

Коллоидный журнал, 2021, T. 83, № 6, стр. 660-675
Свойства гидроколлоидов альгината натрия при сорбционном связывании папаина
С. А. Кокшаров 1, С. В. Алеева 1, *, О. В. Лепилова 1, Г. Е. Кричевский 2, Ю. С. Фидоровская 3
1 Институт химии растворов им. Г.А. Крестова РАН
153045 Иваново, Академическая ул., 1, Россия
2 ООО “НПО Текстильпрогресс Инженерной Академии”
115093 Москва, ул. Павловская, 21, Россия
3 ООО “Колетекс”
115093 Москва, ул. Павловская, 21, Россия
* E-mail: sva@isc-ras.ru
Поступила в редакцию 15.07.2021
После доработки 03.08.2021
Принята к публикации 06.08.2021
Аннотация
Исследованы закономерности адсорбционной иммобилизации папаина сильно- и слабокислыми коллоидными растворами альгината натрия при низкоскоростном режиме перемешивания для обоснования условий получения биополимерных покрытий для очищения и заживления ран. Состояние дисперсной фазы оценено с применением методов электронной сканирующей микроскопии и динамического рассеяния света. Сравнение характеристик вязкого течения растворов проведено в рамках модели Оствальда–де Ваале. Описание сорбционного равновесия с помощью модели Ленгмюра позволило определить термодинамические характеристики взаимодействия и вероятный механизм сорбционного связывания энзима в структуре полимера, который согласуется с результатами анализа ИК-спектров.
ВВЕДЕНИЕ
Альгинат натрия и спустя 140 лет после его открытия британским химиком Стенфордом (E.C.C. Stanford) является широко востребованным водорастворимым биополимерным продуктом, мировое производство которого в 2018 г. превысило 38 тыс. тонн [1]. Высокая способность к загущению и структурированию растворов, стабилизации эмульсий, дисперсий и пен, влагоудерживающие и клеящие свойства обусловливают применение данного полисахарида не только в традиционных сферах пищевой и косметической индустрии, в текстильной и типографской печати, но и в интенсивно развивающихся направлениях биотехнологии и медицины, аддитивных технологий [2–4].
Безвредность, биосовместимость и биодеградируемость альгината натрия в сочетании с выраженной физиологической активностью являются базовыми благоприятными свойствами для его использования в фармацевтике и лечебной практике. А свойственная альгинатам легкость гелеобразования, способность к формированию полиэлектролитных комплексов и покрытий, а также 3D-сшитых структур с регулируемыми упругостью и диффузионной проницаемостью лежат в основе все возрастающего числа технических воплощений, анализу которых посвящены обзоры [5–10].
Однако в большинстве литературных источников основное внимание уделяется исследованию физиологической активности создаваемых биополимерных объектов и терапевтической эффективности их применения. При этом характер взаимодействий, структурная организация и свойства таких систем освещаются недостаточно, либо требуют уточнений.
Актуальной и постоянно совершенствующейся областью биомедицинского использования альгинатов являются ранозаживляющие покрытия [11–14]. В этом случае свойства биополимерных композиций должны быть адаптированы к разнообразию форм материалов, подходящих для лечения ран разной этиологии, включая гидроколлоиды, гелевые повязки, пленки и мембраны, волокнистые композиты и текстильные салфетки. Наряду с выполнением функций по обеспечению атравматичного воздействия и уменьшения бактериального инфицирования ран, общих для всех перечисленных форм лечебных материалов, биополимерные компоненты могут способствовать достижению противоположных эффектов. Например, благодаря различию структурной организации альгинаты и смесевые композиции на их основе могут способствовать осушению раны, поглощению избытка раневой жидкости, либо увлажнять и поддерживать физиологическую влажность среды. Биополимер может извлекать из раны метаболиты, продукты распада тканей и выделений, либо регулировать выход в рану биологически активных веществ и лекарственных препаратов. При этом изменение свойств биополимерных покрытий позволяет оперативно изменять и их функции на разных стадиях ранозаживления.
Особые задачи связаны с обеспечением противовоспалительного и очищающего действия ранозаживляющих покрытий за счет введения протеолитических ферментных препаратов [15–17]. Известно, что некролитическое действие протеаз при непосредственном внесении в рану продолжается не более 30 мин [18]. Активно исследуются возможности повышения стабильности ферментов с применением известных методов их иммобилизации природными биополимерами [19, 20]. Наиболее часто практикуемым вариантом иммобилизации является метод включения, который предполагает создание пространственных ограничений, накладываемых трехмерной структурой полимера без взаимодействия с молекулами фермента. При этом сшитые полимерные матрицы могут создавать помехи и для диффузионного проникновения субстрата, а если им является высокомолекулярное соединение (белок или полипептид), то этот метод считается малоприменимым [21]. Ковалентное связывание обеспечивает наиболее высокую устойчивость фермента, но требует особых мер предупреждения необратимых изменений структуры биокатализатора и защиты его активного центра. Адсорбционная иммобилизация является наиболее простым методом, но приводит к наименьшему выходу связанного фермента.
Относительно механизма иммобилизации ферментов альгинатами у исследователей нет единой точки зрения. Отмечается, что несшитый гидроколлоид альгината натрия обеспечивает адсорбцию ферментного препарата – Протеазы C – за счет слабых водородных и координационных связей [17]. Для ковалентного связывания папаина и трипсина необходима модификация биополимера, например, глутаровым альдегидом, активная карбонильная группа которого взаимодействует с аминогруппой молекулы фермента [22]. Авторы [23] на основании данных ИК-спектроскопии и электронной микроскопии утверждают о химической сшивке папаина в гранулах альгината, хотя общепризнанным является образование ковалентной связи между амино- и карбоксильными группами лишь при взаимодействии аминокислот. Однако аналогичные сведения есть и о возможности ковалентных взаимодействий между хитозаном и пектином [24]. Тем не менее, более распространенным является мнение, что ассоциация альгинатных цепей и белковых молекул происходит, главным образом, за счет электростатических взаимодействий между противоположно заряженными группами полипептидной цепи и макромолекул полисахаридов [25, 26].
Требуют уточнения также и несогласующиеся данные о влиянии иммобилизованного фермента на свойства гидроколлоидов альгината натрия. Авторы [27] наблюдали снижение характеристической вязкости и гидродинамического размера частиц альгината в присутствии папаина, что объясняют нейтрализацией отрицательного заряда карбоксильных групп полимера и уменьшением межцепных сил отталкивания. Вместе с тем для получаемых электростатической сборкой стабильных коллоидных систем альгината с протеолитическим ферментом лизоцимом зафиксировано увеличение размера наночастиц [28], причем в случае полимера с более высокой молекулярной массой этот эффект усиливается. При этом целесообразно учитывать сведения о том, что, участвуя в межчастичных взаимодействиях, макромолекулы альгината могут сохранять исходную конформацию статистического клубка или фиксироваться в развернутой форме, делая доступными внутренние реакционные центры [29, 30].
Настоящая работа направлена на прояснение указанных выше вопросов и развитие существующих представлений о состоянии и взаимодействиях в биополимерных системах на альгинатной основе, содержащих протеолитические ферменты, для решения комплекса научно-практических задач по усовершенствованию материалов для лечения гнойных ран. Основываясь на многолетнем практическом опыте создания биополимерных лечебных депо-композиций для использования в различных областях медицины [31, 32], можно считать наиболее оправданным выбор несшитого альгината натрия в качестве полимерной матрицы. Сохранение текучести гидроколлоида и обеспечение его сорбционных свойств даст возможность рационального распределения активного вещества (фермента) в фазе раневой повязки и на поврежденном участке тела для одновременного расщепления полипептидных веществ гнойного экссудата и эффективного их отведения из очага поражения.
Выбор папаина обусловлен тем, что латекс папайи (Carica papaya) является известным средством для лечения различных кожных заболеваний, в том числе для заживления ран и ожогов [33]. При анализе поведения папаина в растворах альгината натрия предполагается учесть особенности расположения в макромолекуле реакционноспособных аминогрупп.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Объекты исследования
Использовался промышленный препарат альгината натрия (производитель ООО “Архангельский водорослевый комбинат”) с содержанием основного вещества 83.0%. Степень полимеризации альгината оценена по данным о характеристической вязкости, определенной экстраполяционным методом из концентрационных зависимостей приведенной вязкости. Измерения проводили на вискозиметре Уббелоде с диаметром капилляра 0.82 мм при температуре 25°C; фоновый электролит – 0.1 М раствор хлорида натрия. Величина средневязкостной молекулярной массы составляет 202.0 кДа; степень полимеризации – 1020.
Альгиновая кислота (Acros Organics, Бельгия) получена из морских водорослей Macrocystis pyrifera и содержит 91.0% основного вещества с молекулярной массой 207.6 кДа.
Папаин (Tayga Shanghai Co., Ltd., Китай) является ферментом цистеиновой протеазы (EC 3.4.22.2), присутствующим в папайе. Его молекулярная масса 23.4 кДа. Кинематическая вязкость 4%-ных растворов папаина оценена методом капиллярной вискозиметрии и в интервале значений pH 2–8 составляет 1.04–1.07 мм2/с, т.е. сопоставима с вязкостью фонового электролита в гидроколлоидах альгината натрия.
Гидроколлоиды получали растворением полисахаридов при 25°C при скорости вращения якорной мешалки 10 об./мин; перемешивание вели в течение 20 мин. Их кислотно-основные свойства в интервале pH 5–8 регулировали с помощью фосфатных буферных растворов, pH 2 задавали с помощью 0.5 N раствора HCl. Двухкомпонентные гидроколлоиды получали введением навески папаина в раствор полисахарида, перемешиваемый со скоростью вращения мешалки 10 об./мин при 25°C в течение не менее 20 мин для достижения концентрационного равновесия.
Пленки из коллоидных растворов индивидуальных компонентов и их смесей формировали методом полива на тефлоновых шаблонах с сушкой на воздухе. Образцы для исследования в сканирующем электронном микроскопе (СЭМ) получали, равномерно распределяя каплю раствора на предметном стекле и высушивая ее сначала на воздухе при комнатной температуре, а затем при 30°C и остаточном давлении 3 мПа.
Аппаратура и методы исследований
СЭМ-изображения дисперсной фазы гидроколлоидов получены в микроскопе Quattro S (Thermo Fisher Scientific, Нидерланды).
Измерение размера частиц в гидрозолях осуществляли методом динамического рассеяния света на приборе Zetasizer Nano ZS (Malvern Instruments Ltd., Англия). В качестве источника излучения используется He–Ne-лазер с длиной волны λ = 632.8 нм и мощностью 4 мВт. В качестве приемника используется лавинный фотоумножитель с квантовой эффективностью более 50% при λ = 633 нм. Время накопления сигнала в серии из трех измерений составляло 20 мин. Анализ результатов измерений осуществлялся автоматизированной программой на базе решения интегрального уравнения Фредгольма I рода с экспоненциальным ядром для нормированной корреляционной функции [34]. В настройках программы обработки результатов внесены изменения [35] для исследования полидисперсных систем.
Реологические исследования проведены с использованием ротационного вискозиметра Реотест-2 с рабочим узлом “цилиндр в цилиндре” при 25 ± 0.5°C в диапазоне скоростей сдвига 0.167–1.3 × 103 с–1.
Сорбционное равновесие в системе пленка альгината натрия/водный раствор папаина исследовали аналогично [36] в статических условиях методом переменных концентраций фермента в интервале C0 = (0.5–2.2) × 10–3 моль/мл при температуре 25, 30 и 35°C с точностью термостатирования ±0.2°C. Время установления равновесия в системе было определено из предварительного кинетического эксперимента и составило 60 мин. Сорбцию фермента осуществляли из растворов с величиной pH 2 или 6.
Для оценки остаточной концентрации папаина в растворе Се из приповерхностной фазы раствора на анализ отбирали 1 мл жидкости. Содержание фермента в растворе определяли по величине оптической плотности окрашенного комплекса с биуретовым реактивом при длине волны 540 нм [37] с применением спектрофотометра UNICO 2800, используя предварительно построенную градуировочную зависимость. Равновесную сорбционную емкость сорбента qe рассчитывали с учетом соотношения объема раствора фермента V и массы полимерной пленки m: ${{q}_{{\text{e}}}} = {{{\text{(}}{{C}_{{\text{0}}}} - {{C}_{{\text{e}}}}{\text{)}}V} \mathord{\left/ {\vphantom {{{\text{(}}{{C}_{{\text{0}}}} - {{C}_{{\text{e}}}}{\text{)}}V} m}} \right. \kern-0em} m}.$
ИК-спектроскопические исследования проведены на фурье-спектрометре Vertex 80v (Bruker). Образцы пленок, сформированных из индивидуальных или двухкомпонентных гидроколлоидов, после механического размола просеивали через сито с размером ячеек 1 мм и подвергали глубокому обезвоживанию в условиях вакуумной сушки при давлении 66.5 мПа. Затем прессовали таблетки из смеси биополимерного материала и порошка KBr в соотношении 2 : 300. Колебательные спектры записаны в режиме пропускания в диапазоне частот 400–4000 см–1 с разрешением 2 см–1.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В водных растворах альгината натрия в отсутствие сшивающих агентов при нейтральных и слабокислых значениях pH термодинамически выгодным состоянием гидратированных макромолекул является конформация статистического клубка [29, 38]. Этому способствует обилие изломов в цепи основных валентных связей между звеньями β-D-маннуроновой и α-1-гулуроновой кислот (рис. 1а).
Рис. 1.
Блочная структура альгината натрия (а) и концевые аминокислоты в глобуле папаина (б): М и G – остатки маннуроновой и гулуроновой кислот, Asn и Ile – аспарагин и изолейцин.

Большое количество диссоциированных карбоксильных групп обусловливает высокую плотность отрицательного заряда на поверхности частиц, что предопределяет взаимное отталкивание и обособленность макромолекул, невозможность формирования ими единой трехмерной сетки даже при высоких концентрациях полимера. Если растворение альгината проводится при гидродинамическом воздействии низкой интенсивности, макромолекулярные клубки можно зарегистрировать на СЭМ-изображениях полимера после высушивания тонкой пленки раствора непосредственно на предметных стеклах для микроскопа (рис. 2).
Рис. 2.
СЭМ-изображения дисперсной фазы растворов альгината натрия с концентрацией 6 (а) и 0.06 мас. % (б) при увеличении ×1000 и ×10 000 соответственно.

Форма частиц папаина также близка к сферической, поскольку этот фермент относится к группе глобулярных белков. Его молекула (рис. 1б) представляет собой объединенные одной полипептидной цепью два домена, между которыми расположен активный центр.
Глобулы папаина много меньше макромолекул альгината, их невозможно увидеть в СЭМ. Молекулярная масса фермента (23.4 кДа) почти на порядок меньше молекулярной массы исследуемого образца альгината (202 кДа), которую рассчитывали по данным определения характеристической вязкости методом капиллярной вискозиметрии. Кроме того, глобула плотнее макромолекулярного клубка полисахарида, поскольку четвертичная структура фермента стабилизирована межцепными ковалентными связями.
Анализ размера частиц в гидрозоле альгината натрия и папаина
Оценка коллоидного состояния биополимеров проведена в сильно разбавленных растворах для обеспечения необходимого уровня их оптической прозрачности. Тем не менее, даже при концентрации альгината 0.06 мас. % и папаина 0.04 мас. % удельное количество их частиц измеряется колоссальными значениями: 1.78 × 1015 и 1.03 × 1017 мл–1 соответственно. Этого достаточно для обнаружения их взаимодействия.
Для гидрозоля папаина получена мономодальная зависимость распределения числа частиц по размеру с малой шириной пика (кривая 1 на рис. 3а), что свидетельствует о полном переходе фермента в мономолекулярное состояние. Пик практически симметричен, т.е. форма глобулы полипептида близка к сферической. Мода приходится на величину гидродинамического радиуса частиц r = = 4.36 нм.
Рис. 3.
Распределение относительного числа частиц (N) по размеру в растворах папаина (1) и альгината натрия (2) при pH 6 (а) и относительного объема дисперсной фазы (V) в гидроколлоиде альгината натрия (б).

Распределение числа частиц альгината по размеру является бимодальным (кривая 2 на рис. 3а), что обусловлено присутствием в препарате макромолекул с различающейся степенью полимеризации. Этот фактор, по-видимому, проявляется также в увеличении ширины как основного пика, так и “шлейфа” (второго пика), отвечающего крупным частицам, по сравнению с распределением по размеру частиц папаина. Сопоставление представленных в табл. 1 пиковых значений среднего размера частиц альгината и параметров соответствующих мод демонстрирует асимметрию обоих пиков (со смещением верхних границ диапазонов размеров в область бóльших значений). Возможно, это связано с отклонениями формы клубка макромолекулы от сферической и неоднородной степенью его прозрачности при попадании лазерного луча.
Таблица 1.
Параметры распределений числа частиц по размеру в индивидуальных и смешанных гидроколлоидax
| Образец | Параметры основного пика, нм | Параметры шлейфа, нм | |||||
|---|---|---|---|---|---|---|---|
| ширина | среднее значение | мода | ширина | среднее значение | мода | ||
| Папаин | 3.25–5.85 | 4.55 | 4.36 | – | – | – | |
| Альгинат Na | 34–110 | 77 | 45.6 | 308–995 | 652 | 478 | |
| Альгинат–папаин 0.06:0.04 | рН 2 | 45.6–110 | 77.8 | 61.2 | 308–1152 | 730 | 553 |
| рН 5 | 171–742 | 457 | 308 | 860–3220 | 2040 | 1790 | |
| рН 6 | 148–742 | 445 | 308 | 860–3220 | 2040 | 1790 | |
| рН 7 | 82–308 | 195 | 148 | 356–3220 | 1788 | 478 | |
| рН 8 | 82–308 | 195 | 148 | 356–2073 | 1215 | 742 | |
При анализе данных динамического рассеяния света для двухкомпонентной системы альгинат–папаин (рис. 4) следует отметить исчезновение пика, соответствующего ферменту. По-видимому, это обусловлено не экранированием мелких частиц папаина более крупными частицами полимера, а взаимодействием между папаином и альгинатом и их объединением, что подтверждено в дальнейших экспериментах.
Рис. 4.
Распределение относительного числа частиц (N) по размеру в гидроколлоидах альгинат–папаин при соотношении компонентов 0.06 : 0.04 мас. % и разных значениях pH: 1 – 5, 2 – 6, 3 – 7, 4 – 8, 5 – 2.

Наблюдаемые различия в смещении пиков по шкале размеров при варьировании pH двухкомпонентной системы свидетельствуют, что характер взаимодействий в существенной степени зависит от кислотно-основных свойств среды. Анализ совместного поведения биополимеров в сильнокислой среде показал, что небольшой рост размера частиц имеет место и при подавлении диссоциации карбоксильных групп альгината. Величина смещения основной моды соизмерима с размерами связываемых частиц фермента (табл. 1). Адсорбционный слой папаина на частицах альгината формируется в данных условиях, по-видимому, за счет водородных связей с участием электронодонорных и электроноакцепторных группировок, имеющихся в молекулах обоих полимеров. Снижение поверхностного заряда частиц альгината способствует их ассоциации. Как следствие, верхняя граница диапазона размеров сдвигается вправо и для доминирующей фракции, и для шлейфа (фракции больших частиц).
В слабокислой среде размерные параметры возрастают многократно как для основного пика, так и для шлейфа крупных частиц. Значения основной моды при pH 5 и 6 превышают величину этого показателя для индивидуального раствора альгината почти в 7 раз.
При переходе к нейтральной среде и, далее, к слабощелочной прирост размерных параметров на регистрируемых распределениях заметно уменьшается, хотя в этих условиях достигается практически полная диссоциация карбоксильных групп альгината. Очевидно, проявляющаяся тенденция связана с изменениями степени протонирования аминогрупп в глобуле папаина.
Концевые фрагменты полипептидной цепи папаина, состоящей из 212 аминокислотных остатков, расположены в диаметрально противоположных частях глобулы (см. рис. 1б). На N-конце молекулы аминокислотой является изолейцин (Ile), на C-конце − аспарагин (Asn). Ile присоединен к макромолекуле карбоксильной группой, Asn – аминогруппой, связанной с вторичным атомом углерода. При этом оба конца молекулы содержат аминогруппы, протонирование которых в кислой среде обеспечивает взаимодействие с диссоциированными карбоксильными группами альгината. Поскольку аминогруппы разнесены на разные стороны глобулы, фермент может одновременно реагировать с двумя частицами альгината, которые, в свою очередь, могут объединяться молекулами фермента в более крупные ассоциаты.
При переходе к мольному выражению концентрации биополимеров нетрудно определить, что на 1 структурную единицу полисахарида приходится 6 глобул фермента. Очевидно, что увеличение размера частиц в двухкомпонентных растворах при pH 5–8 невозможно объяснить только формированием поверхностного слоя молекул папаина на частицах альгината. Более вероятными являются процессы ассоциации с участием нескольких структурных субъединиц обоих компонентов.
Ранее мы продемонстрировали важность одновременного анализа гидрозоля исследуемых объектов на основании характеристик распределений по размеру частиц как их числа N, так и относительного объема дисперсной фазы V [39–42]. Применительно к исследуемой системе необходимость контроля распределения объема дисперсной фазы по размеру частиц обусловлена тем, что именно фракции крупных частиц могут оказывать наибольшее структурирующее влияние на гидроколлоид (с увеличением размера частиц в 10 раз их объем возрастает, как известно, на три десятичных порядка). Показательна трансформация распределения частиц двухкомпонентной системы альгинат–папаин по размеру в слабокислой среде при переходе к данным об относительном объеме дисперсной фазы (рис. 5). Основной пик смещается в зону 1000–3500 нм. Мода попадает на значение гидродинамического радиуса 2400 нм. По-видимому, частицы именно этого размерного диапазона определяют реологические свойства гидроколлоида.
Рис. 5.
Распределение относительного объема дисперсной фазы (V) по размеру частиц в бинарном гидроколлоиде альгината натрия и папаина при pH 5.

Вместе с тем эффективность протеолитического действия на белковые компоненты гнойного экссудата будет обеспечивать основная масса молекул папаина, взаимодействующих с многочисленными малыми частицами альгината с образованием небольших ассоциатов. Правомерно полагать, что это происходит аналогично “слиянию” частиц крупных фракций, наблюдаемому с помощью электронной микроскопии (рис. 6). При этом в системе с концентрацией альгината 0.03 мас. %, обеспечивающей 12-кратный мольный избыток фермента, вероятность множественных межмолекулярных контактов увеличивается и количество структурных единиц в ассоциатах возрастает (см. рис. 6б).
Рис. 6.
СЭМ-изображения дисперсной фазы растворов альгинат–папаин в соотношениях 0.06 : 0.04 мас. % (а) и 0.03 : 0.04 мас. % (б); увеличение ×10 000.

Следовательно, для максимального инициирования межчастичных взаимодействий в двухкомпонентной системе необходимо обеспечить перевод карбоксильных групп альгината в диссоциированное состояние, а аминогрупп папаина в протонированную форму. Выполнению первого условия способствуют смещение кислотно-основного равновесия в щелочную область pH, а для реализации второго предпочтительно использование кислой дисперсионной среды. Наиболее эффективный вариант, обеспечивающий максимальное число заряженных групп и активацию межчастичных взаимодействий, – получение двухкомпонентных гидроколлоидов с уровнем кислотности 5–6 ед. рН.
Исследование реологических свойств гидроколлоидов
Введение папаина не вызывает принципиальных изменений в характере кривых течения гидроколлоидов альгината натрия, отражающих свойственное псевдопластичным жидкостям уменьшение кажущейся (динамической) вязкости η с увеличением скорости сдвига γ.
Вместе с тем весьма существенное повышение сопротивления вязкому течению в присутствии фермента (рис. 7а) свидетельствует о том, что глобулы папаина при их 6-кратном (кривая 1 *) или 12-кратном (кривая 2 *) мольном избытке не стали своеобразными элементами качения или скольжения, облегчающими формирование потока.
Рис. 7.
Кривые течения гидроколлоидов при рН 6 (а) и их анализ в координатах модели Оствальда–де Ваале (б): 1, 1* – 6 мас. % альгината натрия, 2, 2* – 3 мас. % альгината натрия; 1 *, 2 * – смеси альгината с 4 мас. % папаина.

Совокупность экспериментальных данных (точки на рис. 7а) была аппроксимирована прямыми линиями, построенными по срединным участкам кривых течения, без учета концевых точек. Сравнение систем целесообразно проводить не только при одинаковом значении напряжения сдвига τ, но и в пределах прямолинейных участков зависимостей lg η = f(lg τ). Закрашенные серым цветом на рис. 7а зоны сравнения соответствуют диапазону напряжения сдвига от 100 до 400 Па.
В указанном диапазоне значений τ для двухкомпонентных растворов наблюдается примерно одинаковое повышение динамической вязкости (табл. 2). При этом увеличение значения относительного прироста вязкости ∆η с 1.4 до 1.65 при повышении мольного избытка папаина в 2 раза отражает весомость загущающего эффекта с введением фермента.
Таблица 2.
Влияние папаина на вязкость гидроколлоидов альгината натрия при pH 6
| Концентрация, мас. % | Величина вязкости на границах зоны сравнения, Па с | Прирост вязкости на границах зоны сравнения, отн. ед. | Параметры модели Оствальда–де Ваале | |||||
|---|---|---|---|---|---|---|---|---|
| альгинат | папаин | η100 | η400 | ∆η100 | ∆η400 | k | n | R2 |
| 6 | 0 | 7.04 | 4.58 | 1.42 | 1.35 | 12.5 | 0.765 | 0.9945 |
| 4 | 9.98 | 6.18 | 15.3 | 0.774 | 0.9902 | |||
| 3 | 0 | 2.99 | 1.94 | 1.69 | 1.61 | 8.7 | 0.731 | 0.9776 |
| 4 | 5.05 | 3.13 | 9.9 | 0.770 | 0.9995 | |||
Сравнение исследуемых систем возможно путем анализа полученных данных с применением известных математических моделей, описывающих неньютоновское поведение растворов полимеров [43]. Наиболее широко применяемая модель Оствальда–де Ваале предполагает возможность описания кривых течения для разных видов жидкостей с помощью уравнения
где k – константа, называемая коэффициентом консистенции, n – индекс течения (показатель неньютоновского течения раствора).Коэффициент k при постоянстве вида и размеров применяемой аппаратуры чувствительно реагирует на изменение степени структурированности раствора. Для исследуемых псевдопластичных систем неравенство n < 1 характеризует степень отклонения от ньютоновского течения и уровень усилий, необходимый для преобразования внутренней структуры полимера, распрямления (разворачивания) молекулярных цепей для вовлечения их в формирующийся поток жидкости.
Для определения указанных параметров кривые течения на рис. 7б были аппроксимированы степенным полиномом. Корректность применения модели Оствальда–де Ваале демонстрируют высокие значения коэффициента детерминации R2 (см. табл. 2). Близкие значения показателя n характеризуют принципиальную неизменность структуры загущающего полимера с проявлением двухкомпонентными системами тенденции к уменьшению отклонения от ньютоновского поведения, т.е. вовлечение макромолекул в поток облегчается.
Наблюдаемые изменения могут быть, по-видимому, объяснены эффектом одновременного “разматывания” нескольких макромолекулярных клубков, изначально связанных между собой, что облегчает их кооперированное встраивание в поток при заданной скорости сдвига. С данным предположением согласуется значимый прирост коэффициента k, характеризующий необходимость повышенных напряжений сдвига для возникновения и развития вязкого течения двухкомпонентных растворов.
Анализ изотерм сорбции папаина альгинатом натрия
Изучение сорбционного равновесия в системе раствор папаина/пленка альгината натрия и подбор модели для адекватного его описания проведены для определения величины энтальпии взаимодействия и вероятного механизма иммобилизации фермента в структуре полимера. Оценка термодинамических параметров сорбционного равновесия позволяет не только вскрыть природу взаимодействий в системе сорбат–сорбент, но и количественно проанализировать протекание сложного процесса сорбции с учетом изменяющегося состояния функциональных группировок полисахарида [44–46].
При иммобилизации фермента принципиальное значение может иметь степень диссоциации карбоксильных групп альгината (для остатков D-маннуроновой и α-L-гулуроновой кислот значения pKa составляют соответственно 3.38 и 3.2). Исследования проводили при значениях pH раствора фермента, равных 2 и 6, обеспечивающих соответственно либо полное преобразование альгината натрия в недиссоциированную альгиновую кислоту, либо практически полную ионизацию карбоксильных групп. Величину равновесной сорбции папаина (qe) при варьируемой начальной его концентрации (C0) определяли при температуре 25, 30 и 35°C (рис. 8а).
Рис. 8.
Изотермы сорбции папаина альгинатом натрия (а) и их описание в координатах уравнения Ленгмюра (б) при рН 6 (1, 2, 3) и рН 2 (1*, 2*, 3*) и разных значениях температуры: 1, 1* – 25, 2, 2* – 30, 3, 3* – 35°C.
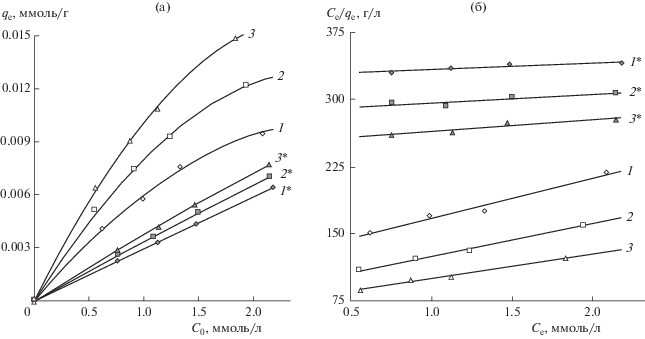
Для экспериментальных зависимостей qe = f(C0) при pH 6 характерен традиционный вид изотермы адсорбции Ленгмюра. В области малой концентрации папаина в растворе наблюдается его интенсивная сорбция. При более высоких значениях C0 система стремится к насыщению, когда вся поверхность адсорбирующих частиц альгината в структуре набухшей полимерной пленки покрывается слоем папаина.
При pH 2 торможения роста значений qe по мере увеличения C0 не наблюдается. Это может быть обусловлено как высокой обратимостью связывания молекул фермента на поверхности частиц альгината, так и возможностью их диффузии в макромолекулярные клубки этого полисахарида. Однако и пропорционального возрастания величины qe с ростом C0 также не происходит.
При увеличении C0 возрастает не только количество сорбированного папаина, но и его равновесная остаточная концентрация в растворе Ce. Описание изотерм равновесной адсорбции в рамках модели Ленгмюра предполагает анализ изменения распределения сорбата между фазами как функции его возрастающей остаточной концентрации сорбата в фазе раствора: ${{{{С}_{{\text{e}}}}} \mathord{\left/ {\vphantom {{{{С}_{{\text{e}}}}} {{{q}_{{\text{e}}}}}}} \right. \kern-0em} {{{q}_{{\text{e}}}}}} = f\left( {{{C}_{{\text{e}}}}} \right).$
Высокая степень аппроксимации экспериментальных данных (рис. 8б) подтверждает применимость модели Ленгмюра для анализа адсорбционного равновесия в исследуемых системах. Наблюдаемое уменьшение показателя Се/qe с ростом температуры отражает повышение “степени полезного использования” папаина при проведении сорбции как в слабо-, так и в сильнокислой среде. Представленные зависимости описываются линейной формой уравнения Ленгмюра [47]
Используя параметр KL, можно рассчитать величину коэффициента KD, который характеризует распределение фермента между раствором и альгинатной пленкой [48]: ${{K}_{{\text{D}}}} = {{K}_{{\text{L}}}}1000{{С}_{0}}.$
Значения параметра KD позволяют определить величину энтальпии процесса адсорбции ΔH по уравнению изобары Вант-Гоффа:
где R – универсальная газовая постоянная, T – температура, K.Высокую степень аппроксимации результатов сорбционного эксперимента в координатах Аррениуса ${\text{ln}}{{K}_{{\text{D}}}} = f{\text{(}}{{T}^{{ - 1}}}{\text{)}}$ подтверждают полученные значения коэффициента детерминации R 2 для каждой группы расчетных параметров (табл. 3). Это обусловливает корректность графического определения значений теплового эффекта сорбции и расчета величин энтропии ΔS и свободной энергии Гиббса ΔG в соответствии с фундаментальными уравнениями термодинамики:
Таблица 3.
Термодинамические параметры сорбции папаина пленками альгината натрия
| рН | ΔH, кДж/моль | ΔS, Дж/(К моль) | ΔG, кДж/моль | ||
|---|---|---|---|---|---|
| 298 К | 303 К | 308 К | |||
| 6.0 | –67.3 | –133.0 | –27.64 | –26.97 | –26.31 |
| 2.0 | –24.0 | 2.3 | –24.64 | –24.65 | –24.66 |
Согласно существующей классификации сорбционных процессов по величине энтальпии [49], значения –ΔH > 100 кДж/моль характеризуют необратимую хемосорбцию с образованием ковалентных связей. Диапазон значений –ΔH < < 100 кДж/моль отражает наличие неспецифических или специфических донорно-акцепторных взаимодействий. При этом энергия наименее сильных химических взаимодействий с образованием водородных связей составляет 8–30 кДж/моль [50, 51]. К этой категории следует отнести, в частности, взаимодействие папаина и альгината натрия в сильнокислой среде.
Взаимодействия в слабокислой среде характеризуются более существенными изменениями внутренней энергии системы, которые, однако, не достигают уровня, соответствующего образованию ковалентных связей. Следовательно, в системе не происходит прочного связывания фермента, например, за счет реакции этерификации с участием карбоксильной группы полипептида. Вместе с тем энергетика электростатических взаимодействий за счет формирования нековалентной солевой связи между протонированными аминогруппами папаина и диссоциированными карбоксильными группами альгината достаточна велика и превосходит, например, энергетику специфических нековалентных взаимодействий между ароматическими соединениями за счет межмолекулярного перекрытия p-орбиталей в π-сопряженных системах бензольных колец. Энергия стэкинг-взаимодействий достигает 50 кДж/моль [52].
ИК-спектроскопия взаимодействий альгината натрия и папаина
Применение колебательной спектроскопии для оценки изменения состояния альгината и папаина в двухкомпонентных системах сопряжено с объективными сложностями. Оба полимера имеют сложные спектры (рис. 9) с наложением характеристических полос как в высокочастотном диапазоне валентных колебаний, так и в диапазоне “отпечатков пальцев”, где спектральный контур для каждого из веществ представляет собой сложно расшифровываемую суперпозицию деформационных колебаний в различных функциональных группах.
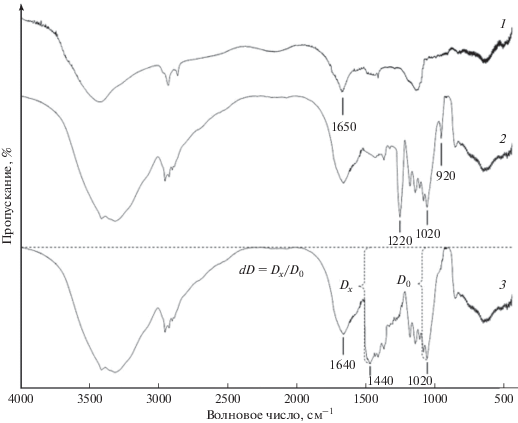
Тем не менее, в спектре альгината имеются полосы поглощения, которые при минимальном фоновом искажении чувствительно реагируют на присутствие папаина. Полоса νC–O при 1440 см–1 обусловлена валентными колебаниями связи C–O в карбоксилах, связанных с Na+ [53]. На спектре альгиновой кислоты такой полосы не наблюдается.
Количественная оценка изменений состояния альгината возможна благодаря наличию неперекрывающейся полосы νC–C при 1020 см–1, соответствующей валентным колебаниям связи С–С в пиранозном цикле. При анализе полисахаридов данный пик принимается в качестве полосы внутреннего стандарта [54–56]. Оценку изменения интенсивности анализируемой полосы осуществляют по величине относительной оптической плотности dD, которая определяется как Dx/D0, т.е. как отношение значений оптической плотности данной полосы Dx и полосы внутреннего стандарта D0 (см. рис. 9). Значения оптической плотности рассчитываются из соотношения $D = {\text{lg}}{{{{I}_{0}}} \mathord{\left/ {\vphantom {{{{I}_{0}}} {I,}}} \right. \kern-0em} {I,}}$ где I0 и I – интенсивность соответственно падающего и прошедшего света с учетом положения базисной линии [45, 57].
Трансформация полосы νC–O при 1440 см–1 по мере увеличения содержания папаина в бинарной системе (рис. 10а) сопровождается прогрессирующим нарастанием поглощения при 1600 см–1 на фоне неизменной интенсивности полосы деформационных колебаний δC=C на частоте 1640 см–1 в карбоксильной группе альгината. Появление новой полосы, очевидно, обусловлено известным фактом батохромного смещения полосы деформационных колебаний связи N–H в первичных аминах δN–H 1650 см–1 при протонировании и образовании солевой связи. Полоса $\delta _{{{\text{N}}{\kern 1pt} - {\kern 1pt} {\text{H}}}}^{ + }$ при 1600 см–1 обнаружена также на спектрах бинарной смеси папаина и альгиновой кислоты (рис. 10б). При этом наблюдается синхронное уменьшение интенсивности полос деформационных колебаний в незамещенных карбоксильных группах: δO–H при 920 см–1 и δC–OH при 1220 см–1.
Рис. 10.
Зона “отпечатка пальцев” на ИК-спектрах продуктов кристаллизации 6%-ных гидроколлоидов альгината натрия (а) и альгиновой кислоты (б) при разном содержании папаина: 1 – 2, 2 – 4, 3 – 6 мас. %.

При оценке изменений интенсивности характеристических полос (табл. 4) изменения значений dD целесообразно сопоставлять только для каждой полосы индивидуально. Нет противоречий в том, что в образцах альгината общие изменения поглощения νs(C–ONa) в исследуемом интервале концентрации папаина составили 36.6% от исходного значения, а амплитуда полосы $\delta _{{{\text{N}}{\kern 1pt} - {\kern 1pt} {\text{Н}}}}^{ + }$ с ростом концентрации фермента увеличилась более чем в 2 раза. Не критично, что для одних и тех же карбоксильных групп в альгиновой кислоте при введении 6 мас. % папаина интенсивность слабой полосы δO–H уменьшается в 3 раза, а сильной полосы δC–OH – не более чем на 40%. Важно, что динамика наблюдаемых изменений синхронна и увеличение количества вводимого фермента сопровождается эквивалентным нарастанием изменений величины dD для каждой из анализируемых полос.
Таблица 4.
Интенсивность полос в ИК-спектрах альгината натрия и альгиновой кислоты при варьируемой концентрации папаина
| Волновое число, см–1 | Группа | Тип колебаний | Относительная оптическая плотность dD в спектрах образцов кристаллизованных гидроколлоидов с разной концентрацией папаина (мас. %) | |||
|---|---|---|---|---|---|---|
| 0 | 2 | 4 | 6 | |||
| альгинат натрия + папаин | ||||||
| 1440 | карбоксильная группа | νs(С−ОNa) | 1.12 | 0.98 | 0.85 | 0.71 |
| 1600 | аминогруппа в ионном взаимодействии | $\delta _{{{\text{N}}{\kern 1pt} - {\kern 1pt} {\text{H}}}}^{ + }$ | − | 0.80 | 1.21 | 1.63 |
| альгиновая кислота + папаин | ||||||
| 920 | карбоксильная группа | δ(O–H) | 0.30 | 0.24 | 0.19 | 0.13 |
| 1220 | карбоксильная группа | δ(C–ОН) | 1.21 | 1.06 | 0.90 | 0.75 |
| 1600 | аминогруппа в ионном взаимодействии | $\delta _{{{\text{N}}{\kern 1pt} - {\kern 1pt} {\text{H}}}}^{ + }$ | − | 0.81 | 1.23 | 1.62 |
Изменения состояния карбоксильных групп полисахаридов в смесях с папаином согласуются с трансформацией колебательных процессов в первичных аминогруппах фермента. Сохранившееся в кристаллизованном продукте протонированное состояние атома азота является достоверным свидетельством электростатической природы взаимодействий, происходящих при введении папаина в гидрозоли альгината натрия или альгиновой кислоты.
Таким образом, результаты анализа колебательных спектров индивидуальных веществ и их бинарных смесей согласуются с заключением о наличии взаимодействия между компонентами, сделанным на основании вискозиметрических, гранулометрических и сорбционных исследований. Введение фермента в полимерную матрицу в нейтральной и слабокислой средах обеспечивает его иммобилизацию по адсорбционному механизму с последующим регулируемым выходом биокатализатора во внешнюю среду, что необходимо для эффективного расщепления полипептидных веществ гнойного экссудата и их отведения из очага поражения при использовании нового ассортимента ранозаживляющих покрытий.
ЗАКЛЮЧЕНИЕ
Близкие значения индекса течения индивидуальных и двухкомпонентных гидроколлоидов и полученные СЭМ-изображения дисперсной фазы свидетельствуют о неизменности конформации статистического клубка альгината натрия при растворении папаина в режиме ламинарного низкоскоростного перемешивания. Результаты измерений размера частиц в гидрозолях подтверждают образование адсорбционного слоя глобул папаина на частицах альгината при рН 2, либо ассоциатов с участием нескольких структурных субъединиц обоих компонентов при рН 5–8. Динамическая вязкость растворов альгината при рН 6 возрастает в присутствии папаина в 1.4–1.65 раза.
Корректное описание изотерм сорбции папаина альгинатом в рамках модели Ленгмюра позволило определить термодинамические параметры взаимодействия. Изменение энтальпии при рН 2 составляет –24 кДж/моль, что соответствует уровню теплового эффекта при образовании водородных связей. Понижение внутренней энергии системы на 67.3 кДж/моль при pH 6 свидетельствует о протекании хемосорбции за счет формирования солевой связи между протонированными аминогруппами папаина и диссоциированными карбоксильными группами альгината. Подтверждение предложенного механизма адсорбции получено при анализе колебательных спектров биополимеров и аддуктов их взаимодействия. Результаты работы позволяют оптимизировать процессы получения нового ассортимента ранозаживляющих покрытий с противовоспалительным действием.
Список литературы
Ишевский А.Л., Успенская М.В. Гунькова П.И., Давыдов И.А., Василевская И.А. // Известия СПбГТИ(ТУ). 2019. Вып. № 51. С. 61.
Alginates: Biology and Applications / Ed. by Bernd H., Rehm A. Berlin: Springer, 2009.
Полимеры в биологии и медицине / Под ред. Дженкинса М., пер. с англ. М.: Научный мир, 2011.
Lewicki J., Bergman J., Kerins C., Hermanson O. // Bioprinting. 2019. V. 16. ID e00053.
Lee K. Y., Mooney D. J. // Progr. Polym. Sci. 2012. V. 37. P. 106.
Sun J., Tan H. // Materials. 2013. V. 6. P. 1285.
Анурова М.Н., Бахрушева Е.О., Демина Н.Б. // Хим.-фарм. журн. 2015. Т. 49. № 9. С. 39.
Alginates in Drug Delivery / Ed. by Nayak A.K., Md Hasnain S. London: Academic Press, 2020.
Олтаржевская Н.Д., Кричевский Г.Е. // Хим.-фарм. журн. 2005. Т. 39. № 3. С. 42.
Олтаржевская Н.Д., Кричевский Г.Е., Коровина М.А., Гусев И.В. // Биофармацевтический журн. 2017. Т. 9. № 2. С. 3.
Saco M., Howe N., Nathoo R., Cherpelis B. // Dermatol. Online J. 2016. V. 22. № 8. P. 1.
Aderibigbe B.A., Buyana B. // Pharmaceutics. 2018. V. 10. P. 42.
Zhang M, Zhao X. // Int. J. Biol. Macromol. 2020. V. 162. P. 1414.
Stoica A.E., Chircov C., Grumezescu A.M. // Molecules. 2020. V. 25. P. 2699.
Sinclair R.D., Ryan T.J. // Australas. J. Dermatol. 1994. V. 35. P. 35.
Юданова Т.Н., Решетов И.В. // Хим.-фарм. журн. 2006. Т. 40. № 8. С. 24.
Верниковский В.В., Степанова Э.Ф. // Росс. хим. журн. 2010. Т. 54. № 6. С. 94.
Перцев И.М., Даценко Б.М., Гунько В.Г. // Фармация. 1990. № 5. С. 73.
Datta S., Christena L. R., Rajaram Y.R.S. // 3Biotech. 2013. V. 3. P. 1.
Bilal M., Iqbal H.M.N. // Int. J. Biol. Macromol. 2019. V. 130. P. 462.
Березин И.В., Клячко Н.Л., Левашов А.В., Мартинек К., Можаев В.В., Хмельницкий Ю.Л. Иммобилизованные ферменты / Биотехнология. Т. 7. М.: Высшая школа, 1987.
Flores-Maltos A., Rodríguez-Durán L.V., Renovato J., Contreras J.C., Rodríguez R., Aguilar C.N. // Enzyme Res. 2011. V. 2011. ID 768183.
Kanbargi K.D, Sonawane S.K., Arya S.S. // Int. J. Food Prop. 2017. V. 20. P. 3215.
Сигаева Н.Н., Вильданова Р.Р., Султанбаев А.В., Иванов С.П. // Коллоид. журн. 2020. Т. 82. С. 363.
Bastos L.P.H., Carvalho C.W.P., Rojas E.E.G. // Int. J. Biol. Macromol. 2018. V. 120. Pt. A. P. 332.
Zhao Y., Li F., Carvajal M.T., Harris M. // J. Colloid Interface Sci. 2009. V. 332. P. 345.
Rocha M.V., Di Giacomo M., Beltramino S., Loh W., Romanini D., Nerli B.B. // Sep. Purif. Technol. 2016. V. 168. P. 168.
Fuenzalida J.P., Nareddy P.K., Moreno-Villoslada I., Moerschbacher B.M., Swamy M.J., Pan S., Ostermeier M., Goycoolea F.M. // Food Hydrocoll. 2016. V. 53. P. 239.
Щипунов Ю.А., Постнова И.В. // Высокомолек. соед. А. 2006. Т. 48. С. 285.
Feng L., Cao Y., Xu D., You S., Han F. // Ultrason. Sonochem. 2016. V. 32. P. 224.
Олтаржевская Н.Д., Коровина М.А. // Росс. хим. журн. 2011. Т. 55. № 3. С. 97.
Олтаржевская Н.Д., Швец В.И., Коровина М.А., Липатова И.М., Хлыстова Т.С. // Биотехнология. 2016. № 1. С. 43.
Gurung S., Skalko-Basnet N. // J. Ethnopharmacol. 2009. V. 12. P. 338.
Yan Y.D., Clarke J.H.R. // Adv. Colloid Interface Sci. 1989. V. 29. P. 277.
Кокшаров С.А. // Изв. вузов. Химия и хим. технология. 2015. Т. 58. № 1. С. 33.
Котова Д.Л., Крысанова Т.А., Васильева С.Ю. // Коллоид. журн. 2020. Т. 82. С. 334.
Nowotny A. Basic Exercises in Immunochemistry: A Laboratory Manual. Berlin: Springer, 1979. P. 168.
Rees D.A., Morris E.R., Thon D., Madden J.K. // The Polysaccharides / Ed. by Aspinall G.O. New York: Academic Press, 1982. P. 195.
Kornilova N., Bikbulatova A., Koksharov S., Aleeva S., Radchenko O., Nikiforova E. // Coatings. 2021. V. 11. 616.
Лепилова О.В., Кокшаров С.А., Алеева С.В. // Журн. прикл. химии. 2018. Т. 91. С. 68.
Koksharov S.A., Kornilova N.L., Nikiforova E.N. // Solid State Phenom. 2021. V. 316. P. 68.
Кокшаров С.А., Алеева С.В., Корнилова Н.Л., Калинин Е.Н. // Перспективные материалы. 2021. № 4. С. 37.
Малкин А.Я., Исаев А.И. Реология: концепции, методы, приложения. СПб: Профессия, 2007.
Koksharov S.A., Aleeva S.V, Lepilova O.V. // J. Mol. Liq. 2019. V. 283. P. 606.
Алеева С.В., Чистякова Г.В. Лепилова О.В, Кокшаров С.А. // Журн. физ. хим. 2018. Т. 92. С. 1308.
Алеева С.В., Лепилова О.В., Кокшаров С.А. // Физикохимия поверхности и защита материалов. 2021. Т. 57. С. 41.
Magdy Y.H., Daifullah A.A.M. // Waste Manage. 1998. V. 18. P. 219.
Khotimchenko M., Kovalev V., Khotimchenko Yu. // J. Hazard. Mater. 2007. V. 149. P. 693.
Никифорова Т.Е., Козлов В.А. // Физикохимия поверхности и защита материалов. 2011. Т. 47. С. 23.
Курс органической химии / Под ред. Дрюк В.Г., Малиновского М.С. Киев: Вища шк., 1987.
Карапетьянц М.Х., Дракин С И. Общая и неорганическая химия. М.: Химия, 1981.
Steed J.W., Atwood J.L. Supramolecular Chemistry. Chichester: Wiley, 2000.
Беллами Л. Инфракрасные спектры сложных молекул / Пер. с англ. под ред. Пентина Ю.А. М.: Изд-во иностр. лит-ры, 2000.
Тарутина Л.И., Позднякова Ф.О. Спектральный анализ полимеров. Л.: Химия, 1986.
Алеева С.В., Чистякова Г.В., Кокшаров С.А. // Изв. вузов. Химия и хим. технология. 2009. Т. 52. № 10. С. 119.
Koksharov S., Aleeva S., Lepilova O. // Autex Res. J. 2015. V. 15. P. 215.
Алеева С.В., Лепилова О.В., Кокшаров С.А. // Журн. прикл. спектроскопии. 2020. Т. 87. С. 694.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал