

Коллоидный журнал, 2022, T. 84, № 1, стр. 23-33
Наноэмульсии с инкорпорированным липофильным лекарственным веществом фелодипином и микрогетерогенные полимерные адгезивные матрицы на их основе
Н. М. Задымова 1, *, В. В. Куруленко 1
1 Московский государственный университет имени М.В. Ломоносова,
химический факультет
119991 Москва, Ленинские горы, 1, стр. 3, Россия
* E-mail: zadymova@colloid.chem.msu.ru
Поступила в редакцию 19.12.2021
После доработки 25.12.2021
Принята к публикации 26.12.2021
- EDN: SYCCXQ
- DOI: 10.31857/S0023291222010141
Аннотация
Получены прямые наноэмульсии (НЭ) простого состава, в которых липофильное немицеллообразующее неионогенное ПАВ (Твин 85) является и дисперсной фазой, и стабилизатором. НЭ длительное время (несколько месяцев) остаются в метастабильном состоянии, имеют высокоразвитую удельную поверхность (Sуд ≅ 63 м2/г), высокую солюбилизационную емкость по отношению к липофильному гипотензивному лекарственному веществу фелодипину (Фл). Наноразмерные капли (dср ≅ 95 нм) НЭ эффективно транспортируют Фл в водной среде. На основе прямых НЭ Твин 85 с инкорпорированным Фл и раствора полимерного адгезива (смесь полиизобутиленов с различной молекулярной массой и полибутена) в гептане получены двойные эмульсии масло1/вода/масло2, использованные в качестве премикса полимерных микрогетерогенных матриц. Полимерные пленки характеризуются хорошей адгезией к коже, отсутствием кристаллизации Фл и постоянной скоростью его высвобождения в течение нескольких часов.
1. ВВЕДЕНИЕ
Липофильные лекарственные вещества (ЛВ) преобладают на мировом фармацевтическом рынке, однако они характеризуются низкой биодоступностью вследствие плохой растворимости в воде. Создание новых лекарственных форм таких ЛВ с целью повышения их биодоступности является актуальной и сложной коллоидно-химической задачей, связанной с изучением возможности инкорпорирования липофильных ЛВ в различные типы наноразмерных носителей в водной среде. Иммобилизация в наноконтейнеры не только увеличивает растворимость липофильных ЛВ в воде, но и защищает от гидролиза, других типов деградации, включая ферментативное разложение, и обеспечивают их пролонгированную доставку. Дисперсные системы с водной дисперсионной средой, содержащие наноразмерные носители ЛВ, можно разделить на термодинамически устойчивые (мицеллярные растворы ПАВ [1–5] и микроэмульсии [6–11]) и термодинамически неустойчивые, но характеризующиеся длительной кинетической стабильностью, т.е. квазиравновесные (липосомы [12–19], ниосомы [20–27], наноэмульсии (НЭ) [28–32]).
В последние годы возрос интерес к изучению прямых НЭ, что связано с перспективами их применения в медицине и фармацевтике. НЭ – это дисперсии жидкость/жидкость с узким распределением частиц по размерам и их средним радиусом от 10 до 200 нм [33–35]. В отличие от прямых микроэмульсий IV типа по Винзору, пригодных для инкорпорирования липофильных ЛВ, но которые можно получить лишь при высоких концентрациях ПАВ и со-ПАВ (обычно > 30 мас. %), для стабилизации НЭ требуются существенно меньшие концентрации эмульгатора (примерно от 5 до 10 мас. %) [28]. При этом НЭ имеют гораздо менее сложный компонентный состав по сравнению с микроэмульсиями. Кроме того, НЭ характеризуются более высокой солюбилизационной емкостью (СЕ) по отношению к липофильным ЛВ, нежели мицеллы ПАВ. Важно также, что НЭ имеют преимущество и перед липосомами, вследствие большей стабильности, возможности жесткой стандартизации сырья и условий получения, меньших производственных затрат. Однако НЭ могут быть подвержены процессам коагуляции, коалесценции и изотермической перегонки (оствальдово созревание). Как правило, основным механизмом деградации НЭ является изотермическая перегонка [36, 37], которая представляет собой диффузионный массоперенос вещества дисперсной фазы от мелких капель к более крупным, вследствие чего концентрация мелких капель уменьшается (вплоть до полного их исчезновения), а концентрация крупных капель возрастает. Причиной оствальдова созревания, как известно, являются разные химические потенциалы (следовательно, и разная растворимость в дисперсионной среде) капель разных размеров. С уменьшением размеров капель растет их растворимость в дисперсионной среде (закон Кельвина).
Для создания агрегативно устойчивых в течение длительного времени НЭ необходимо ингибировать изотермическую перегонку. Для этого можно в качестве дисперсной фазы выбрать вещество, плохо растворимое в дисперсионной среде [33, 36, 38], или же добавить в дисперсную фазу небольшое количество компонента, который гораздо хуже растворим в дисперсионной среде, нежели основной компонент [39–41]. В последнем случае основной компонент быстрее диффундирует в крупные капли. При этом с течением времени изменяется состав капель разных размеров: концентрация малорастворимого компонента в мелких каплях растет, а в крупных падает. Это делает термодинамически невыгодным дальнейшее уменьшение размеров мелких капель вследствие роста их химического потенциала за счет концентрирования второго компонента. В результате возникает квазиравновесное состояние, при котором капиллярный и концентрационный эффекты компенсируют друг друга.
Следует иметь в виду, что при использовании мицеллообразующих ПАВ для стабилизации прямых НЭ в дисперсионной среде будут присутствовать мицеллы, которые солюбилизируют вещество дисперсной фазы. При этом диффузия мицелл, содержащих солюбилизат, увеличивает скорость изотермической перегонки [36, 42, 43].
Сказанное выше относится к НЭ, в которых дисперсная фаза представляет собой масла различной природы (предельные и непредельные углеводороды, терпены и т.д.). В литературе отсутствуют сведения о прямых НЭ простого состава, в которых немицеллообразующее липофильное ПАВ является и дисперсной фазой, и стабилизатором. Согласно нашей гипотезе, такие НЭ не будут подвержены изотермической перегонке и могут находиться в квазиравновесном состоянии в течение длительного времени, что является ключевым условием их практического применения.
К новым эффективным лекарственным формам принадлежат трансдермальные пластыри, способные обеспечить контролируемое поступление ЛВ через кожу в кровоток с терапевтически оптимальной скоростью в течение длительного времени (от 1 до 7 сут) [44, 45]. Использование пластырей безболезненно и удобно, исключает потери, сопряженные с метаболизмом ЛВ в желудочно-кишечном тракте и печени, позволяет варьировать дозу и прекращать лечение в любой момент. Применение пластырей снижает риск побочных эффектов за счет нивелирования скачков концентрации ЛВ в крови. Для пациентов с хроническими заболеваниями, нуждающихся в постоянном и многократном пероральном применении ЛВ, и особенно для страдающих заболеваниями желудочно-кишечного тракта и печени, трансдермальная терапия особенно показана.
В большинстве случаев главной составной частью трансдермальных пластырей является полимерная пленка (матрица), содержащая ЛВ в молекулярной (реже диспергированной) форме, усилитель проницаемости кожи и полимер (адгезив), обеспечивающий хорошую адгезию пленки к ее поверхности. Как правило, такие полимерные матрицы получают на основе расплавов или растворов, когда ЛВ и полимерный адгезив присутствуют в одной и той же фазе (обычно липофильной). Патчи такого типа подходят для препаратов с оптимальным гидрофильно-липофильным балансом (ГЛБ), но неэффективны для липофильных ЛВ с низкой растворимостью в воде, поскольку затруднен их диффузионный массоперенос в глубинные гидрофильные слои кожи, где расположены кровеносные капилляры, что снижает биодоступность.
Трансдермальные пластыри, в которых полимерная матрица содержит жидкие микродомены с ЛВ (микрорезервуарный тип), считаются наиболее перспективными для доставки липофильных ЛВ в биодоступной форме [46]. Такие матрицы визуально однородны (микродомены видны только под микроскопом), компактны, удобны, безопасны, расширяют круг трансдермально доставляемых ЛВ, позволяют инкорпорировать целевые компоненты различной полярности. Пластыри микрорезервуарного типа даже можно разрезать с целью уменьшения дозы ЛВ [46]. В настоящее время насчитывается лишь несколько исследований, посвященных патчам микрорезервуарного типа, когда в гидрофобную полимерную матрицу инкорпорируют микродомены, представляющие собой жидкое ЛВ или его раствор в органическом растворителе, способном усиливать проницаемость кожи [47–49]. Разработки основаны на эмульгировании, причем ЛВ и полимерный адгезив находятся в разных фазах. Однако присутствие в микродоменах органических растворителей может вызывать раздражение и аллергические реакции кожи.
Мотивацией данного исследования послужила гипотеза о перспективности использования двойных эмульсий (ДЭ) масло1/вода/масло2, внешней дисперсионной средой (масло2) которых является раствор полимерного адгезива в летучем неполярном растворителе, а первичной эмульсией (масло1/вода) − прямая НЭ с ЛВ, в качестве основы микрогетерогенных полимерных матриц для доставки липофильных ЛВ. В первоочередные задачи входила разработка агрегативно устойчивых в течение длительного времени прямых НЭ простого состава, в которых липофильное немицеллообразующее неионогенное ПАВ (НПАВ) является дисперсной фазой и стабилизатором, и которые также обладают высокими СЕ и транспортными свойствами относительно модельного липофильного ЛВ.
2. ОБЪЕКТЫ И МЕТОДЫ ИССЛЕДОВАНИЯ
2.1. Объекты
Фелодипин (Фл) в форме основания (3-этил-5-метил-4-(2,3-дихлорофенил)-2,6-диметил-1,4-дигидропиридин-3,5-дикарбоксилат) – современное ЛВ гипотензивного действия. Фл, который при комнатной температуре находится в твердом агрегатном состоянии (Tпл = 145°С) и практически не растворим в воде (Sв = 2.2 × 10–6 М [4]), что обуславливает его низкую биодоступность, не превышающую 20% при пероральном применении, был выбран в качестве модельного гидрофобного ЛВ. Молекулярная масса Фл равна 384.2 Да, его структурная формула представлена на рис. 1а. В работе использовался препарат фирмы PCAS (Финляндия) со степенью чистоты 99%.
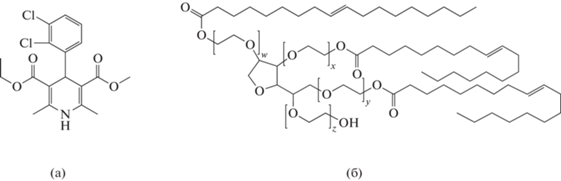
Твин 85 (полиоксиэтилированный(20)триолеат сорбитана) − не образующее мицелл в водной среде НПАВ (ГЛБ = 11) – при комнатной температуре представляет собой жидкость с плотностью 1.028 × 103 кг/моль. Молекулярная масса Твин 85 равна 1836 Да, его структурная формула приведена на рис. 1б. Данное НПАВ плохо растворимо в воде, безопасно, биоразлагаемо и одобрено для использования в пищевой промышленности и в медицине. В работе использовали препарат марки “ч” от Sigma-Aldrich.
Применяли дважды дистиллированную воду с удельной электропроводностью 1.9 мкСм/см (22°С). В воду добавляли 0.01 мас. % азида натрия для защиты НЭ от биодеградации.
Этанол и н-пропанол были марки “х.ч.” от Sigma-Aldrich, а гептан от фирмы Химмед (Россия) со степенью чистоты 99%. Все органические растворители дополнительной очистке не подвергали.
В качестве полимерного адгезива использовали смесь полибутена (Indopol H-1900, Mw = 2.5 кДа, INEOS Oligomers) и полиизобутиленов различной молекулярной массы (Oppanol B100, Mw = 250 кДа и Oppanol B12SFN, Mw = 51 кДа, BASF) в соотношении 4/1/5 (по массе). Смесь полимеров такого состава, обозначаемая ниже как ПИБ, характеризуется хорошей адгезией к различным субстратам, включая кожу человека. ПИБ относится к классу адгезивов, чувствительных к давлению [50].
Системы, содержащие Фл, хранились в защищенном от света месте.
2.2. Методы исследования
Прецизионная турбидиметрия. Растворимость Твин 85 в водном растворе азида натрия (0.01 мас. %) была определена с помощью прецизионной турбидиметрии, методика которой предложена нами ранее [51] и подробно описана в работах [52, 53]. Измерения оптической плотности проводили с помощью однолучевого спектрофотометра Agilent 8453 (США). Спектры получали относительно водного раствора азида натрия. Использовались кварцевые кюветы толщиной 1 см. Точность определения оптической плотности составляла ±1 × 10–4.
Дисперсионный анализ. Исследование распределения частиц по размерам проводилось с помощью динамического рассеяния света на высокоскоростном анализаторе Zetatrac NPA152 (Microtrac Inc.). Данный прибор работает по принципу лазерной дифракции. Источником излучения (λ = 730 нм) служит лазерный диод. Средой сравнения служил 0.01% водный раствор азида натрия, который использовался в качестве водной фазы. Для каждого образца проводились систематические измерения во времени, начиная с момента приготовления дисперсий. Обработка полученных данных осуществлялась с помощью программного обеспечения Microtrac FLEX Software, по итогам обработки были получены дифференциальные кривые распределения, характеризующие объемную долю (или об. %) частиц каждого диаметра. В качестве среднего размера частиц при данной технике измерения принято использовать медианный диаметр, соответствующий такому размеру частиц, что 50% частиц оказываются равного или большего размеров, а 50% частиц имеют меньшие размеры.
УФ-спектроскопия применялась при исследовании растворимости (солюбилизации) Фл в водных дисперсиях Твин 85. Пробу НЭ с ЛВ растворяли в н-пропиловом спирте; в качестве образца сравнения использовали н-пропиловый спирт, содержащий такое же количество НЭ без Фл. Растворимость Фл рассчитывали по формуле
где Amax – оптическая плотность в максимуме поглощения, Emax – молярный коэффициент экстинкции. Для Фл в н-пропиловом спирте λmax = = 363 нм, E363 = 6795 ± 20 М–1 см–1 [5].Измерения проводились на однолучевом спектрофотометре Agilent 8453 (США), использовались кварцевые кюветы толщиной 1 см. Точность определения оптической плотности составляла ±1 × 10–4.
Диффузионный массоперенос ЛВ из жидких образцов и пленок исследовали с помощью УФ-спектроскопии и диффузионных ячеек Франца, представленных на рис. 2.
Рис. 2.
Диффузионные ячейки Франца для определения скорости массопереноса ЛВ из жидких образцов (а) и пленок (б).

Между донорной и акцепторной частями ячейки находится отверстие площадью 0.71 см2, на него помещали мембрану, через которую ЛВ диффундирует в приемную среду. Использовали мембрану MF-Millipore толщиной 150 мкм из биологически инертной смеси эфиров целлюлозы с диаметром пор 800 нм и пористостью 75%; площадь мембраны составляла 5.3 см2. В акцепторную ячейку помещали магнитную мешалку.
При исследовании жидких образцов в донорную часть ячейки (рис. 2а) помещали известный объем исследуемой НЭ. В случае пленок образец наклеивали на мембрану (рис. 2б). В акцепторную часть ячейки помещали известный объем приемной среды, в качестве которой использовали бинарный растворитель этанол/вода (40/60, по массе), рекомендованный для исследования трансдермального массопереноса Фл [54]. Концентрацию Фл в приемной среде оценивали спектрофотометрически через определенные промежутки времени. Расчеты проводили по формуле (1), для Фл в данном бинарном растворителе λmax = 364 нм, E364 = = 6950 ± 20 М–1 см–1 [5].
Количество Фл Q (мкг/см2), прошедшего через мембрану за время t, оценивали по формуле
где CФл – концентрация Фл (мкг/см3) в приемной среде в момент времени t, Vпр – объем приемной среды (7.5 см3) и ao – площадь, через которую идет диффузия, равная площади отверстия в ячейке Франца (0.71 см2).Опыты повторяли несколько раз и вычисляли средние значения Q. Из зависимости Q(t) определяли скорость диффузионного переноса ЛВ.
Оптическая микроскопия. Морфологию объектов (ДЭ и полимерных матриц) изучали с помощью микроскопа Axioskop 40 (Carl Zeiss, Германия) в проходящем свете (неполяризованном и поляризованном). Микроскоп снабжен цифровым фотоаппаратом Canon PowerShot A620. Масштабирование полученных изображений осуществлялось с помощью программы Adobe Photoshop CS.
В данной работе все измерения проводились при температуре 22 ± 2°С.
Методика получения полимерных матриц основана на использовании ДЭ масло1/вода/масло2 в качестве премикса, которые, в свою очередь, получали инкорпорированием прямых НЭ Твин 85 в растворы полимерного адгезива в гептане (перемешивание палочкой).
Полимерные матрицы из ДЭ формировали методом полива. В качестве подложки использовали полиэтилентерефталат марки Loparex 7300A, который применяется в трансдермальных пластырях в качестве удаляемой перед употреблением пленки. ДЭ наносили с помощью ракли на силиконизированную поверхность подложки. Как правило, толщина наносимого слоя составляла 625 мкм. Сушка пленок осуществлялась при 40−50°C в течение 30−60 мин в сушильном шкафу Binder (Германия) с горизонтальным потоком воздуха.
Остаточное содержание гептана в полимерных матрицах определяли с помощью газовой хроматографии. Анализ проводили на модифицированном хроматографе ЛХМ-8МД с использованием капиллярной колонки длиной 28 м и внутренним диаметром 0.32 мм со слоем неподвижной жидкой фазы SE-54 толщиной 3.5 мкм. Газ-носитель – гелий, давление на входе в колонку составляло 0.9 атм. Использовался дозатор-инжектор с делением потока газа-носителя 1 : 50. Детектор – пламенно-ионизационный, шкала электрометра (5–20) × 10–12 А. Содержание гептана в пленке после сушки во всех случаях не превышало 0.3 мас. %.
Высушенные полимерные матрицы ламинировали базовой пленкой (Scotch Pak 9732). Толщину полимерных матриц после сушки определяли с помощью микрометра с учетом толщины базовой и удаляемой пленок. Полученные пленки хранили в запаянных фольгированных пакетиках.
3. РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
3.1. Прямые наноэмульсии Твин 85 для инкорпорирования фелодипина
Для оценки растворимости Твин 85 в 0.01 мас. % водном растворе азида натрия при разбавлении базовой эмульсии с известной молярной концентрацией, полученной ультразвуковым методом (частота 20 кГц, 10 с), была приготовлена серия ультрадисперсных эмульсий Твин 85 в растворе азида натрия с концентрацией этого НПАВ (CTв) от 5.61 × × 10–5 до 3.93 × 10–4 М). По данным динамического рассеяния света средний диаметр капель составлял dср = 94 ± 1 нм, что позволяет данные дисперсии классифицировать как НЭ. Они были агрегативно устойчивы в течение двух недель и более.
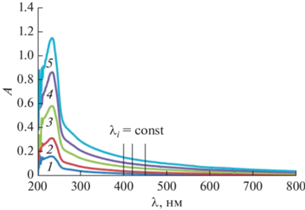
Дифференциальные спектры экстинкции НЭ (относительно водного раствора азида натрия) регистрировали в широком диапазоне длин волн (λ = 200–800 нм) через определенные интервалы времени t с момента их приготовления.
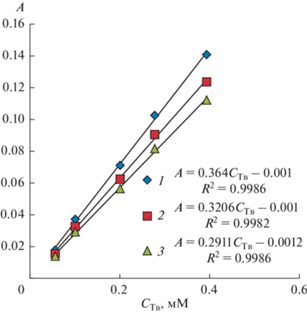
На рис. 3 в качестве примера представлены спектры для t = 12 сут; видно, что в УФ-области наблюдается максимум поглощения. Поскольку НЭ бесцветны, то в видимой области спектра наблюдается исключительно рассеяние света частицами дисперсной фазы, тогда как в УФ-области спектр представляет собой суперпозицию поглощения и рассеяния. При выбранном в видимой области спектра постоянном значении длины волны λ зависимость оптической плотности от молярной концентрации Тв является линейной: A = –k + bCТв (k и b – положительные численные коэффициенты). Экстраполяция зависимости A(CТв) к нулевому значению оптической плотности (A = 0) позволяет определить растворимость (S) Твин 85 для момента времени t при выбранном значении λ: S = CA → 0 = k/b.
Рис. 3.
Спектры экстинкции для серии разбавленных НЭ Твин 85 в водном 0.01 мас. % растворе азида натрия спустя 12 сут после приготовления. Концентрация Твин 85: 5.61 × 10–5 (1), 1.01 × 10–4 (2), 2.01 × 10–4 (3), 2.78 × 10–4 (4), 3.93 × 10–4 M (5).
Аналогичным образом рассчитывали значения S (соответствующие времени t) при нескольких длинах волн и вычисляли среднее значение растворимости (рис. 4). Равновесная растворимость Твин 85 в водном растворе азида натрия, равная (3.3 ± 0.2) × 10–6 М, достигалась за 12 сут. Можно ожидать, что, вследствие такой низкой растворимости, НЭ, в которых Твин 85 является дисперсной фазой, а водный раствор азида натрия − дисперсионной средой, не будут подвержены изотермической перегонке.
Рис. 4.
Зависимости оптической плотности НЭ от концентрации Твин 85 в 0.01 мас. % водном растворе азида натрия при различных длинах волн (1 − 420, 2 − 450, 3 − 480 нм) спустя 12 сут после приготовления.
Эмульсии Твин 85 в водном растворе азида натрия готовили путем добавления к точной навеске этого НПАВ известного объема раствора азида натрия. Массовую концентрацию дисперсной фазы эмульсии (CТв в мас. %, т.е. в г/100 см3 эмульсии) рассчитывали с учетом плотности обоих компонентов. Дополнительных стабилизирующих ПАВ не использовали.
Способ приготовления подбирали на примере дисперсии с 1 мас. % Твин 85. Использовали два способа: самоэмульгирование, т.е. незначительное механическое воздействие в отсутствие диспергирующих приборов (перемешивание при помощи стеклянной палочки), и ультразвуковое эмульгирование (22 кГц, в течение 10 с) при охлаждении. Дисперсионный анализ показал, что самоэмульгирование приводит к образованию субмикронной эмульсии с бимодальным распределением частиц по размерам, стабильной в течение нескольких дней. В то же время незначительное ультразвуковое воздействие обеспечивает получение прямой НЭ Твин 85 с мономодальным распределением частиц по размерам (dср = 96 ± 5 нм), которая сохраняет агрегативную устойчивость в течение длительного времени (рис. 5). При этом удельная поверхность НЭ Sуд = 64.0 м2/г, что почти в 2 раза превышает значение Sуд субмикронной эмульсии, полученной при самоэмульгировании.
Рис. 5.
Дифференциальные кривые распределения частиц по размерам для эмульсий с 1 мас. % Твин 85 при различных способах приготовления и сроках хранения: 1 – самоэмульгирование (спустя 2 дня после приготовления), 2 − ультразвуковое диспергирование (спустя 1 месяц).

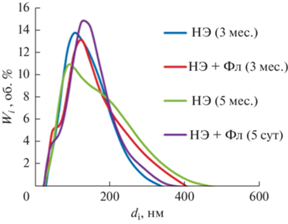
Серия НЭ Твин 85 с различным содержанием дисперсной фазы (CТв от 1 до 5 мас. %, с шагом 1 мас. %) была приготовлена при апробированном нами ультразвуковом воздействии (10 с). НЭ каждой концентрации делили на две равные части. Одну часть насыщали Фл при его избытке, а другую использовали для сравнения. Для всех образцов в течение длительного времени (нескольких месяцев) проводили дисперсионный анализ и оценивали растворимость Фл. Типичные результаты дисперсионного анализа на примере прямой НЭ Твин 85 (4 мас. %) приведены на рис. 6.
Рис. 6.
Дифференциальные кривые распределения частиц по размерам для прямой НЭ Твин 85 (4 мас. %) с Фл (2, 4) и без него (1, 3) спустя 5 сут (3, 4) и 3 месяца (1, 2) после приготовления.
Серия НЭ характеризуется следующими свойствами:
• практически неизменным мономодальным распределением частиц по размерам (dср = 93 ± 4 нм) в течение нескольких месяцев,
• распределение частиц по размерам и стабильность НЭ не изменяются при инкорпорировании Фл,
• удельная поверхность не зависит от концентрации дисперсной фазы и равна 63.4 ± 0.4 м2/г;
• межфазная поверхность в расчете на 1 л НЭ линейно возрастает с концентрацией дисперсной фазы (от 586.9 до 3457.0 м2/л).
Перечисленные выше свойства позволяют отнести полученные прямые НЭ Твин 85 к псевдолиофильным дисперсным системам [37]. Действительно, очень низкая растворимость Твин 85 в дисперсионной среде ингибирует изотермическую перегонку в НЭ. Кроме того, для этих НЭ выполняется условие термодинамической устойчивости дисперсной системы к коагуляции (а значит, и к коалесценции) [37]:
где k – константа Больцмана, T – абсолютная температура, uк − энергия сцепления пары частиц в контакте, которая может быть оценена из соотношения [37] где A* – сложная константа Гамакера, rср – средний радиус капель, h0 − расстояние наибольшего сближения капель, которое принимают равным 0.2 нм.Если принять более жесткие условия, то с учетом соотношений (3) и (4), можно записать:
По данным дисперсионного анализа rcp ≈ 47 нм, что на основании неравенства (5) позволяет заключить, что сложная константа Гамакера A*, характеризующая взаимодействия капель через прослойки дисперсионной среды в исследованных псевдолиофильных НЭ, не превышает 2.1 × 10–21 Дж.
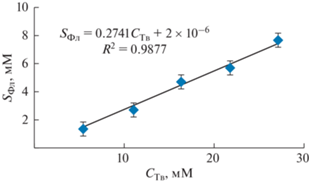
Растворимость (солюбилизацию) Фл в НЭ Твин 85 изучали с помощью УФ-спектроскопии. Пробы отбирали периодически и разбавляли н-пропиловым спиртом (см. раздел 2). Во всех случаях примерно через неделю устанавливаются неизменные значения солюбилизации. Квазиравновесная изотерма солюбилизации Фл в НЭ Твин 85 разной концентрации, SФ(CТв), представлена на рис. 7, видно, что изотерма линейна. Этот результат аналогичен закономерностям, наблюдаемым при солюбилизации липофильных веществ в водных мицеллярных растворах ПАВ [4, 5], и позволяет по тангенсу угла наклона изотермы SФ(CТв) определить СЕ капель НЭ по отношению к Фл. Значение СЕ составило 0.27 моль/моль, что в 2.7 раза превышает СЕ мицелл Твин 80 по отношению к Фл [5]. Таким образом, НЭ Твин 85 более эффективно инкорпорируют Фл по сравнению с мицеллами Твин 80. При этом повышение CТв в исследованном интервале позволило увеличить концентрацию Фл в водной среде по сравнению с его растворимостью в воде в 680–3400 раз.
Рис. 7.
Квазиравновесная изотерма солюбилизации Фл в НЭ Твин 85 с различным содержанием дисперсной фазы.
При известных значениях СЕ и растворимости Фл в дисперсионной среде (Sw) стандартная энергия Гиббса солюбилизации была рассчитана из соотношения [4]
(5)
$\Delta G^\circ = - RT{\kern 1pt} {\text{ln}}\left( {{{k}_{0}}CE{\text{/}}\left( {\left( {CE + 1} \right){{S}_{{\text{w}}}}} \right)} \right),$Исходя из того, что и солюбилизация Фл (рис. 7), и площадь межфазной поверхности НЭ линейно возрастают с ростом концентрации дисперсной фазы, можно предположить, что Фл локализован на поверхности капель. Для проверки этой гипотезы была использована УФ-спектроскопия, которая эффективна при определении полярности микроокружения солюбилизата, следовательно, и места его локализации [55, 56]. В связи с этим необходимо сопоставить спектры поглощения Фл, солюбилизированного в прямой НЭ Твин 85 (рис. 8), и в растворителях различной полярности (табл. 1). Максимум поглощения Фл, инкорпорированного в НЭ, наблюдается при 364 нм (рис. 8), что соответствует максимуму поглощения этого ЛВ, солюбилизированного в мицеллах Твин 80 (табл. 1). Таким образом, Фл находится в гидратированном слое полиоксиэтилированных цепей НПАВ, окружающем капли НЭ. Фл, являясь дигидропиридином, образует водородные связи (N–H⋅⋅⋅O) с оксиэтильными группами Твин 85, что удерживает его в гидратированном слое полиоксиэтилированных цепей. По-видимому, основной вклад в величину энергии Гиббса солюбилизации Фл в НЭ Твин 85 обусловлен именно водородными связями.
Рис. 8.
Спектр Фл, инкорпорированного в НЭ Твин 85 (CТв = 1 мас. %). Образец разбавлен водным раствором азида натрия в 10.17 раз. Спектр получен относительно НЭ с той же концентрацией Твин 85, но без Фл, разбавленной аналогичным образом.
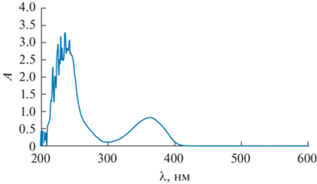
Таблица 1.
Значения длины волны (λмакс), соответствующие максимуму поглощения Фл в разных средах [5]
| Растворитель | λмакс, нм |
|---|---|
| Вода | 366 |
| Водные растворы ПЭГ400 (4–22 мас. %) | 366 |
| 5 мМ водный мицеллярный раствор Твин 80 | 364 |
| ПЭГ400 | 361 |
| Гептан | 345 |
Поскольку Фл фактически адсорбирован на поверхности капель НЭ, его адсорбцию (Г) и степень заполнения поверхности капель (θ) можно рассчитать, зная площадь межфазной поверхности НЭ (S, м2/л) и величину солюбилизации Фл: Г = SФ/S и θ = Г/Гmax (где предельная адсорбция Фл Гmax = = (3.5 ± 0.1) × 10–6 М [57]). Результаты расчетов для НЭ с различной концентрацией дисперсной фазы приведены в табл. 2, при этом средние значения адсорбции Фл и степени заполнения поверхности капель соответственно составили Г = (2.3 ± ± 0.15) × 10–6 моль/м2 и θ = 0.67 ± 0.04.
Таблица 2.
Адсорбция Фл и степень заполнения межфазной поверхности НЭ Твин 85
| CТв, мас. % | Г × 106, моль/м2 | θ |
|---|---|---|
| 1 | 2.39 | 0.68 |
| 2 | 2.05 | 0.59 |
| 3 | 2.57 | 0.73 |
| 4 | 2.42 | 0.69 |
| 5 | 2.23 | 0.64 |
Таким образом, разработаны стабильные в течение длительного времени НЭ простого состава, в которых Твин 85 является и дисперсной фазой, и стабилизатором. Эти НЭ солюбилизируют Фл более эффективно, чем мицеллы НПАВ. При этом почти 70% межфазной поверхности НЭ заполнено Фл вследствие формирования его молекулами водородных связей (N–H⋅⋅⋅O) с оксиэтильными группами Твин 85.
Кинетику массопереноса Фл частицами НЭ Твин 85 исследовали с помощью диффузионной ячейки Франца и УФ-спектроскопии. Мембрана, разделяющая донорную и акцепторную части ячейки, имела размер пор, на порядок превосходящий размер капель НЭ, и не лимитировала скорость массопереноса. Из акцепторной части ячейки периодически в течение нескольких часов отбирались пробы и анализировались методом УФ-спектроскопии без разбавления. Приемную среду (бинарная смесь вода/этанол, 60/40 мас. %) использовали в качестве раствора сравнения при спектрофотометрических измерениях.
В качестве примера на рис. 9 (кривая 1) представлен спектр прямой НЭ Твин 85 (3 мас. %) с солюбилизированным Фл, измеренный через 30 мин после начала эксперимента. В видимой области спектра наблюдается рассеяние (рис. 9, кривая 1). Очевидно, что в УФ-области спектр представляет собой сумму вкладов поглощения и рассеяния света. Для количественного определения концентрации Фл в приемной среде необходимо выделить вклад поглощения света. Для этого необходимо вычесть вклад рассеяния света в соответствии с процедурой, описанной ниже.
Рис. 9.
Спектры пробы, взятой из ячейки Франца через 30 мин после начала эксперимента, для НЭ Твин 85 (3 мас. %) с солюбилизированным Фл: 1 – эксперимент, 2 – рассеяние света, 3 – поглощение света.

• Для видимой области спектра, где наблюдается только рассеяние света, получали зависимость ln A от ln λ, которая, как показали расчеты, хорошо описывается линейным уравнением lnA = = −a ln λ + b. Коэффициенты для разных значений времени приведены в табл. 3.
Таблица 3.
Численные коэффициенты уравнения ln A = −a ln λ + b и коэффициент корреляции (R2) для НЭ Твин 85 (3 мас. %) с инкорпорированным Фл
| t, ч | a | b | R2 |
|---|---|---|---|
| 0.25 | 1.5191 | 7.3031 | 0.9956 |
| 0.5 | 1.5752 | 8.1098 | 0.9968 |
| 0.75 | 1.5780 | 8.3515 | 0.9976 |
| 1.0 | 1.5896 | 8.5481 | 0.9973 |
| 1.25 | 1.5939 | 8.7972 | 0.9977 |
| 2.5 | 1.6334 | 9.1983 | 0.9970 |
| 3.25 | 1.6233 | 9.2104 | 0.9969 |
• Полученную функцию A(λ) экстраполировали на УФ-область спектра (рис. 9, кривая 2), оценивая тем самым вклад рассеяния света.
• Вычитая из экспериментального спектра (рис. 9, кривая 1) вклад рассеяния (рис. 9, кривая 2), определяли вклад поглощения (рис. 9, кривая 3), которое может быть результатом поглощения света не только Фл, но и Твин 85.
• С целью оценки вклада Твин 85 в абсорбцию света при λ = 364 нм (максимум поглощения Фл в приемной среде) получали спектры для НЭ Твин 85 различной концентрации в водно-спиртовой смеси (в отсутствие Фл), в которых наблюдалось рассеяние света в видимой области и суперпозиция вкладов рассеяния и поглощения в УФ-области. Описанным выше способом исключали вклад рассеяния и получали спектры поглощения Твин 85 в данном бинарном растворителе, который оказался пренебрежимо малым при длине волны, соответствующей максимуму поглощения Фл (рис. 10).
Рис. 10.
Спектры поглощения Твин 85 в бинарном растворителе этанол/вода (40/60, мас. %) при различной концентрации этого НПАВ: 1 − 6.85 × 10–6, 2 − 1.02 × 10–5, 3 − 1.36 × 10–5, 4 − 2.35 × 10–5, 5 − 1.00 × × 10–4, 6 − 2.00 × 10–4 M.

• Концентрацию Фл рассчитывали на основе спектра поглощения для НЭ Твин 85 с солюбилизированным Фл (рис. 9, кривая 3).
Описанная выше процедура была выполнена для каждой пробы из акцепторной части ячейки Франца. Спектры поглощения Фл в приемной среде в различные моменты времени представлены на рис. 11.
Рис. 11.
Спектры поглощения, измеренные для оценки массопереноса Фл частицами прямой НЭ Твин 85 (3 мас. %) при различных значениях времени отбора проб (в часах), которые указаны в поле рисунка.

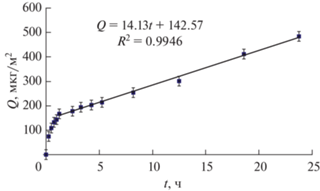
По результатам спектрофотометрических измерений было рассчитано количество этого ЛВ, которое переносится через единицу площади поверхности при различных значениях t (рис. 12). Видно, что при t > 1 ч скорость массопереноса Фл остается неизменной в течение всего времени наблюдения (25 ч). Таким образом, частицы НЭ являются эффективными переносчиками Фл в водной среде.
3.2. Ультрамикрогетерогенные полимерные матрицы с инкорпорированным фелодипином на основе прямых наноэмульсий Твин 85
С целью получения полимерных матриц для доставки Фл в качестве премикса использовали ДЭ масло1/вода/масло2, полученные при инкорпорировании прямой НЭ Твин 85, содержащей Фл, в раствор полимерного адгезива ПИБ в гептане (CПИБ = 40 мас. %). ДЭ готовили весовым методом при осторожном механическом перемешивании. Содержание НЭ Твин 85 (CТв = 5 мас. %) в ДЭ составляло 30 мас. %.
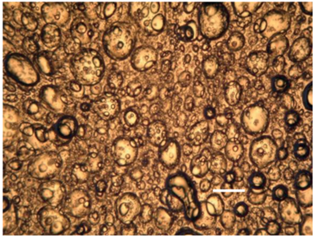
Из ДЭ методом полива с последующей сушкой получали полимерную пленку (см. раздел 2.2). Пленка обладала хорошей адгезией к поверхности кожи, была оптически прозрачна, ее ультрамикрогетерогенная структура наблюдалась только под микроскопом (рис. 13). Данные оптической микроскопии в поляризованном свете подтвердили отсутствие кристаллизации Фл в полимерной матрице. Содержание Фл в полимерной матрице рассчитывали, исходя из плотности ДЭ, толщины наносимого слоя и концентрации Фл в ДЭ. При толщине наносимого слоя 625 мкм (что соответствует 0.0625 см3 эмульсии/см2) содержание Фл в пленке составило 48.2 мкг/см2.
Рис. 13.
Микрофотография полимерной матрицы в неполяризованном свете (длина масштабного отрезка равна 200 мкм).
Кинетика выделения Фл из полимерной матрицы исследовалась с помощью ячейки Франца и УФ-спектроскопии (см. раздел 2.2). Пробы анализировали, не разбавляя, и каждый раз возвращали в ячейку. Спектры поглощения проб приемной среды, взятые через определенные промежутки времени, использовали для определения концентрации Фл в приемной среде (см. раздел 2.2). Количество Фл Q (мкг/см2), прошедшее через мембрану за время t, оценивали по формуле (2). Зависимость Q(t) приведена на рис. 14, который демонстрирует, что уже через полчаса устанавливается постоянная скорость высвобождения Фл, равная 5.26 мкг/(см2 ч). При этом практически все ЛВ высвобождается из пленки в течение 5 ч.

4. ЗАКЛЮЧЕНИЕ
Получены прямые наноэмульсии простого состава, в которых липофильное немицеллообразующее неионогенное ПАВ (Твин 85) является и дисперсной фазой, и стабилизатором. НЭ (dср ≅ 95 нм) длительное время (несколько месяцев) остаются в метастабильном состоянии, имеют высокоразвитую удельную поверхность (Sуд ∼ 63 м2/г), при этом площадь их межфазной поверхности (м2/л НЭ) линейно возрастает с увеличением концентрации дисперсной фазы. Солюбилизационная емкость НЭ Твин 85 по отношению к фелодипину составляет 0.27 моль/моль, что примерно в 3 раза превышает СЕ мицелл Твин 80 в водной среде. Установлено, что Фл локализован в окружающем капли НЭ гидратированном слое полиоксиэтилированных цепей Твин 85 за счет формирования его молекулами водородных связей N–H⋅⋅⋅O с оксиэтильными группами НПАВ, при этом молекулы ЛВ занимают примерно 70% площади межфазной поверхности НЭ. Количество инкорпорированного Фл можно увеличивать путем повышения концентрации дисперсной фазы. Исследование кинетики массопереноса Фл, инкорпорированного в НЭ, с использованием ячейки Франца и УФ-спектроскопии подтвердило эффективность наноразмерных капель Твин 85 в качестве переносчиков ЛВ в водной среде.
На основе прямых НЭ Твин 85 с инкорпорированным Фл и раствора полимерного адгезива (смеси полиизобутиленов с различной молекулярной массой и полибутена) в гептане получены двойные эмульсии масло1/вода/масло2, использованные в качестве премикса полимерных микрогетерогенных матриц. Полимерные пленки характеризуются хорошей адгезией к коже, отсутствием кристаллизации Фл и постоянной скоростью его высвобождения в течение нескольких часов.
Список литературы
Rupp C., Steckel H., Muller B.W. // Int. J. Pharm. 2010. V. 395. P. 272.
Rosen M.J. Surfactants and Interfacial Phenomena. New York: Wiley, 2004.
Ishii S., Ishikawa S, Mizuno N., Matsuoka K., Honda C., Endo K. // J. Colloid Interface Sci. 2008. V. 317. P. 115.
Zadymova N.M., Ivanova N.I. // Mosc. Univ. Chem Bull. 2013. V. 68. P. 110.
Задымова Н.М., Иванова Н.И. // Коллоид. журн. 2013. Т. 75. С. 179.
Malmsten M. // Handbook of Microemulsion Science and Technology / Eds. Kumar P., Mittal K.L. New York: Marcel Dekker, 1999. P. 755.
Lawrence M.J., Rees G.D. // Adv. Drug Deliv. Rev. 2000. V. 45. P. 89.
Gasco M.R. // Handbook of Microemulsion Science and Technology / Eds. Kumar P., Mittal K.L. New York: Marcel Dekker, 1999. P. 97.
Kreilgaard M. // Adv. Drug Deliv. Rev. 2002. V. 54. P. 77.
Patel M.R. Shen L., Hou X., Wang Z. et al. // Drug Deliv. 2021. V. 28. P. 2062.
Saini J.K., Nautiyal U., Kumar S. et al. // Int. J. Pharm. Med. Res. 2014. V. 2. P. 15.
Ali M.H., Moghaddam B., Kirby D.J. et al. // Int. J. Pharm. 2013. V. 453. P. 225.
Kragh-Hansen U., Maire M., Moller J.V. // Biophys. J. 1998. V. 75. P. 2932.
Immordino M.L., Dosio F., Cattel L. // Int. J. Nanomedicine. 2006. V. 1. P. 297.
Швец В.И., Краснопольский Ю.М. // Провизор. 2008. Вып. 3. С. 18.
Goyal P., Goyal K., Kumar S.G.V. et al. // Acta Pharm. 2005. V. 55. P. 1.
Yadav D., Sandeep K., Pandey D., Dutta R.K. // J. Biotechnol. Biomater. 2017. V. 7. № 4. https://doi.org/10.4172/2155-952X.1000277
Kim J.S. // J. Pharm. Investig. 2016. V. 46. P. 387.
Sharma D., Ali A.A.E., Trivedi L.R. // PharmaTutor. 2018. V. 6. № 2. P. 50.
Uchegbu F.I. // Int. J. Pharm. 1998. V. 172. P. 33.
Moghassemi S., Hadjizadeh A. // J. Control. Release. 2014. V. 185. P. 22.
Kumar P., Gannu R.P. // Acta Pharm. Sin. B. 2011. V. 1. P. 208.
Mandal S., Banerjee C., Ghosh S. et al. // J. Phys. Chem. 2013. V. 117. P. 6957.
Makeshwar K.B. // Asian J. Pharm. Res. 2013. V. 3. P. 16.
Muzzalupo R., Tavano L., Mesa C. // Int. J. Pharm. 2013. V. 458. P. 224.
Tavano L., Muzzalupo R., Mauro L. et al. // Langmuir. 2013. V. 29. P. 12638.
Pardakhty A., Moazeni E. // Nanomed. J. 2013. V. 1. P. 1.
Lovelyn C., Attama A.A. // J. Biomater. Nanobiotechnol. 2011. V. 2. P. 626.
Shah P., Bhalodia D., Shelat P. // Sys. Rev. Pharm. 2010. V. 1. P. 24.
Ravi T.P.U., Padma T. // Res. Biotechnol. 2011. V. 2. № 3. P. 1.
Kotta S., Khan A.W., Pramod K. et al. // Expert Opin. Drug Deliv. 2012. V. 9. № 5. P. 585.
Shakeel F., Ramadan W., Gargum H.M., Singh R. // Sci. Pharm. 2009. V. 78. P. 47.
Tadros T., Izquierdo P., Esquena J., Solans C. // Adv. Colloid Interface Sci. 2004. V. 108–109. P. 303.
Solans C., Izquierdo P., Nolla J. et al. // Curr. Opin. Colloid Interface Sci. 2005. V. 10. P. 102.
Mason T.G., Wilking J.N., Meleson K. et al. // J. Phys.: Condens. Matter. 2006. V. 18. P. 635.
Taylor P. // Adv. Colloid Interface Sci. 1998. V. 75. P. 107.
Щукин Е.Д., Перцов А.В., Амелина Е.А. Коллоидная химия. 4-ое изд. М.: Высш. школа, 2006.
Buscall R., Davis S.S., Potts D.S. // Colloid. Polym. Sci. 1979. V. 257. P. 636.
Higuchi W.I., Misra J. // J. Pharm. Sci. 1962. V. 51. P. 459.
Кабальнов А.С., Перцов А.В., Апросин Ю.Д., Щукин Е.Д. // Коллоид. журн. 1985. Т. 47. С. 1048.
Dario M.F., Soledade M., Santos C.S. et al. // Colloids Surf. A. 2016. V. 489. P. 256.
Capek I. // Adv. Colloid Interface Sci. 2004. V. 107. P. 125.
Weiss J., Canceliere C., McClements D.J. // Langmuir. 2000. V. 16. P. 6833.
Chien Y.W. // Transdermal Controlled Systemic Medications / Ed. by Chien Y.W. New York: Marcel Dekker, 1976. P. 1.
Williams A. Transdermal and Topical Drug Delivery. London: Pharmaceutical Press, 2003.
Huynh J., Aebi C. // Oregon DUR Board Newsletter. 2008. V. 10. № 8. P. 1.
Chein Y.W., Tojo K. Patent 4690683 USA. 1987.
Aguadisch L.M.J., Rankin F.S. Patent 4814184 USA. 1989.
Мюллер В. Патент РФ RU 2311908C2. 2002 // Б. И. 2007. № 34.
Handbook of Pressure-Sensitive Adhesives and Products. Applications of Pressure-Sensitive Products / Ed. by Benedek I., Feldstein M.M. Boca Raton: CRC Press, 2008. V. 1−3.
Задымова Н.М., Кармашева Н.В., Потешнова М.В., Цикурина Н.Н. // Коллоид. журн. 2002. Т. 64. С. 449.
Задымова Н.М., Цикурина Н.Н., Потешнова М.В. // Коллоид. журн. 2003. Т. 65. С. 347.
Задымова Н.М., Цикурина Н.Н., Руделев Д.С., Потешнова М.В. // Коллоид. журн. 2004. Т. 66. С. 175.
Tavares L., Shevchuk I., Alfonso M. et al. Patent 7018649 B2 USA. 2006.
Мицеллообразование, солюбилизация и микроэмульсии / Под ред. Миттела К.М.: Мир, 1980.
Bhat P.A., Rather G.M, Dar A.A. // J. Phys. Chem. B. 2009. V. 113. P. 997.
Задымова Н.М., Должикова В.Д., Харлов А.Е. // Коллоид. журн. 2020. Т. 82. С. 423.
Дополнительные материалы отсутствуют.
Инструменты
Коллоидный журнал