

Координационная химия, 2019, T. 45, № 1, стр. 58-63
Метилпропиолатный кластерный комплекс (Ph4P)2[W6I8(C≡C–C(O)OCH3)6]М. А. Михайлов, П. А. Абрамов, А. Д. Миронова, М. Р. Галлямов, Д. Г. Шевень, В. В. Первухин, М. Н. Соколов
М. А. Михайлов 1, *, П. А. Абрамов 1, 2, А. Д. Миронова 1, М. Р. Галлямов 1, Д. Г. Шевень 1, В. В. Первухин 1, М. Н. Соколов 1, 2, 3
1 Институт неорганической химии им. А.В. Николаева СО РАН
, Россия
2 Новосибирский государственный университет
, Новосибирск, Россия
3 Казанский федеральный университет
, Казань, Россия
* E-mail: mikhajlovmaks@yandex.ru
Поступила в редакцию 15.03.2018
После доработки 15.06.2018
Принята к публикации 10.05.2018
Аннотация
В реакции (Bu4N)2[W6I14] с метилпропиолатом серебра AgC≡C–C(O)OCH3 получен иодидный металлорганический кластерный комплекс (Bu4N)2[W6I8(C≡C–C(O)OCH3)6] (I), аналог ранее описанного (Bu4N)2[Mo6I8(C≡C–C(O)OCH3)6]. Кристаллическая структура установлена для тетрафенилфосфониевой соли (Ph4P)2[W6I8(C≡C–C(O)OCH3)6] (II). По данным РСА (СIF file CCDC № 1829205), атомы вольфрама в II координированы концевыми атомами углерода метилпропиолатных лигандов на расстояниях W–C 2.220(12)–2.268(14) Å. Метилпропиолатные комплексы охарактеризованы данными электроспрей масс-спектрометрии, ЯМР на ядрах 1Н и 13С, элементного анализа и ИК-спектроскопии.
Октаэдрические галогенидные кластерные комплексы [{M6(μ3-I)}8L6] (M = Mo, W; X = I; L = = органический или неорганический лиганд) обладают рекордными фотофизическими свойствами среди координационных соединений [1, 2]. Сюда относятся относительно узкая полоса эмиссии в красной области спектра, возможность возбуждения люминесценции видимым светом, высокие квантовые выходы, высокая конверсия триплетного кислорода в синглетный. Синтезирован достаточно представительный набор иодидных кластерных комплексов молибдена с концевыми лигандами L различной природы (галогениды, О-, N-, S-, C-доноры). Предложено несколько перспективных областей применения для люминесцентных материалов и реагентов на основе иодидных кластеров молибдена [3–22]. Октаэдрические кластеры вольфрама изучены в гораздо меньшей степени, чем кластеры молибдена, при этом иодидные кластеры вольфрама изучены в минимальной степени. Помимо ожидаемого сходства в фотофизических свойствах [23] иодидные кластеры вольфрама вызывают интерес в силу относительной стабильности парамагнитного 23-электронного состояния (потенциал пары [W6I14]2–/[W6I14]– в ацетонитриле составляет всего 0.57 В относительно стандартного каломельного электрода [24]), а также уникального сочетания 14 тяжелых атомов в кластерном ядре {W6I8}4+, что обеспечивает высокий фактор рассеяния рентгеновских лучей на данных атомах. Подобное обстоятельство делает данные кластерные комплексы перспективными рентгеноконтрастными веществами [25]. Тем не менее к настоящему времени известно лишь ограниченное количество октаэдрических иодидных кластерных комплексов вольфрама. Сюда относятся, в первую очередь, сам W6I12, смешанный иодид-хлорид W6I8Cl4 [26, 27] и галогенидные комплексы [{W6I8}X6]2– (X = Cl, Br, I) [28]. Замещение концевых атомов иода позволяет получить трифлатные [25], тозилатные [29] и карбоксилатные комплексы, для которых подробно изучены фотофизические свойства [30, 31].
В настоящей работе мы сообщаем о получении первого металлоорганического производного {W6I8}4+ – пропиолатного комплекса [W6I8(C≡C–C(O)OCH3)6]2–, выделенного и охарактеризованного в виде тетрабутиламмонийной и тетрафенилфосфониевой солей.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исходный комплекс (Bu4N)2[W6I14] синтезировали согласно оптимизированной методике [31]. Метилпропиолат серебра AgC≡C–C(O)OCH3 получали в виде белого порошка по обменной реакции между коммерчески доступными HC≡C–C(O)OCH3 и AgNO3 в присутствии Et3N в растворе ацетонитрила [16]. Органические растворители ацетонитрил и хлористый метилен очищали по стандартным процедурам. Синтезы проводили в атмосфере аргона, перекристаллизацию можно осуществлять на воздухе.
Синтез (Bu4N)2[W6I8(C≡C–C(O)OCH3)6] (I). (Bu4N)2W6I14 (0.33 г, 0.10 ммоль) растворяли в 10 мл CH2Cl2 в небольшой конической колбе, предварительно обмотанной алюминиевой фольгой для изоляции от воздействия света. К полученному светло-оранжевому раствору добавляли десятикратный мольный избыток твердого AgC≡C–C(O)OCH3 (0.186 г, 0.98 ммоль). Реакционную смесь перемешивали на магнитной мешалке 2 сут, фильтровали через бумажный фильтр. Фильтрат упаривали на роторном испарителе, образовавшееся густое масло растворяли в ацетонитриле. При медленном насыщении ацетонитрила парами диэтилового эфира выкристаллизовывается продукт в виде светло-оранжевых кристаллов. Выход 30 мг (10%).
ИК-спектр (KBr; ν, см–1): 2958 ср, 2873 ср, 2234 сл, 2089 с, 1865 сл, 1678 с, 1511 сл, 1478 ср, 1456 ср, 1430 ср, 1378 сл, 1209 с, 1108 ср, 1059 ср, 1006 ср, 862 ср, 795 сл, 756 ср, 743 ср, 648 сл, 554 ср. Электроспрей-масс-спектр (MeCN, m/z): средн. 1309.1 ([W6I8(C≡C–C(O)OCH3)6]2–; выч. 1309.0), средн. 2859.3 ({(Bu4N)[W6I8(C≡C−C(O)OCH3)6]}–; выч. 2859.3). Спектр ЯМР 13C ((CD3)2SO; δ, м.д.): 152.14 (С=O-группировка лиганда), 132.49 и 112.93 (С≡С-группировка лигандов), 58.47 (катионов (Bu4N)+) 52.58 (OCH3-группировка лигандов), 23.99, 20.14, 14.42 (катионов (Bu4N)+ соответственно). Спектр ПМР ((CD3)2SO; δ, м.д., нормировано на 24 протона СН3-групп Bu4N+): 3.58 (с., 18 H, ОСН3-группы координированного пропиолата).
Качественные монокристаллы для проведения РСА получали в виде Рh4Р-соли II диффузией паров диэтилового эфира в ацетонитрильный раствор (Bu4N)2[W6I8(C≡C–C(O)OCH3)6] и Ph4PBr.
ИК-спектры записывали на спектрометрах Scimitar FTS 2000 и Specord IR 75 в области 4000–400 см–1. Спектр ЯМР 13С записывали на спектрометре Bruker Avance 500 при комнатной температуре c Si(CH3)4 в качестве внутреннего стандарта. Элементный анализ выполняли в Аналитической лаборатории ИНХ СО РАН.
Масс-спектрометрические данные получали на жидкостном хроматографе – масс-спектрометре (LC-MC) фирмы Agilent (6130 Quadrupole MS, 1260 infinity LC). Анализ проводили в диапазоне масс 350–3000 а.е.м. как для положительно, так и для отрицательно заряженных ионов в режиме SCAN. В качестве источника ионизации использовали электрораспыление. Осушителем служил поток газообразного азота, температура 350°C, скорость потока 7 л/мин; давление на распылителе (азот) 60 фунтов на кв. дюйм; напряжение на капилляре 4000 В. С целью сохранить слабо связанные формы в масс-спектрах, напряжение на фрагменторе было нулевым во всех экспериментах. Раствор исследуемого соединения (5 мкл) в дейтерированном ацетонитриле с концентрацией ~10–4 г/мл вводили в подвижную фазу (ацетонитрил “ос. ч.”) со скоростью потока 0.4 мл/мин, распыляли и ионизировали. Экспериментальные пики сравнивали с расчетными, в том числе и по изотопному распределению. Для расчетов использовали программу Molecular Weight Calculator by Matthew Monroe.
РCA. Строение соединения II установлено по стандартной методике на дифрактометре Bruker-Apex Duo (MoKα, λ = 0.71073 Å, графитовый монохроматор). Интенсивности отражений измерены методом φ- и ω-сканирования узких (0.5°) фреймов при T = 150 K. Поглощение учтено эмпирически по программе SADABS [32]. Структура расшифрована прямым методом и уточнена полноматричным МНК по F 2 в анизотропном для неводородных атомов приближении по алгоритму SHELX 2014\7 в программе ShelXle [33]. Длины связей C≡C (1.2 Å) были зафиксированы из-за некорректного учета поглощения, что привело к аномальным размерам тепловых эллипсоидов атомов углерода. Основные длины связей в кластерном анионе комплекса II (Å) приведены в табл. 1. Кристаллографические данные и результаты уточнения структуры комплекса II приведены в табл. 2. Координаты атомов и другие параметры структур депонированы в Кембриджском банке структурных данных (CCDC № 1829205; http://www.ccdc.cam.ac.uk/data_request/cif).
Таблица 1.
Основные длины связей в кластерном анионе комплекса II
| Связь | d, Å | Cвязь | d, Å |
|---|---|---|---|
| W(1)–W(2) | 2.6876(7) | I(2)–W(3) | 2.8038(10) |
| W(1)–W(2)i | 2.6934(7) | I(3)–W(1) | 2.7951(9) |
| W(1)–W(3)i | 2.6886(7) | I(3)–W(2)i | 2.8041(9) |
| W(1)–W(3) | 2.6906(7) | I(3)–W(3)i | 2.8144(10) |
| W(2)–W(3) | 2.6767(7) | I(4)–W(1) | 2.8123(9) |
| W(2)–W(3)i | 2.6920(7) | I(4)–W(2)i | 2.8082(10) |
| I(1)–W(1) | 2.7890(9) | I(4)–W(3) | 2.8052(10) |
| I(1)–W(2) | 2.8107(9) | C(2)–W(3) | 2.247(13) |
| I(1)–W(3)i | 2.7881(10) | C(3)–W(2) | 2.268(14) |
| I(2)–W(1) | 2.7989(9) | C(4)–W(1) | 2.220(12) |
| I(2)–W(2) | 2.8155(10) |
Таблица 2.
Кристаллографические характеристики и детали дифракционного эксперимента для соединения II
| Параметр | Значение |
|---|---|
| Брутто-формула | C72H58O12P2I8W6 |
| М | 3295.42 |
| Температура, K | 150.0(2) |
| Сингония | Триклинная |
| Пр. гр. | P1 |
| а, Å | 11.5267(11) |
| b, Å | 13.0563(12) |
| c, Å | 13.4785(15) |
| α, град | 87.802(4) |
| β, град | 88.936(4) |
| γ, град | 75.980(3) |
| V, Å3 | 1966.5 (3) |
| Z | 1 |
| μ, мм–1 | 11.976 |
| Размеры кристалла (мм) | 0.30 × 0.04 × 0.02 |
| Область сбора данных по θ, град | 1.6–26.4 |
| Интервалы индексов отражений | –14 ≤ h ≤ 14, –16 ≤ k ≤ 16, –16 ≤ l ≤ 16 |
| Измерено отражений | 22 051 |
| Независимых отражений (Rint) | 8058 (0.057) |
| Отражений с I ≥ 2σ(I) | 4869 |
| Число уточняемых параметров | 451 |
| GOOF | 0.98 |
| R1 (по |F|2 для отражений с I > 2σ(I)) | 0.043 |
| wR2 (по |F|2 для всех отражений) | 0.103 |
| Остаточная электронная плотность (min/max), e/Å3 | –1.42/1.87 |
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Кластерная комплексная соль I получена в результате обменной реакции (Bu4N)2[W6I14] с солью серебра AgC≡C–C(O)OCH3 при комнатной температуре в хлористом метилене по аналогии с молибденовым кластером (Bu4N)2[Мо6I8(C≡C–C(O)OCH3)6] [16]. Образование нерастворимого AgI обеспечивало необходимое смещение равн-овесия химической реакции. Использование AgC≡C–C(O)OCH3, генерированного in situ [16], в обменной реакции не дает никаких преимуществ. Выход продукта не превышает 10–15%, независимо от того, используется ли избыток AgC≡C–C(O)OCH3 или же берется стехиометрическое количество (6 экв.). Это существенно ниже, чем для молибденового кластера; причины этого неясны. Возможно, низкий выход I можно объяснить образованием нерастворимых побочных продуктов реакции из-за связывания кластерных комплексных анионов с ионами серебра, которые выпадают в осадок совместно с AgI. Действительно, в некоторых образцах в электроспрей-масс- спектрах наблюдались пики, отвечающие анионам {Ag[W6I8(C≡C–C(O)OCH3)6]}–.
Избыток соли серебра против стехиометрии (10 экв. вместо 6) способствует более полному и более быстрому прохождению реакции, полноту прохождения которой удобно контролировать с помощью электроспрей-масс-спектрометрии, фиксируя сигнал, соответствующий молекулярному дианиону целевого соединения [W6I8(C≡C–C(O)OCH3)6]2–. В электроспрей-масс-спектре ацетонитрильного раствора I наблюдаются сигналы от двухзарядных анионов [W6I8(C≡C–C(O)OCH3)6]2– и от аддуктов этих частиц с тетрабутиламмонийными катионами {(Bu4N)[W6I8(C≡C–C(O)OCH3)6]}–, для которых теоретические изотопные распределения хорошо накладываются на экспериментально найденные с совпадением соответствующих “центров масс” сигналов: пику с m/z = 1309.1 соответствует частица [W6I8(C≡C–C(O)OCH3)6]2– (m/z(выч.) = 1309.0); пику с m/z = 2859.3 соответствует аддукт {(Bu4N)[W6I8(C≡C–C(O)OCH3)6]}– (m/z(выч.) = 2859.3). Помимо указанных интенсивных сигналов, в неперекристаллизованных образцах могут наблюдаться минорные сигналы от [W6I8(C≡C–C(O)OCH3)5Cl]2– и аддукта {TBA[W6I8(C≡C–C(O)OCH3)5Cl]}–, источником которых может быть обменная реакция с хлористым метиленом или следами HCl, от которых трудно избавиться в этом растворителе. Соединения I и II – умеренно устойчивые на воздухе вещества, растворимые в обычных органических растворителях (кроме углеводородных).
Спектр ЯМР 13С раствора I в (CD3)2SO (рис. 1а) содержит сигналы от С-координированных пропиолатных лигандов и от катионов тетрабутиламмония. На этом рисунке приведен спектр ЯМР 13С для НC≡C–C(O)OCH3 в (CD3)2SO (рис. 1б). Как видно из сопоставления спектров, координация пропиолатных лигандов приводит к изменению положения всех химических сдвигов. При этом наиболее сильное смещение (в область сильного поля) наблюдается для атомов при тройной связи (δ = 132.49 и 112.93 м.д. (С≡С-группировка координированных лигандов) в сравнении с δ = = 79.14 и 74.93 м.д. (С≡С-группировка свободного пропиолата)), тогда как смещение химических сдвигов (в область слабого поля) для карбонильной и метоксидных групп осуществляется не столь сильно при координации (δ = 152.14 и 52.58 м.д. (=С=O и –OMe-группировки координированных лигандов соответственно) и δ = 153.19 и 53.33 м.д. (=С=O и –OMe-группировки свободного пропиолата)). Это согласуется с тем, что наиболее сильному изменению электронной плотности подвержены атомы углерода при тройной связи из-за образования связи W–C и сопряжения по тройной связи. Смещение химических сдвигов влево означает уменьшение электронной плотности на атомах С≡С и ее делокализацию при участии кластерного ядра {W6I8}4+, которое ведет себя как сильная кислота Льюиса. Спектр ПМР раствора I в (CD3)2SO содержит химический сдвиг δ = 3.58 м.д. от протонов метоксидной группы лигандов (против δ = 3.74 м.д. – для свободного пропиолата) (рис. 2).
Рис. 1.
Cпектры ЯМР 13С растворов: комплекса I в (CD3)2SO (а), раствора HC≡C–C(O)OCH3 в (CD3)2SO (б).
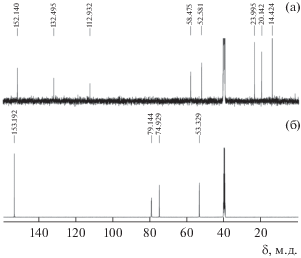
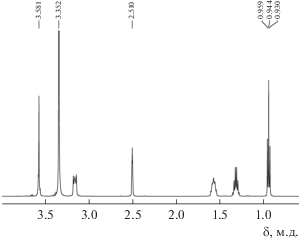
Рис. 2.
Cпектр ПМР раствора комплекса I в (CD3)2SO. Указаны хим. сдвиги протонов (δ, м.д.): CH3O-групп пропиолатных лигандов (3.58), воды (3.35), растворителя (2.51 м.д.) метильных групп катионов Bu4N+ (триплет справа c центром 0.94 м.д.). Остальные хим. сдвиги соответствуют протонам от трех метиленовых групп тетрабутиламмония.
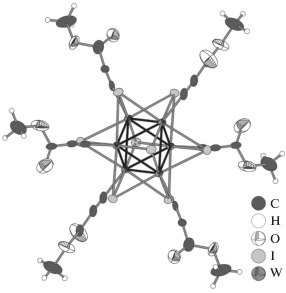
Монокристаллы, пригодные для РСА, удалось получить при замене катионов Bu4N+ на стерически более жесткие и менее склонные к разупорядочению катионы Ph4P+ при медленном насыщении парами диэтилового эфира смеси I и Ph4PBr в ацетонитриле. Согласно данным РСА, пропиолатные лиганды координированы к кластерному ядру концевыми атомами углерода при тройной связи вдоль прямой линии. Вид аниона [W6I8(C≡C–C(O)OCH3)6]2– и кристаллической упаковки комплекса II показан на рис. 3, 4 соответственно. Кристаллы II изоструктурны кристаллам кластерного соединения (Ph4P)2[Mo6I8(C≡C–C(O)OCH3)6] (III) [16].
Рис. 3.
Строение кластерного аниона [W6I8(C≡C–C(O)OCH3)6]2– (эллипсоиды атомных смещений 50%-ной вероятности).
Расстояния М–С при переходе от комплекса {Mo6I8}4+ к {W6I8}4+ несколько увеличиваются – c 2.180(5)–2.194(2) (cредн. 2.188) [16] до 2.220(12)–2.268(14) Å (средн. 2.245) Å (табл. 1) соответственно. Расстояния W–C в II больше, чем Мo–С в III и в металлорганическом комплексе с координированным этиленом в транс-[{Mo6Cl8}Cl2(C2H5)2 (PBu3)2] · 2PhCH3 (2.21(3) Å) [34] и сопоставимы с межатомными расстояниями Mo–C в комплексе с координированными бензильными лигандами в транс-[{Mo6Cl8}(PBu3)2(CH2Ph)4] (2.239(14)–2.271(15) Å) [35]. Сообщалось о получении термически устойчивых, но светочувствительных кластерных металлорганических комплексов транс-[{W6Cl8}Cl2(C2H5)2(PR3)2] (R = н-C4H9, н-C5H11) и транс,мер-[{W6Cl8}Cl(C2H5)3(PR3)2] (R = н-C4H9, н-C5H11) с η1-координированным этиленом (по данным ЯМР 13С), для которых, однако, РСА не проводили [36]. Межатомные расстояния W–W, W–(μ3-I) кластерных ядер в II характерны для ядер {W6I8}4+ в комплексах [{W6I8}L6] c различной природой лигандов [24–31] и приведены в табл. 1.
Таким образом, в настоящей работе получено первое металлорганическое производное для о-ктаэдрических галогенидных кластеров вольфрама.
Работа выполнена при финансовой поддержке ФАНО России (Программа фундаментальных научных исследований пункт V.44.4.5).
Список литературы
Sokolov M.N., Mihailov M.A., Peresypkina E.V. et al. // Dalton Trans. 2011. V. 40. P. 6375.
Elistratova J., Mikhailov M., Burilov V. et al. // RSC Adv. 2014. V. 4. P. 27922.
Kirakci K., Kubat P., Dusek M. et al. // Eur. J. Inorg. Chem. 2012. P. 3107.
Efremova O.A., Shestopalov M.A., Chirtsova N.A. et al. // Dalton Trans. 2014. V. 43. P. 6021.
Kirakci K., Šícha V., Holub J. et al. // Inorg. Chem. 2014. V. 53. P. 13012.
Kirakci K., Fejfarová K., Kučeráková M., Lang K. // Eur. J. Inorg. Chem. 2014. P. 2331.
Cordier S., Grasset F., Molard Y. et al. // J. Inorg. Organomet. Polym. Mater. 2015. V 25. P. 189.
Elistratova J., Burilov V., Mustafina A. et al. // Polymer. 2015. V. 72. P. 98.
Prevot M., Amela-Cortes M., Manna S.K. et al. // J. Mater. Chem. C. 2015. V. 3. P. 5152.
Amela-Cortes M., Paofai S., Cordier S. et al. // Chem. Commun. 2015. V. 51. P. 8177.
Prevot M., Amela-Cortes M., Manna S.K. et al. // Adv. Funct. Mater. 2015. V. 25. P. 4966.
Robin M., Kuai W., Amela-Cortes M. et al. // ACS Appl. Mater. Interfaces. 2015. V. 7. P. 21975.
Solovieva A.O., Vorotnikov Y.A., Trifonova K.E. et al. // J. Mater. Chem. B. 2016. V. 4. P. 4839.
Kirakci K., Kubát P., Fejfarová K. et al. // Inorg. Chem. 2016. V. 55. P. 803.
Vorotnikov Y.A., Efremova O.A., Vorotnikova N.A. et al. // RSC Adv. 2016. V. 6. P. 43367.
Sokolov M.N., Mikhailov M.A., Brylev K.A. et al. // Inorg. Chem. 2013. V. 52. P. 12477.
Sokolov M.N., Mikhailov M.A., Virovets A.V. et al. // Russ. Chem. Bull. 2013. V. 62. P. 1764.
Mikhailov M.A., Brylev K.A., Abramov P.A. et al. // Inorg. Chem. 2016. V. 55. P. 8437.
Mikhailov M.A., Brylev K.A., Virovets A.V. et al. // New J. Chem. 2016. V. 40. P. 1162.
Beltran A., Mikhailov M., Sokolov M.N. et al. // J. Mater. Chem. B. 2016. V. 4. P. 5975.
Felipe-Leon C., Arnau del Valle C., Perez-Laguna V. et al. // J. Mater. Chem. B. 2017. V. 5. P. 6058.
Mikhaylov M.A., Abramov P.A., Komarov V.Yu., Sokolov M.N. // Polyhedron. 2017. V. 122. P. 241.
Zietlow T.C., Schaefer W.P., Sadeghi B. et al. // Inorg. Chem. 1986. V. 25. P. 2195.
Mussell R.D., Nocera D.G. // Inorg. Chem. 1990. V. 29. P. 3711.
Franolic J.D., Long J.R., Holm R.H. // J. Am. Chem. Soc. 1995. V. 117. P. 8139.
Strobele M., Meyer H.-J. // Inorg. Chem. 2017. V. 56. P. 5880.
Strobele M., Enseling D., Jüstel T., Meyer H.-J. // Z. Anorg. Allg. Chem. 2016. V. 642. P. 1435.
Hogue R.D., McCarley R.E. // Inorg. Chem. 1970. V. 9. P. 1354.
Mikhailov M.A., Gushchin A.L., Gallyamov M.R. et al. // Russ. J. Coord. Chem. 2017. V. 43. P. 172.
Riehl L., Seyboldt A., Ströbele M. et al. // Dalton Trans. 2016. V. 45. P. 15500.
Sokolov M.N., Brylev K.A., Abramov P.A. et al. // Eur. J. Inorg. Chem. 2017. P. 4131.
Sheldrick G.M. SADABS. Göttingen (Germany): Univ. of Göttingen, 1996.
Hübschle C.B., Sheldrick G.M., Dittrich B. // J. Appl. Cryst. 2011. V. 44. P. 1281.
Saito T., Nishida M., Yamagata T. et al. // Inorg. Chem. 1986. V. 25. P. 1111.
Yamagata T., Okiyama H., Imoto H., Saito T. // Acta Crystallogr. C. 1997. V. 53. P. 859.
Saito T., Manabe H., Yamagata T., Imoto H. // Inorg. Chem. 1987. V. 26. P. 1362.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия