

Коллоидный журнал, 2021, T. 83, № 4, стр. 382-393
Молекулярная динамика солюбилизации декана и диффузии агрегатов из молекул ПАВ и декана в водных растворах
Н. А. Волков 1, *, Ю. А. Ерошкин 1, А. К. Щёкин 1, И. Н. Кольцов 2, Н. Ю. Третьяков 3, Е. А. Турнаева 3, С. С. Волкова 3, А. А. Громан 4
1 Санкт-Петербургский государственный университет
199034 Санкт-Петербург, Университетская набережная, 7/9, Россия
2 ООО “ГАЗПРОМНЕФТЬ – Технологические Партнерства”
625048 Тюмень, ул. 50 лет Октября, 14, Россия
3 ЦКП “Рациональное природопользование и физико-химические исследования”,
Тюменский государственный университет
625003 Тюмень, ул. Перекопская, 15а, Россия
4 ООО “ГАЗПРОМНЕФТЬ – Технологические Партнёрства”
190000 Санкт-Петербург, ул. Почтамтская, 3–5, Россия
* E-mail: nikolay.volkov@spbu.ru
Поступила в редакцию 05.03.2021
После доработки 12.03.2021
Принята к публикации 12.03.2021
Аннотация
Метод полноатомной молекулярной динамики применен для изучения процессов самоагрегации и солюбилизации в водных растворах, содержащих декан, ионное и неионное поверхностно-активные вещества (ПАВ), а также добавки солей. В частности, проведено моделирование мицеллообразования в водном растворе анионного ПАВ (додецилсульфата натрия) в присутствии углеводорода (декана) при заданных температуре и давлении и разных начальных концентрациях ПАВ и углеводорода в растворе. Также проведено моделирование самоагрегации в системах, содержащих воду, декан и смесь анионного (додецилсульфат натрия) и неионного ПАВ (монодециловый эфир гексаэтиленгликоля, C10E6), как в бессолевом растворе, так и в присутствии хлорида натрия, хлорида кальция или смеси этих солей. Рассчитаны коэффициенты диффузии агрегатов, состоящих из молекул углеводорода и ПАВ, и произведена оценка вязкости соответствующих водных растворов. Расчеты вязкости произведены в ячейках моделирования, содержащих как один, так и несколько агрегатов.
ВВЕДЕНИЕ
Исследование сложных молекулярных систем, к которым относятся смеси, содержащие углеводороды, ионные и неионные поверхностно-активные вещества (ПАВ), воду, солевые добавки является неполным без привлечения полного арсенала научных методов: лабораторных исследований, аналитической теории и молекулярного моделирования. Как аналитическая теория, так и молекулярное моделирование имеют дело с моделями физических систем, а любые модели описывают природные явления и технологические процессы упрощенно. Очевидно, что ближе всего к точному описанию реальных явлений и процессов находится лабораторный эксперимент, и любые данные, полученные теоретическими методами, в конечном итоге, должны быть согласованы с имеющимися экспериментальными данными. В то же время, не все экспериментальные условия могут быть воспроизведены при помощи существующего в настоящее время оборудования, поэтому аналитическая теория и вычислительные методы могут успешно дополнять лабораторные исследования, тем более что в ряде случаев применение теоретических методов требует меньших временны́х и/или финансовых ресурсов по сравнению с проведением лабораторных экспериментов.
Молекулярное моделирование помогает совершенствованию и проверке теоретических моделей и позволяет наблюдать на уровне отдельных атомов за поведением сложных молекулярных систем, в том числе систем, содержащих углеводороды, ионные и неионные поверхностно-активные вещества. Для кинетики и термодинамики агрегации и солюбилизации [1–5] молекулярная динамика дает возможность детального анализа динамики зарождения и роста агрегатов, их формы и структуры при различных числах агрегации молекул ПАВ и солюбилизата, позволяет рассчитать кинетические коэффициенты, связанных с коэффициентами диффузии молекул и агрегатов и с вязкостью раствора. В отличие от теории, тяготеющей к малым концентрациям, молекулярное моделирование с легкостью позволяет переходить к концентрированным системам [6]. Таким образом, методы молекулярного моделирования дополняют теорию и эксперимент при изучении водных растворов, содержащих углеводороды и ПАВ. Молекулярное моделирование успешно применялось для исследования мицеллярных растворов как ионных, так и неионных ПАВ (см. обзоры [6–10]), в частности, для вычисления коэффициентов диффузии различных ионов [10–14], мономеров ПАВ [11, 15, 16], молекул растворителя [12, 13, 17–19] и агрегатов [11–14, 20–24]. В то же время, структурные и транспортные свойства мицелл ПАВ с солюбилизированными молекулами углеводородов в зависимости от состава и температуры раствора, а также от размера и состава самих мицелл, требуют более широких исследований в рамках молекулярного моделирования.
В данной работе был использован метод полноатомной молекулярной динамики для описания процессов агрегации и солюбилизации в водных растворах, содержащих углеводород, ионное и неионное ПАВ и добавки солей. Рассчитаны коэффициенты диффузии агрегатов, состоящих из молекул углеводорода и ПАВ, и произведена оценка вязкости соответствующих водных растворов. Расчеты вязкости произведены в ячейках моделирования, содержащих как один, так и несколько агрегатов.
1. МОДЕЛИ И МЕТОДЫ
Для проведения молекулярно-динамического моделирования нами был использован программный пакет Gromacs 2020.1 [25–32]. Процесс моделирования включал в себя создание начальной конфигурации системы, минимизацию ее энергии, уравновешивание системы и получение молекулярно-динамической траектории необходимой длины, с помощью анализа которой были определены интересующие физико-химические свойства системы. Для создания начальных конфигураций молекулярных систем была использована программа packmol [33].
Для изучения самоагрегации мицелл и солюбилизации в них углеводорода, а также транспортных свойств мицеллярного раствора проводилось полноатомное молекулярно-динамическое моделирование ряда систем в изотермо-изобарическом (NPT) статистическом ансамбле, т.е. при заданных температуре, давлении и числе молекул в ячейке моделирования. Использовались периодические граничные условия в трех пространственных направлениях. Шаг по времени при численном решении уравнений движения составил 2 фс. Электростатические взаимодействия учитывались при помощи гладкого метода частица–сетка по Эвальду [34] с обрезанием в реальном пространстве на расстоянии 1 нм. Леннард-джонсовские взаимодействия обрезались на расстоянии 1 нм при помощи схемы Верле. Все рассмотренные системы моделировались при температуре T = 300 K (27°C) и давлении P = 1 бар (105 Па) с использованием термостата [35] и баростата [36]. Для описания взаимодействий между молекулами и ионами использовалось силовое поле CGenFF 4.4 [37, 38], являющееся представителем семейства силовых полей CHARMM. При моделировании была использована трехцентровая модель воды TIP3P [39], которая часто используется совместно с силовым полем CHARMM. Для визуального анализа молекулярно-динамических траекторий применялась программа VMD v.1.9.4a38 [40], для получения мгновенных снимков рассмотренных систем – программа Avogadro v.1.2.0 [41].
2. РЕЗУЛЬТАТЫ РАСЧЕТОВ
Как уже отмечалось во введении, важная особенность методов молекулярного моделирования заключается в том, что они дают возможность изучать наноразмерные объекты, которые сложно (а подчас практически невозможно) наблюдать в лабораторных экспериментах. В качестве примера можно привести водные растворы ПАВ, в том числе и с солюбилизированными в мицеллах молекулами углеводородов. Известно, что в реальных растворах могут образовываться агрегаты, состоящие из различного числа молекул ПАВ и углеводородов. При этом устойчивые агрегаты с характерными числами агрегации, присутствующие в растворе в большом количестве, дают основной вклад в результаты, полученные экспериментальными методами. Наряду с такими агрегатами в растворе образуются агрегаты и с другими числами агрегации, время жизни и количество которых может быть существенно меньше, что затрудняет их экспериментальное изучение. В свою очередь, методы молекулярного моделирования позволяют конструировать устойчивые агрегаты в широком диапазоне чисел агрегации и изучать их структурные, транспортные и термодинамические свойства. Устойчивость агрегатов с различными числами агрегации может достигаться за счет ограниченности количества вещества в ячейке моделирования [42] и за счет характерных взаимодействий в системе, например, электростатического отталкивания агрегатов друг от друга [14].
В табл. 1 представлены параметры систем, для которых нами было проведено молекулярно-динамическое моделирование молекулярной самоагрегации. Системы содержали указанное в табл. 1 число молекул углеводорода, анионного ПАВ, воды и солевых добавок. В качестве модельного углеводорода, ПАВ и соли были выбраны декан, додецилсульфат натрия (ДСН), хлорид натрия и хлорид кальция. Соли вводились в системы в виде набора ионов (Ca2+, Na+, Cl–), ДСН – в виде поверхностно-активного иона и противоиона (Na+). В начальный момент времени все молекулы и ионы были распределены в ячейке моделирования случайным образом.
Таблица 1.
Перечень систем, для которых проводилось моделирование молекулярной самоагрегации. В таблице приведены числа молекул, составляющих моделируемые системы; tsim – время моделирования
| № | ДСН | C10H22 | H2O | NaCl | CaCl2 | tsim, нс |
|---|---|---|---|---|---|---|
| 1 | 150 | 150 | 25 000 | – | – | 50 |
| 2 | 150 | 300 | 25 000 | – | 40 | 50 |
| 3 | 150 | 300 | 25 000 | – | 75 | 50 |
| 4 | 150 | 300 | 25 000 | 100 | 40 | 50 |
| 5 | 150 | 300 | 25 000 | – | 150 | 75 |
| 6 | 150 | 600 | 25 000 | 150 | – | 50 |
| 7 | 150 | – | 25 000 | – | – | 50 |
| 8 | 150 | – | 25 000 | 150 | – | 50 |
| 9 | 150 | 75 | 25 000 | – | – | 50 |
Мгновенные снимки установившихся после 50 нс моделирования молекулярных конфигураций рассмотренных систем приведены на рис. 1–9 (молекулы воды на рисунках не показаны, ячейки моделирования выделены черным цветом). Номера рисунков соответствуют нумерации систем в табл. 1. На рисунках видно, что в процессе моделирования в ячейке с периодическими граничными условиями образуется один или несколько молекулярных агрегатов, количество, размер и состав которых зависят от состава исходной смеси.
Рис. 1.
Мгновенный снимок системы, состоящей из 150 молекул ДСН, 150 молекул декана и 25 000 молекул воды, спустя 50 нс после начала моделирования.
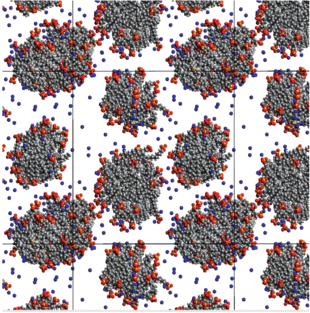
Рис. 2.
Мгновенный снимок системы, состоящей из 150 молекул ДСН, 300 молекул декана, 40 молекул хлорида кальция и 25 000 молекул воды, спустя 50 нс после начала моделирования.
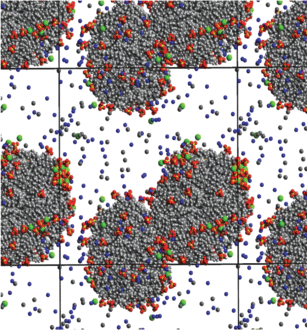
Рис. 3.
Мгновенный снимок системы, состоящей из 150 молекул ДСН, 300 молекул декана, 75 молекул хлорида кальция и 25 000 молекул воды, спустя 50 нс после начала моделирования.

Рис. 4.
Мгновенный снимок системы, состоящей из 150 молекул ДСН, 300 молекул декана, 100 молекул хлорида натрия, 40 молекул хлорида кальция и 25 000 молекул воды, спустя 50 нс после начала моделирования.
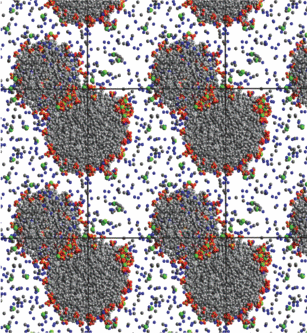
Рис. 5.
Мгновенный снимок системы, состоящей из 150 молекул ДСН, 300 молекул декана, 150 молекул хлорида кальция и 25 000 молекул воды, спустя 50 нс после начала моделирования.
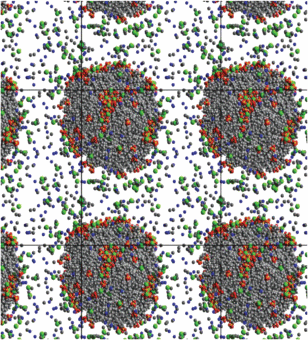
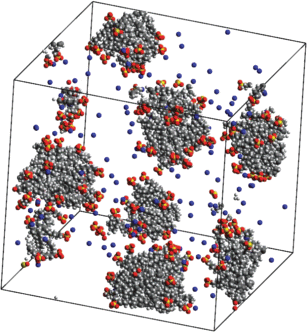
Рис. 6.
Мгновенный снимок системы, состоящей из 150 молекул ДСН, 600 молекул декана, 150 молекул хлорида натрия и 25 000 молекул воды, спустя 50 нс после начала моделирования.

Рис. 7.
Мгновенный снимок системы, состоящей из 150 молекул ДСН и 25 000 молекул воды, спустя 50 нс после начала моделирования.
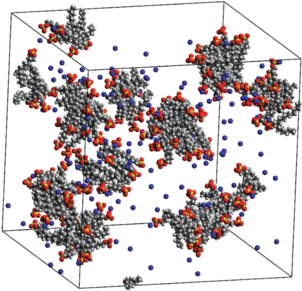
Рис. 8.
Мгновенный снимок системы, состоящей из 150 молекул ДСН, 150 молекул хлорида натрия и 25 000 молекул воды, спустя 50 нс после начала моделирования.
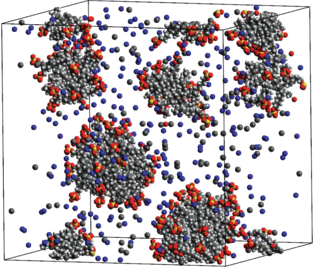
Рис. 9.
Мгновенный снимок системы, состоящей из 150 молекул ДСН, 75 молекул декана и 25 000 молекул воды, спустя 50 нс после начала моделирования.
Анализируя рис. 1–9, можно сделать вывод, что наличие декана в водном растворе ДСН уменьшает число агрегатов в ячейке моделирования и увеличивает их средний размер по сравнению с раствором ДСН без молекул углеводорода. Например, в системе, содержащей 150 молекул ДСН, 600 молекул декана, 150 молекул хлорида натрия и 25 000 молекул воды, образовался единственный агрегат (см. рис. 6), включивший в себя все поверхностно-активные ионы и молекулы декана. При этом в системе, изображенной на рис. 7 и содержащей 150 молекул ДСН и 25 000 молекул воды, образовалось несколько агрегатов. Отметим, что при наличии декана в растворе размер агрегатов увеличивается как за счет солюбилизации молекул декана в мицеллах, так и за счет того, что слияние агрегатов, по-видимому, происходит в этом случае легче. Вероятно, увеличение площади гидрофобных участков на поверхности агрегатов, состоящих из молекул декана и поверхностно-активных анионов ДСН, по сравнению с мицеллами без солюбилизата, приводит, с одной стороны, к усилению гидрофобного эффекта, с другой стороны, уменьшает электростатическое отталкивание отрицательно заряженных агрегатов.
При добавлении хлорида кальция CaCl2 в систему, содержащую 150 молекул ДСН, 300 молекул декана и 25 000 молекул воды, могут наблюдаться различные эффекты в зависимости от концентрации CaCl2. Когда в систему было добавлено 150 молекул CaCl2, то образовался единственный агрегат (см. рис. 5). В этом случае на каждый поверхностно-активный ион приходилось по одному иону кальция. Наличие большого числа двухзарядных противоионов, по-видимому, привело к электростатическому связыванию отрицательно заряженных головных групп поверхностно-активных ионов и, в итоге, существенно облегчило и ускорило процесс самоагрегации. При добавлении в систему 40 молекул CaCl2 (см. рис. 2) или смеси солей, состоящей из 40 молекул CaCl2 и 100 молекул NaCl (см. рис. 4), в системе образовались устойчивые агрегаты в форме “гантели”. Формирование гантелевидных агрегатов произошло за счет образования на поверхностях сферических агрегатов “островков” из нескольких головных групп анионов ДСН со связанными с ними ионами Ca2+. Такие островки на поверхностях двух отдельных сферических агрегатов связывают их и приводят к образованию одного гантелевидного агрегата.
Оценить влияние хлорида натрия на процесс самоагрегации в водном растворе ДСН сложнее, чем в случае с хлоридом кальция, так как это влияние выражено слабее. Тем не менее, вероятно, что наличие хлорида натрия в растворе также облегчает процесс самоагрегации (см. рис. 7 и 8) за счет экранирования электростатического отталкивания между отрицательно заряженными агрегатами ДСН.
Нами был проведен ряд молекулярно-динамических расчетов для определения коэффициентов диффузии и размеров агрегатов, состоящими из молекул декана, поверхностно-активных ионов ДСН и молекул неионного ПАВ, монодецилового эфира гексаэтиленгликоля (C10E6) в разных пропорциях. Полученные значения коэффициентов диффузии и радиусов сферических агрегатов с различными числами агрегации позволили нам при помощи формулы Стокса–Эйнштейна оценить вязкость рассмотренных растворов. В частности, было проведено моделирование системы, состоящей из 150 молекул ДСН, 300 молекул декана и 25 000 молекул воды, полное время моделирования составило 250 нс. Во временнóм диапазоне от 0 до 100 нс образовались 4 агрегата (см. рис. 10а), числа агрегации которых оставались неизменными на протяжении последующих 150 нс. Для вычисления интересующих нас величин использовался временнóй промежуток от 100 до 250 нс. Были определены коэффициенты диффузии и размеры агрегатов, также была оценена вязкость раствора.
Рис. 10.
(а) Мгновенный снимок системы, состоящей из 150 молекул ДСН, 300 молекул декана и 25 000 молекул воды (молекулы воды на снимке не показаны); (б) зависимости от времени (с точностью до коэффициента 1/6) средних квадратов смещения четырех агрегатов, образовавшихся в системе. Полное время моделирования составило 250 нс.
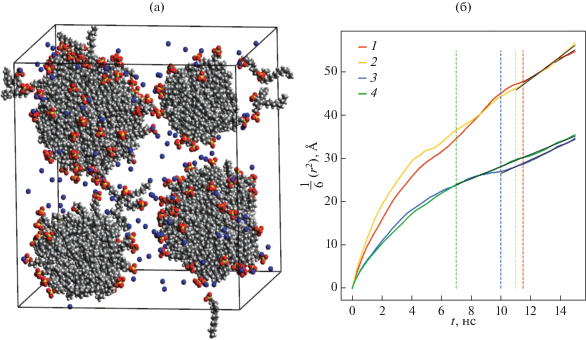
Образовавшиеся в системе агрегаты имели следующие числа агрегации: агрегат № 1 – 86 мономеров (29 ионов ДСН и 57 молекул C10H22), агрегат № 2 – 65 мономеров (23 иона ДСН и 42 молекулы C10H22), агрегат № 3 – 125 мономеров (44 иона ДСН и 81 молекула C10H22), агрегат № 4 – 174 мономеров (54 иона ДСН и 120 молекул C10H22). Для определения характерных радиусов агрегатов Rs применялась формула, предложенная в работе [43] и использованная нами в работах [12–14, 24] для оценки размеров ионных мицелл и предмицеллярных агрегатов,
(1)
${{R}_{{\text{s}}}} = {{\left( {\frac{5}{3}} \right)}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}{{R}_{{\text{g}}}},$Для вычисления коэффициентов диффузии агрегатов, в первую очередь, были найдены положения их центров масс (ЦМ) на том участке молекулярно-динамической траектории, который был выбран для анализа свойств рассматриваемой системы. В данном случае положения ЦМ агрегатов были определены для времен в диапазоне от 100 до 250 нс. В указанном диапазоне времен было выбрано так называемое “окно” размером 15 нс, для которого строилась временнáя зависимость среднего квадрата смещения ЦМ каждого из агрегатов. Для улучшения статистики начало отсчета времени для этого “окна” сдвигалось от 100 нс до 235 нс с шагом в 1 пс. Полученные зависимости для каждого из агрегатов были усреднены по разным значения начала отсчета времени. Результаты расчетов представлены на рис. 10б. Для вычисления коэффициентов диффузии агрегатов были использованы участки, находящиеся правее соответствующих вертикальных линий на рис. 10б, где зависимости средних квадратов смещения ЦМ агрегатов от времени можно аппроксимировать линейными функциями. Коэффициенты диффузии были вычислены по формуле Эйнштейна
(2)
$D = \mathop {\lim }\limits_{t \to \infty } \frac{{\left\langle {\Delta r{{{\left( t \right)}}^{2}}} \right\rangle }}{{6t}},$Для оценки вязкости раствора, содержащего коллоидные частицы, размер которых значительно превышает размер молекул растворителя, а форма близка к сферической, может быть использована формула Стокса–Эйнштейна D = = kBT/6πηR (kB – постоянная Больцмана), связывающая коэффициент диффузии сферической частицы D, ее радиус R, температуру T и вязкость среды η, в которой частица находится. Этот подход был ранее использован нами при вычислении вязкости мицеллярного раствора анионного ПАВ, где на основе данных полноатомной молекулярной динамики были получены средние радиусы предмицеллярных агрегатов и их коэффициенты диффузии. В итоге, формула Стокса–Эйнштейна позволила получить приближенное значение вязкости водного раствора ДСН [24]. Метод вычисления вязкости по данным молекулярного моделирования, основанный на формуле Стокса–Эйнштейна, также применялся в работе [44].
Зная радиусы сферических агрегатов, образовавшихся в системе, состоящей из 150 молекул ДСН, 300 молекул декана и 25 000 молекул воды, и коэффициенты диффузии этих агрегатов, мы, используя формулу Стокса–Эйнштейна, получили следующие оценки для вязкости раствора: η1 = = (5.8 ± 1.7) мПа с, η2 = (5.3 ± 1.4) мПа с; η3 = = (7.0 ± 4.0) мПа с, η4 = (7.1 ± 1.8) мПа с. Точность вычисления коэффициентов диффузии агрегатов (и, как следствие, точность вычисления вязкости) может быть повышена путем увеличения длины молекулярно-динамической траектории, используемой для анализа, при условии, что числа агрегации в системе останутся неизменными.
Использование периодических граничных условий может оказывать существенное влияние на значения вычисляемых в рамках молекулярного моделирования величин, в частности, на значения коэффициентов диффузии агрегатов. В работах [45, 46] приводится формула для коррекции значений коэффициентов диффузии, полученных в системах с периодическими граничными условиями:
(3)
${{D}_{\infty }} = {{D}_{{{\text{PBC}}}}} + 2.837297{{{{k}_{{\text{B}}}}T} \mathord{\left/ {\vphantom {{{{k}_{{\text{B}}}}T} {6\pi \eta L,}}} \right. \kern-0em} {6\pi \eta L,}}$В рамках метода молекулярной динамики могут быть независимо определены размеры и коэффициенты диффузии коллоидных частиц, присутствующих в системе, что дает возможность оценить вязкость раствора по каждой из этих частиц. Коэффициент диффузии коллоидной частицы зависит от ее размера, т.е. является индивидуальным свойством конкретной частицы. Вязкость же представляет собой коллективное свойство, характеризующее раствор как целое. Таким образом, можно было бы ожидать, что значения вязкости раствора, полученные по формуле Стокса–Эйнштейна для различных коллоидных частиц, находящихся в одной и той же системе, должны совпадать. К сожалению, при вычислении коэффициентов диффузии молекулярных агрегатов в ячейках с периодическими граничными условиями, последние вносят систематическую ошибку, для устранения которой необходимо либо увеличивать размер ячейки моделирования до достижения приемлемой сходимости результатов, либо использовать аналитическую коррекцию в духе формулы (3). При вычислении вязкости раствора, по-видимому, можно провести усреднение по значениям вязкости, полученным при рассмотрении разных агрегатов. В случае системы, состоящей из 150 молекул ДСН, 300 молекул декана и 25 000 молекул воды, среднее значение вязкости, полученное при рассмотрении четырех различных агрегатов, составило η = (6.3 ± 2.2) мПа с.
В рамках данной работы нами также было проведено моделирование системы, состоящей из 75 молекул ионного ПАВ (ДСН), 75 молекул неионного ПАВ (C10E6), 300 молекул декана и 25 000 молекул воды, полное время моделирования составило 225 нс. Во временнóм диапазоне от 0 до 25 нс в системе образовался единственный агрегат (см. рис. 11а), число агрегации которого сохранилось неизменным на протяжении последующих 200 нс. Для вычисления интересующих нас величин использовался временнóй промежуток от 125 до 225 нс, были определены коэффициент диффузии и размер агрегата, а также оценена вязкость раствора.
Рис. 11.
(а) Мгновенный снимок системы, состоящей из 75 молекул ДСН, 75 молекул C10E6, 300 молекул декана и 25 000 молекул воды (молекулы воды на снимке не показаны); (б) временнáя зависимость (с точностью до коэффициента 1/6) среднего квадрата смещения агрегата. Полное время моделирования составило 225 нс.
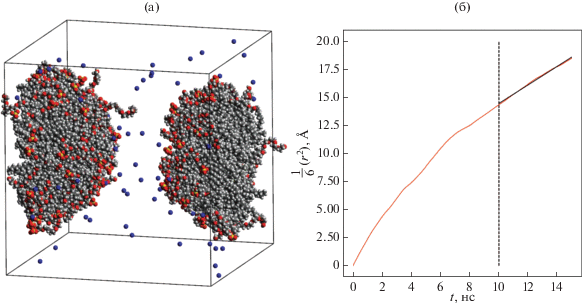
Образовавшийся агрегат состоял из 75 поверхностно-активных анионов ДСН, 75 молекул C10E6 и 300 молекул декана. Радиус агрегата оказался равным R = (31.36 ± 0.09) Å. Малая величина дисперсии, полученная для радиуса, говорит о том, что форма агрегата близка к сферической.
Сравнивая рис. 10а и 11а, можно сделать вывод, что замена определенного числа мономеров ионного ПАВ на мономеры неионного ПАВ приводит к увеличению чисел агрегации и размеров агрегатов. По всей видимости, это связано с уменьшением электростатического отталкивания между агрегатами при уменьшении числа отрицательно заряженных поверхностно-активных ионов, приходящихся на единицу поверхности.
При вычислении коэффициента диффузии агрегата, изображенного на рис. 11а, было выбрано “окно” величиной 15 нс, для которого строилась временнáя зависимость среднего квадрата смещения ЦМ агрегата (см. рис. 11б). Определение среднего значения коэффициента диффузии производилось по интервалу от 10 до 15 нс внутри выбранного временнóго “окна”, полученное значение коэффициента диффузии составило D = = (0.83 ± 0.12) × 10–7 см2/с. Значение вязкости раствора, полученное по формуле Стокса–Эйнштейна, составило η = (8.4 ± 1.2) мПа с.
Было проведено моделирование системы, состоящей из 150 молекул ДСН, 300 молекул декана, 150 молекул хлорида кальция и 25 000 молекул воды. Полное время моделирования составило 225 нс. Во временнóм диапазоне от 0 до 25 нс в системе образовался единственный агрегат (см. рис. 12а), число агрегации которого сохранилось неизменным на протяжении последующих 200 нс. Для вычисления коэффициента диффузии и размера образовавшегося агрегата и вязкости раствора использовался временнóй промежуток от 125 до 225 нс.
Рис. 12.
(а) Мгновенный снимок системы, состоящей из 150 молекул ДСН, 300 молекул декана, 150 молекул CaCl2 и 25 000 молекул воды (молекулы воды на снимке не показаны, ионы кальция, натрия и хлора обозначены зеленым, синим и серым цветами соответственно); (б) временнáя зависимость (с точностью до коэффициента 1/6) среднего квадрата смещения агрегата. Полное время моделирования составило 225 нс.
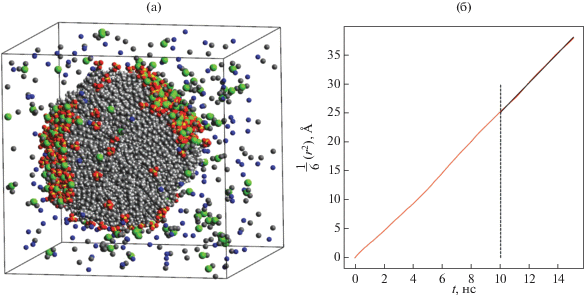
На рис. 12а видно, что на поверхности агрегата образовались участки, которые плотно покрыты головными группами анионов ПАВ и на которых адсорбировано значительное число ионов кальция. Можно сделать вывод, что образование этих участков происходит за счет электростатического взаимодействия отрицательно заряженных головных групп мономеров ДСН с положительно заряженными ионами Ca2+. Отметим также, что при имеющемся соотношении между числом молекул углеводорода и ПАВ на поверхности агрегата присутствуют обширные гидрофобные участки.
Радиус агрегата, состоящего из 150 мономеров ДСН и 300 молекул декана, оказался равным R = = (29.51 ± 0.08) Å. При вычислении коэффициента диффузии агрегата было выбрано “окно” величиной 15 нс, для которого строилась временнáя зависимость среднего квадрата смещения ЦМ агрегата (см. рис. 12б).
Определение среднего значения коэффициента диффузии агрегата производилось по интервалу от 10 до 15 нс внутри выбранного временнóго “окна”, полученное значение коэффициента диффузии составило D = (2.6 ± 0.2) × 10–7 см2/с; значение вязкости раствора составило η = (2.8 ± ± 0.2) мПа с.
Нами было также проведено моделирование систем, содержащих как смесь ионного и неионного ПАВ, так и добавки солей. На рис. 13а представлена система, состоящая из 75 молекул ДСН, 75 молекул C10E6, 300 молекул декана, 100 молекул NaCl и 25 000 молекул воды. Полное время моделирования составило 275 нс. В течение первых 25 нс после начала моделирования в системе образовался один агрегат. Для вычисления коэффициента диффузии и размера образовавшегося агрегата и вязкости раствора использовался временнóй промежуток от 175 до 275 нс. Радиус агрегата оказался равным R = (31.36 ± 0.09) Å. При вычислении коэффициента диффузии агрегата, изображенного на рис. 13а, было выбрано “окно” величиной 15 нс, для которого строилась временнáя зависимость среднего квадрата смещения ЦМ агрегата (см. рис. 13б). Определение среднего значения коэффициента диффузии агрегата производилось по интервалу от 10 до 15 нс внутри выбранного временнóго окна, полученное значение коэффициента диффузии составило D = (2.64 ± ± 0.12) × 10–7 см2/с; значение вязкости раствора, вычисленное по формуле Стокса–Эйнштейна, составило η = (2.6 ± 1.2) мПа с.
Рис. 13.
(а) Мгновенный снимок системы, состоящей из 75 молекул ДСН, 75 молекул C10E6, 300 молекул декана, 100 молекул NaCl и 25 000 молекул воды (молекулы воды на снимке не показаны); (б) временнáя зависимость (с точностью до коэффициента 1/6) среднего квадрата смещения агрегата. Полное время моделирования составило 275 нс.
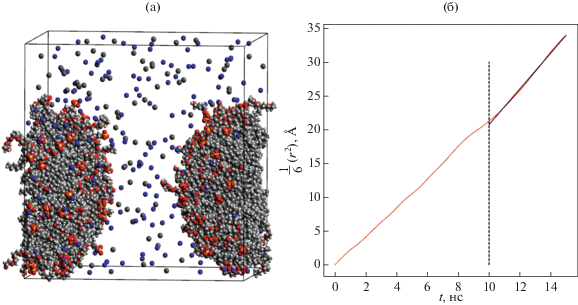
На рис. 14а представлена система, отличающаяся от предыдущей добавкой 40 молекул CaCl2, т.е. она содержит 75 молекул ДСН, 75 молекул C10E6, 300 молекул декана, 100 молекул NaCl, 40 молекул CaCl2 и 25 000 молекул воды. В этой системе тоже образовался единственный агрегат, правда, уже в течение первых 50 нс после начала моделирования. Полное время моделирования составило 400 нс. Для вычисления коэффициента диффузии и размера образовавшегося агрегата и вязкости раствора использовался временнóй промежуток от 250 до 400 нс. Средний радиус агрегата оказался равным R = (31.33 ± 0.09) Å.
Рис. 14.
(а) Мгновенный снимок системы, состоящей из 75 молекул ДСН, 75 молекул C10E6, 300 молекул декана, 100 молекул NaCl, 40 молекул CaCl2 и 25 000 молекул воды (молекулы воды на снимке не показаны); (б) временнáя зависимость (с точностью до коэффициента 1/6) среднего квадрата смещения агрегата. Полное время моделирования составило 400 нс.
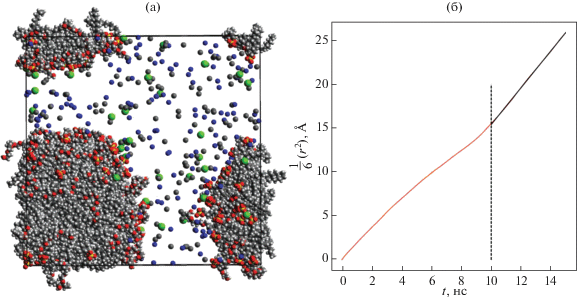
При вычислении коэффициента диффузии агрегата, изображенного на рис. 14а, было выбрано “окно” величиной 15 нс, для которого строилась временнáя зависимость среднего квадрата смещения ЦМ агрегата (см. рис. 14б). Определение среднего значения коэффициента диффузии агрегата производилось по интервалу от 10 до 15 нс внутри выбранного временнóго “окна”, полученное значение составило D = (2.1 ± 0.13) × 10–7 см2/с. Значение вязкости раствора, вычисленное по формуле Стокса–Эйнштейна, равно η = (3.3 ± ± 1.2) мПа с.
Системы, изображенные на рис. 11а и рис. 13а, различаются тем, что во второй из них дополнительно присутствуют 100 молекул хлорида натрия. Средние радиусы агрегатов, сформировавшихся в этих системах и состоящих из 75 молекул ДСН, 75 молекул C10E6 и 300 молекул декана, одинаковы и имеют значение R = (31.36 ± 0.09) Å. Также для этих систем оказались одинаковыми средние расстояния от атомов водорода, находящихся на конце головных групп молекул неионного ПАВ, до ЦМ агрегатов. Значение указанных расстояний составило 36.6 Å, усреднение проводилось как по всем молекулам неионного ПАВ, так и по времени. При этом коэффициенты диффузии агрегатов и, как следствие, значения вязкости, определенные на основе коэффициентов диффузии и размеров агрегатов, существенно различаются. Вязкость системы, содержащей хлорид натрия, оказалась приблизительно в три раза меньше, чем системы без соли. Все сказанное свидетельствует о том, что значительные различия в значениях коэффициентов диффузии агрегатов и вязкости растворов, по-видимому, связаны не с изменением структуры агрегатов при добавлении соли, а с изменением свойств раствора, в котором эти агрегаты находятся.
ЗАКЛЮЧЕНИЕ
В предыдущих разделах статьи представлены результаты полноатомного молекулярно-динамического моделирования водного раствора анионного ПАВ (додецилсульфата натрия), а также водного раствора, содержащего ДСН и углеводород (декан), при заданных температуре и давлении и разных концентрациях ПАВ и углеводорода. Оценено влияние декана и солевых добавок на процесс самоагрегации в водных растворах ДСН. В процессе моделирования растворов с добавлением хлорида кальция на поверхностях агрегатов наблюдалось образование “островков”, состоящих из отрицательно заряженных головных групп ДСН и положительно заряженных ионов Ca2+, что приводило к формированию устойчивых гантелеобразных агрегатов при определенных концентрациях CaCl2 в растворе.
Также в работе представлены результаты моделирования самоагрегации в системах, содержащих воду, декан и смесь анионного (ДСН) и неионного (C10E6) ПАВ, как в бессолевом растворе, так и при добавлении в раствор хлорида натрия или смеси хлоридов натрия и кальция.
Исследование характеристик молекулярных агрегатов, самопроизвольно образующихся в указанных выше системах, позволило определить их структуру, рассчитать их числа агрегации и коэффициенты броуновской диффузии в соответствующих водных растворах. На основе формулы Стокса–Эйнштейна проведены расчеты вязкости для указанных модельных водных растворов, содержащих декан, анионное ПАВ или смесь анионного и неионного ПАВ, а также добавки солей. Расчеты вязкости произведены в ячейках моделирования, содержащих как один, так и несколько агрегатов. Характерные радиусы рассмотренных агрегатов находились в диапазоне от 16 до 31 Å, полученные коэффициенты диффузии агрегатов составили от 0.8 × 10–7 до 2.6 × 10–7 см2/с, а значения вязкости растворов – от 2.6 до 8.4 мПа с. Проведенные расчеты продемонстрировали существенное влияние солей на значения коэффициентов диффузии агрегатов, состоящих из молекул декана, поверхностно-активных анионов ДСН и молекул неионного ПАВ, в водных растворах.
Список литературы
Русанов, Щёкин А.К. Мицеллообразование в растворах поверхностно-активных веществ. 2-е изд. СПб.: Лань, 2016.
Щёкин А.К., Аджемян Л.Ц., Бабинцев И.А., Волков Н.А. // Коллоид. журн. 2018. Т. 80. С. 115.
Self-Assembly: From Surfactants to Nanoparticles / Ed. by Nagarajan R. Hoboken: Wiley, 2019.
Русанов А.И. // Коллоид. журн. 2021. Т. 83. С. 98.
Щёкин А.К., Волков Н.А, Кольцов И.Н., Третьяков Н.Ю., Волкова С.С., Турнаева Е.А. // Коллоид. журн. 2021. Т. 83. № 4.
Русанов А.И., Щёкин А.К., Волков Н.А. // Успехи химии. 2017. Т. 86. С. 567.
Shelley J.C., Shelley M.Y. // Curr. Opin. Colloid Interface Sci. 2000. V. 5. P. 101.
Бродская Е.Н. // Коллоид. журн. 2012. Т. 74. С. 167.
Ladanyi B.M. // Curr. Opin. Colloid Interface Sci. 2013. V. 18. P. 15.
Bruce C.D., Berkowitz M.L., Perera L., Forbes M.D.E. // J. Phys. Chem. B. 2002. V. 106. P. 3788.
Jardat M., Durand-Vidal S., Da Mota N., Turq P. // J. Chem. Phys. 2004. V. 120. P. 6268.
Volkov N.A., Divinskiy B.B., Vorontsov-Velyaminov P.N., Shchekin A.K. // Colloids Surf. A. 2015. V. 480. P. 165.
Volkov N.A., Tuzov N.V., Shchekin A.K. // Fluid Phase Equilib. 2016. V. 424. P. 114.
Волков Н.А., Посысоев М.В., Щёкин А.К. // Колл-оид. журн. 2018. Т. 80. С. 264.
Braun R., Engelman D.M., Schulten K. // Biophys. J. 2004. V. 87. P. 754.
Yuan F., Wang S., Larson R.G. // Langmuir. 2015. V. 31. P. 1336.
Kuhn H., Rehage H. // Progr. Colloid Polym. Sci. 1998. V. 111. P. 158.
Bruce C.D., Senapati S., Berkowitz M.L., Perera L., Forbes M.D.E. // J. Phys. Chem. B. 2002. V. 106. P. 10902.
Dastidar S.G., Mukhopadhyay C. // Phys. Rev. E. 2004. V. 70. 061901.
Dahirel V., Ancian B., Jardat M., Meriguet G., Turq P., Lequin O. // Soft Matter. 2010. V. 6. P. 517.
Marrink S.J., Tieleman D.P., Mark A.E. // J. Phys. Chem. B. 2000. V. 104. P. 12165.
Mohan G., Kopelevich D.I. // J. Chem. Phys. 2008. V. 128. 044905.
Aoun B., Sharma V.K., Pellegrini E., Mitra S., Johnson M., Mukhopadhyay R. // J. Phys. Chem. B. 2015. V. 119. P. 5079.
Volkov N.A., Shchekin A.K., Tuzov N.V., Lebedeva T.S., Kazantseva M. // J. Mol. Liq. 2017. V. 236. P. 414.
GROMACS development team. GROMACS Documentation. Release 2020.1. https://doi.org/10.5281/zenodo.3685920
Berendsen H.J.C., van der Spoel D., van Drunen R. // Comput. Phys. Commun. 1995. V. 91. P. 43.
Lindahl E., Hess B., van der Spoel D. // J. Mol. Mod. 2001. V. 7. P. 306.
van der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A.E., Berendsen H.J.C. // J. Comput. Chem. 2005. V. 26. P. 1701.
Hess B., Kutzner C., van der Spoel D., Lindahl E. // J. Chem. Theory Comput. 2008. V. 4. P. 435.
Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M.R., Smith J.C., Kasson P.M., van der Spoel D., Hess B., Lindahl E. // Bioinformatics. 2013. V. 29. P. 845.
Páll S., Abraham M.J., Kutzner C., Hess B., Lindahl E. // Solving Software Challenges for Exascale. Ed. by Markidis S., Laure E. 2015. V. 8759. P. 3.
Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindahl E. // SoftwareX. 2015. V. 1. P. 19.
Martínez L., Andrade R., Birgin E.G., Martínez J.M. // J. Comput. Chem. 2009. V. 30. P. 2157.
Essmann U., Perera L., Berkowitz M.L., Darden T.A., Lee H., Pedersen L. // J. Chem. Phys. 1995. V. 103. P. 8577.
Bussi G., Donadio D., Parrinello M. // J. Chem. Phys. 2007. V. 126. 014101.
Parrinello M., Rahman A. // J. Appl. Phys. 1981. V. 52. P. 7182.
Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J., Darian E., Guvench O., Lopes P., Vorobyov I., MacKerell A.D., Jr. // J. Comput. Chem. 2010. V. 31. P. 671.
Yu W., He X., Vanommeslaeghe K., MacKerell A.D., Jr. // J. Comput. Chem. 2012. V. 33. P. 2451.
Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. // J. Chem. Phys. 1983. V. 79. P. 926.
Humphrey W., Dalke A., Schulten K. // J. Mol. Graph. 1996. V. 14. P. 33.
Hanwell M.D., Curtis D.E., Lonie D.C., Vandermeersch T., Zurek E., Hutchison G.R. // J. Cheminform. 2012. V. 4. P. 17.
Shchekin A.K., Koga K., Volkov N.A. // J. Chem. Phys. 2019. V. 151. 244903.
Bogusz S., Venable R.M., Pastor R.W. // J. Phys. Chem. B. 2000. V. 104. P. 5462.
Kondratyuk N.D., Orekhov M.A. // J. Phys. Conf. Ser. 2020. V. 1556. 012048.
Dünweg B., Kremer K. // J. Chem. Phys. 1993. V. 99. P. 6983.
Yeh I.C., Hummer G.J. // J. Phys. Chem. B. 2004. V. 108. P. 15873.
Singh S.P., Huang C.-C., Westphal E., Gompper G., Winkler R.G. // J. Chem. Phys. 2014. V. 141. 084901.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал