в мицеллах $x_{2}^{\alpha }$. Наша система состоит из трех компонентов, но если считать, что вода входит в мицеллы только в виде гидратных оболочек ионов ПАВ, то число компонентов мицелл сокращается до двух (I и ПАВ). Ввиду того, что мольная доля I в мицеллах очень мала, она практически равна солюбилизационной емкости (5) и мы можем оценить $x_{2}^{\alpha }$ примерно равной ее величине (табл. 3). Что касается мольной доли I в растворе (за вычетом мицелл) $x_{2}^{\beta }$, опять же очень малой, то здесь нужно учесть, что концентрация ПАВ, хоть и не настолько, но все же достаточно мала, чтобы ее вкладом в $x_{2}^{\beta }$ можно было пренебречь. Тогда мольную долю можно оценить по растворимости I в воде, которую мы определили как 7.38 мкМ [7]. Делением этого числа на 55.56 М находим $x_{2}^{\beta } \approx $ ≈ 1.33 × 10–7. Рассчитанные по формулам (6) и (7) значения коэффициента распределения ${{K}_{x}}$ и стандартного сродства солюбилизации ${{A}_{{i\operatorname{s} }}}$ для II, III и IV приведены в табл. 3. Значение ${{A}_{{i\operatorname{s} }}}$ представлено в единицах kT, что создает определенные удобства с физической точки зрения.
Таблица 3.
Термодинамические параметры солюбилизации I в мицеллярных растворах II, III и IV
| Параметры | ПАВ | ||
|---|---|---|---|
| II | III | IV | |
| ККМ, мМ | 3.8 | 1 | 0.17 |
| $ККМ{\kern 1pt} '$, мМ | 3.25 | 0.88 | 0.083 |
| s, 10–3 | 2.4 | 3.3 | 43.6 |
| n (z = 0) | 67 | 90 | 18 |
| n (z = 1) | 417 | 303 | 23 |
| $x_{2}^{\alpha }$, 10–3 | 2.394 | 3.289 | 41.778 |
| $x_{2}^{\beta }$, 10–7 | 1.33 | 1.33 | 1.33 |
| Kx | 18 045 | 24 812 | 327 820 |
| –Ais, kT | 9.8 | 10.1 | 12.7 |
При просмотре табл. 3 бросаются в глаза две примечательности. Первая относится к значениям солюбилизационной емкости и числам агрегации. В дополнение к сказанному выше о сравнении данных для гомологов II и III, особо выделяется ПАВ IV. Имея ту же длину углеводородного радикала, что и III, IV резко отличается своими характеристиками, что указывает на исключительную роль громоздкой полярной группы молекулы IV. Вторая примечательность видна в последней строке табл. 4 и состоит в том, что, несмотря на все различия, стандартная работа внесения молекулы I в мицеллы всех трех ПАВ почти одинакова. Исключительность IV в этой строке проявляется разве что в наибольшей величине этой работы.
ЗАКЛЮЧЕНИЕ
Главный вывод, который можно сделать из представленного материала, состоит в том, что особенности и закономерности солюбилизации I в мицеллах ПАВ, установленные в [7] для анионного ПАВ ДСН, подтверждаются и для катионных ПАВ. Это относится, например, к такому феномену как связанное с влиянием ПАВ повышение растворимости димеров I прежде чем они распадутся. Так же как и для ДСН, найдены весьма большие числа агрегации в мицеллах II и III с солюбилизированными мономерами I. Возможные варианты объяснения, уже рассмотренные в [7], приложимы и к этому случаю. Однако для вынесения окончательного суждения необходимы дополнительные исследования.
Список литературы
Гольдшлегер Н.Ф., Черняк А.В., Калашникова И.П., Баулин В.Е., Цивадзе А.Ю. // Журн. общей химии. 2012. Т. 82. С. 856.
Гольдшлегер Н.Ф., Лобач А.С., Гак В.Ю., Калашникова И.П., Баулин В.Е., Цивадзе А.Ю. // Физикохимия поверхности и защита материалов. 2014. Т. 50. С. 496.
Гольдшлегер Н.Ф., Черняк А.В., Лобач А.С., Калашникова И.П., Баулин В.Е., Цивадзе А.Ю. // Физикохимия поверхности и защита материалов. 2015. Т. 51. С. 160.
Овсянникова Е.В., Ширяев А.А., Калашникова И.П., Баулин В.Е., Цивадзе А.Ю., Андреев В.Н., Алпатова Н.М. // Макрогетероциклы. 2013. Т. 6. С. 274.
Мовчан Т.Г., Аверин А.А., Баулин Д.В., Плотникова Е.В., Баулин В.Е., Цивадзе А.Ю. // Коллоид. журн. 2018. Т. 80. С. 528.
Мовчан Т.Г., Чернядьев А.Ю., Плотникова Е.В., Аверин А.А., Цивадзе А.Ю., Баулин В.Е. // Коллоид. журн. 2018. Т. 80. С. 694.
Мовчан Т.Г., Русанов А.И., Плотникова Е.В. // Коллоид. журн. 2021. Т. 83. С. 75.
Мовчан Т.Г., Русанов А.И., Плотникова Е.В. // Коллоид. журн. 2016. Т. 78. С. 750.
Dubey N. // Fluid Phase Equilib. 2014. V. 368. P. 51.
Мовчан Т.Г., Русанов А.И., Плотникова Е.В., Соболева И.В. // Коллоид. журн. 2018. Т. 80. С. 308.
Tikariha D., Ghosh K.K., Quagliotto P., Ghosh S. // J. Chem. Eng. Data. 2010. V. 55. P. 4162.
Prasad M., Moulik S.P., MacDonald A., Palepu R. // J. Phys. Chem. B. 2004. V. 108. P. 355.
Крюкова Г.Н., Касаикин Б.А., Синева А.В., Маркина З.Н. // Коллоид. журн. 1978. Т. 40. С. 457.
Rusanov A.I. // Langmuir. 2014. V. 30. P. 14443.
Русанов А.И. // Коллоид. журн. 2016. Т. 78. С. 358.
Русанов А.И. // Коллоид. журн. 2016. Т. 78. С. 613.
Werawatganone P., Wurster D.E. // J. Pharm. Sci. 2007. V. 96. P. 448.
Русанов А.И. // Коллоид. журн. 2021. Т. 83. С. 98.
Evans D.F., Mukherjee S., Mitchell D.J., Ninham B.W. // J. Colloid Interface Sci. 1983. V. 93. P. 184.
Lianos P., Zana R. // J. Colloid Interface Sci. 1981. V. 84. P. 100.
Jadhao M., Mukherjee S., Joshi R., Kumar H., Ghosh S.K. // RSC Adv. 2016. V. 6. P. 77 161.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал