

Координационная химия, 2019, T. 45, № 10, стр. 635-640
Разрыв связей B−C и образование аниона [PhBH3]− в реакции гидридного комплекса Yb(II) с BPh3
Д. М. Любов 1, И. В. Басалов 1, А. С. Шавырин 1, А. В. Черкасов 1, А. А. Трифонов 1, 2, *
1 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
2 Институт элементоорганических соединений им. А.Н. Несмеянова РАН
Москва, Россия
* E-mail: trif@iomc.ras.ru
Поступила в редакцию 28.03.2019
После доработки 04.04.2019
Принята к публикации 22.04.2019
Аннотация
Реакция амидинатгидридного комплекса двухвалентного иттербия [(Amd)YbII(μ2-H)]2 (Amd = = {трет-BuC(NC6H3-изо-Pr2-2,6)2}) с BPh3 протекает с разрывом связей B−C и приводит к образованию комплекса Yb(II)[(Amd)YbII(μ2-H3BPh)]2, содержащего фенилтригидроборатный анион [PhBH3]−. Методом РСА (СIF file CCDC № 1902290) показано, что в комплексе анионы [PhBH3]− мостиковые, при этом для амидинатного лиганда сохраняется “неклассический” κ1-амидо-η6-ареновый тип координации на ион Yb2+.
Уникальная реакционная способность гидридных комплексов лантанoидов в стехиометрических реакциях наряду с впечатляющей каталитической активностью в целом ряде превращений ненасыщенных субстратов дали мощный толчок стремительному развитию этой области химии [1–7]. Первые сообщения о синтезе бис(циклопентадиенил)моногидридных производных лантаноидов появились в 1980-х гг. [8–10]. Позднее были получены их дигидрдные аналоги полу-сэндвичевого ряда [11–15]. К настоящему времени известно большое число моногидридных [16–20], дигидридных [21–29], катионных гидридных [30–34], а также алкилгидридных [35–40] производных редкоземельных металлов, стабилизированных различными типами нециклопентадиенильных лигандов [41–43]. Однако все эти успехи в области химии гидридов относятся к производным трехвалентных лантанoидов, в то время как гидридные комплексы этих металлов в двухвалентном состоянии до сих пор остаются редкими, а их реакционная способность практически неисследованной.
К настоящему времени известно всего три гидридных комплекса лантанoидов в степени окисления +2. В [44] приведен первый пример – гидридный комплекс [(Tp-трет-Bu)Yb(μ2-H)]2 (Tp-трет-Bu = HB(C3HN2-трет-Bu-3-Me-5)3), стабилизированный объемным трис(пиразолил)боратным лигандом. Авторы исследовали реакционную способность полученного соединения в ряде стехиометрических реакций с CH-кислотами (терминальные ацетилены, дипивалоилметан, замещенный циклопентадиен), а также продемонстрировали возможность присоединения связей Yb−H по кратным связям С–С и C–O. В [45] синтезирован гидридный комплекс [{HC(C(Me)NC6H3-изо-Pr2-2,6)2}Yb(μ2-H) (THF)]2, содержащий объемный β-дикетиминатный лиганд, оказавшийся активным катализатором гидросилилирования олефинов. Несколько позднее были опубликованы данные о синтезе амидинатгидридного комплекса [(Amd)Yb(µ2-H)]2 (I), где Amd = {трет-BuC(NC6H3-изо-Pr2-2,6)2} [46], в котором обнаружен “неклассический” κ1‑амидо-η6-ареновый тип координации амидинатного лиганда на ион Yb2+, отличающийся удивительно устойчивым взаимодействием Yb(II)−η6-арен. На примере реакций гидридного комплекса [(Amd)Yb(µ2-H)]2 с одноэлектронными окислителями было показано, что гидридный лиганд – более сильный восстановитель по сравнению с ионом Yb2+ [46, 47]. Совсем недавно были получены первые катионные гидридные комплексы Yb(II) $[({\text{M}}{{{\text{e}}}_{{\text{4}}}}{\text{TACD}}){\text{Yb}}(\mu {\text{ - H}})]_{{\text{2}}}^{{{\text{2}} + }}[{\text{BA}}{{{\text{r}}}_{{\text{4}}}}]_{{\text{2}}}^{ - }$ (Me4TACD = 1,4,7,10-тетраметил-1,4,7,10-тетраазациклододекан) [48], оказавшиеся эффективными катализаторами гидрирования и гидросилилирования гексена-1, а также обмена между H2 и D2 [48].
Однако основные свойства связи Yb(II)−H остаются практически неизученными. С целью исследования способности гидридного аниона в комплексах лантанoидов к координации с кислотами Льюиса как возможного синтетического подхода к “расщеплению” их димерной структуры и образованию мономерных гидридных частиц, в настоящей работе исследована реакция димерного гидридного комплекса Yb(II) [(Amd)Yb(µ2-H)]2 с кислотой Льюиса BPh3. Оказалось, что данное взаимодействие протекает с разрывом связей С–В и приводит к образованию димерного комплекса [(Amd)YbII(μ2-H3BPh)]2, содержащего мостиковые фенилтригидроборатные анионы.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез комплексов проводили в условиях, исключающих контакт с кислородом и влагой воздуха, по стандартной технике Шленка. Толуол сушили над бензофенонкетилом натрия, гексан – над металлическим натрием, затем тщательно дегазировали и конденсировали в вакууме в реакционную ампулу непосредственно перед использованием. Комплекс I получали в соответствии с ранее опубликованной методикой [46].
Синтез [(Amd)Yb(µ-H3BPh)]2 (II). Раствор BPh3 (0.163 г, 0.674 ммоль) в 10 мл толуола добавляли при перемешивании к I (0.400 г, 0.337 ммоль) в 20 мл толуола. Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Цвет раствора менялся с темно-фиолетового на темно-красный. Толуол удаляли в вакууме, а к твердому остатку добавляли гексан (20 мл). Нерастворимый осадок отделяли от растворимых в гексане продуктов центрифугированием и растворяли в свежей порции толуола. Медленное концентрирование полученного раствора в толуоле при комнатной температуре привело к образованию красно-коричневых кристаллов комплекса II. Маточный раствор отделяли декантацией, а кристаллы сушили в вакууме в течение 20 мин. Выход комплекса II 0.070 г (15%). Концентрирование гексанового раствора при комнатной температуре приводило к образованию изумрудно-зеленых кристаллов комплекса (Amd)2Yb (III) с выходом 0.068 г (10% в расчете на количество металла, взятого в реакцию).
| Найдено, %: | C 61.88, | H 7.79, | N 4.01, | Yb 24.87. |
| Для C70H102B2N4Yb2 (M = 1367.32) | ||||
| вычислено, %: | C 61.49, | H 7.52, | N 4.10, | Yb 25.31. |
ЯМР 1H для II (400 МГц, бензол-d6, 293 K; δ, м.д.): 0.92 (уш. с., 18H, CH-трет-Bu), 1.34 (м., 24H, CH3-изо-Pr), 1.60–2.23 (уш. м., 6H, BH3), 3.11 (м., 2H, CH-изо-Pr), 3.41 (м., 2H, CH-изо-Pr), 6.90–7.32 (м., 22H, CHAr и BPh). ЯМР 13С{1H} для II (100 МГц, бензол-d6, 293 K; δ, м.д.): 20.4 (с., CH3-изо-Pr), 23.0 (с., CH3-изо-Pr), 26.0 (с., CH3-изо-Pr), 28.4 (с., CH-изо-Pr), 29.5 (с., CH3-изо-Pr), 31.5 (с., CH3-трет-Bu), 42.7 (с., C-трет-Bu), 118.6 (с., CHAr), 123.0 (с., CHAr), 123.5 (с., CHAr), 124.2 (с., CHAr), 126.2 (уш. с., CHBPh), 133.0 (уш. с., CHBPh), 137.7 (уш. с., CHBPh), 142.5 (с., CAr), 143.4 (с., CAr), 144.2 (с., CAr), 145.7 (уш. с., CBPh), 147.6 (с., CAr), 169.8 (с., NCN). ЯМР 11B{1H} для II (128.4 МГц, бензол-d6, 293 K; δ, м.д.): −21.0.
Спектры ЯМР 1H и 13С{1H} регистрировали на приборах Bruker DPX 200 MHz и BrukerAvance III 400 MHz. Химические сдвиги приведены в миллионных долях по отношению к известным сдвигам остаточных протонов дейтерированных растворителей. Двумерные 11B–1H корреляционные эксперименты проводили с использованием стандартной ge-HSQC последовательности (hsqcetgp) с параметрами, оптимизированными для проведения 11B–1H эксперимента. Элементный анализ выполняли на приборе Perkin-ElmerSeries II CHNS/O Analyser 2400. Содержание лантанида определяли методом комплексонометрического титрования (Трилон Б) с использованием ксиленолового оранжевого в качестве индикатора.
РСА монокристаллического образца II (0.25 × × 0.21 × 0.09 мм) проведен на дифрактометре Bruker Smart Apex (МоКα-излучение, λ = 0.71073 Å, ω-сканирование, T = 100 K, θ = 1.71°–26.04°). Измерение и интегрирование экспериментальных наборов интенсивностей, учет поглощения и уточнение структур проведены с использованием программных пакетов Smart [49], SADABS [50] и SHELX [51]. Кристаллы II (C70H102B2N4Yb2, M = 1367.25) моноклинные, пр. гр. P21/n, a = 10.8191(5), b = = 14.7527(7), c = 20.3110(10) Å, β = 92.7530(10)°, V = 3238.1(3) Å3, Z = 2, ρ(выч.) = 1.402 г/см3, μ = = 2.913 мм–1. Измерено 27 388 отражений, 6383 независимых (Rint = 0.0351) использованы для уточнения 372 параметров структуры полноматричным МНК по F2hkl в анизотропном приближении для неводородных атомов. Атомы водорода фенилтригидроборатных групп найдены из разностного синтеза Фурье электронной плотности и уточнены с некоторыми геометрическими ограничениями (расстояния B–H и H...H вырoвнены между собой с помощью инструкций DFIX и SADI). Остальные атомы водорода помещены в геометрически рассчитанные положения. Все атомы Н уточнены изотропно с фиксированными тепловыми параметрами U(H)изо = 1.2U(C)экв (U(H)изо = = 1.5U(C)экв для метильных групп). После финального уточнения wR2 = 0.0695 и S(F 2) = 1.028 для всех отражений (R1 = 0.0274 для 5048 отражений, удовлетворяющих условию I > 2σ(I)). Остаточные максимум и минимум электронной плотности составили 1.51/–0.44 e/Å3.
Структура II зарегистрирована в Кембриджском банке структурных данных (CCDC № 1902290; ccdc.cam.ac.uk/getstructures).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Большинство известных к настоящему времени гидридных производных редкоземельных металлов в степенях окисления как +3, так и +2 являются димерными или кластерными соединениями в силу легкости образования мостиковых связей Ln−H−Ln. Нуклеарность гидридных производных редкоземельных металлов в первую очередь определяется стерическими параметрами “вспомогательного” лигандного окружения. Более того, использование объемных циклопентадиенильных лигандов позволило получить ряд мономерных гидридных производных Cp2LnH (Cp = = C5H2-трет-Bu3 [3, 52]; C5Me4SiMe3 [53, 54]). В ряде работ также было показано, что диссоциация димерных фрагментов Ln2H2 может проходить под действием оснований Льюиса [55, 56]. Альтернативным методом получения мономерных гидридных соединений может быть диссоциация димерного комплекса на мономерные производные за счет координации связи Ln–H с кислотой Льюиса. Ранее было показано, что мономерный гидридный комплекс церия [C5H2-трет-Bu3]2CeH в растворе может обратимо взаимодействовать с такой кислотой Льюиса, как трифенилбор [52].
С целью получения мономерного гидридного комплекса Yb(II), а также изучения реакционной способности связи Yb−H по отношению к кислотам Льюиса была проведена реакция димерного гидридного комплекса I с двумя эквивалентами трифенилбора BPh3. Реакцию проводили в толуоле при комнатной температуре (схема 1 ). Установлено, что продуктом реакции является новый амидинатный комплекс Yb(II) II, содержащий мостиковые фенилтригидроборатные анионы [PhBH3]−. Интересно, что реакция не останавливается на взаимодействии кислоты и основания Льюиса, а протекает глубже, с разрывом связи C−B, переносом гидридных анионов и образованием мостикового аниона [PhBH3]−. Среди продуктов реакции также был обнаружен бис(амидинатный) комплекс Yb(II) III [46], выделенный в виде изумрудно-зеленых кристаллов с выходом 10%. Образование бис(амидинатного) производного III может являться следствием диспропорционирования смешанолигандных комплексов Yb(II), образующихся в ходе реакции. Выделить и идентифицировать другие иттербий содержащие продукты из реакционной смеси не удалось.
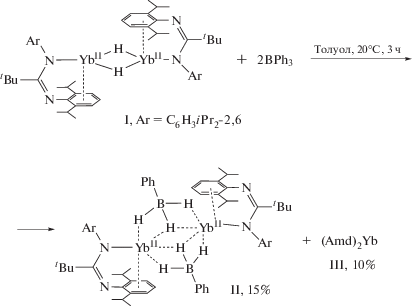
Схема 1 .
Монокристаллические образцы комплекса II, пригодные для РСА, были получены путем медленного концентрирования раствора комплекса II в толуоле при комнатной температуре. Согласно данным РСА, молекула комплекса II в кристаллической ячейке находится в частном положении в центре инверсии; таким образом, кристаллическая ячейка содержит две молекулы комплекса. Основные длины связей и углы указаны в табл. 1. Комплекс II является димером (рис. 1), в котором два амидинатиттербиевых фрагмента (Amd)Yb связаны двумя мостиковыми фенилтригидроборатными анионами [PhBH3]−. В ранее известных комплексах d-переходных металлов (Ti, Zr [57–59], Ru [60], Ir [61]) фенилтригидроборатные лиганды являются терминальными и связаны с ионами переходных металлов через два или три гидридных атома, тогда как мостиковая координация анионов [PhBH3]−, приводящая к образованию димеров, ранее была обнаружена в комплексах лития [62, 63].
Таблица 1.
Основные длины связей (d) и валентные углы (ω) в комплексе II
| Cвязь | d, Å |
|---|---|
| Yb(1)–N(1) | 2.377(3) |
| Yb(1)–B(1) | 2.749(4) |
| Yb(1)−B(1A) | 2.898(4) |
| N(1)−C(1) | 1.359(4) |
| N(2)−C(1) | 1.313(4) |
| Yb(1)···Yb(1A) | 4.0418(3) |
| Yb(1)−C(18) | 2.750(3) |
| Yb(1)−C(19) | 2.798(3) |
| Yb(1)−C(20) | 2.853(3) |
| Yb(1)−C(21) | 2.868(3) |
| Yb(1)−C(22) | 2.870(3) |
| Yb(1)−C(23) | 2.835(3) |
| B(1)−C(30) | 1.579(5) |
| Угол | ω, град |
| Yb(1)B(1)Yb(1A) | 91.4(2) |
| B(1)Yb(1)B(1A) | 88.6(2) |
| N(1)C(1)N(2) | 119.8(3) |
Рис. 1.
Молекулярная структура комплекса II. Тепловые эллипсоиды приведены с 30%-ной вероятностью. Метильные фрагменты 2,6-диизопропилфенильных и трет-бутильных заместителей, а также все атомы водорода за исключением атомов водорода фенилтригидроборатных групп не изображены для ясности.
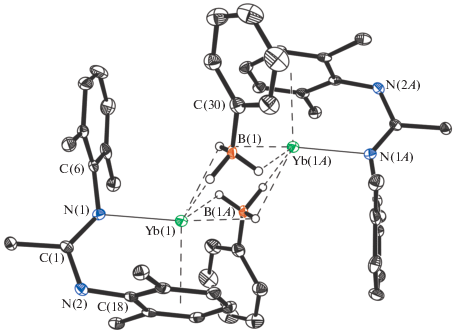
Расстояния Yb−B 2.749(4) и 2.898(4) Å) в II существенно больше соответствующих расстояний в боргидридных производных Yb(II), содержащих терми-нальную боргидридную группу ${\text{BH}}_{4}^{ - }$: [(DIP2Рyr)-Yb(BH4) (THF)3] (Yb−B 2.595(9) Å) [64], [(Tp-трет-Bu)Yb(BH4)(THF)]2 (Yb−B 2.596(5) Å) [65], [(Dipp)2NacNacYb(BH4)(THF)2] (Yb−B 2.576(8) Å) [66], [{CH(PPh2=NSiMe3)(PPh2=S)}Yb(BH4)(THF)2] (Yb−B 2.611(7) Å) [67]. Более того, расстояния Yb−B в II превышают соответствующие значения, найденные в (циклооктан-1,5-диил)дигидроборатных производных Yb(II) Yb{[(µ-H)2BC8H14]2K(Solv)n}2 (Yb−B 2.703(5)–2.738(5) Å) [68], в которых (циклооктан-1,5-диил)дигидроборатные группы [C8H14BH2]− являются мостиковыми между ионами Yb2+ и калия. Расстояния Yb−H в II лежат в широком интервале значений 2.22(3)–2.57(3) Å. Наличие более объемных мостиковых анионов [PhBH3]− в II приводит к увеличению расстояния между катионами металла (Yb···Yb 4.0418(3) Å) в димере по сравнению с исходным гидридом I (Yb···Yb 3.3553(4) Å), однако при этом сохраняется “неклассический” κ1-амидо,-η6-арено тип координации амидинатного лиганда. Длина связи Yb−N 2.377(3) Å и расстояния Yb−Cарил 2.750(3)–2.870(3) Å и Yb−Arцентр 2.458(2) Å в комплексе II несколько больше, чем в исходном гидриде I (Yb−N 2.329(3) Å; Yb−Cарил 2.696(2)–2.838(2) Å; Yb–Arцентр 2.410(5) Å) и сопоставимы с расстояниями в ранее известных амидинатных комплексах Yb(II), в которых реализуется координация амидинатных лигандов по κ1-амидо,-η6-ареновому типу [46, 47, 69–71].
В спектре ЯМР 1H комплекса II, записанном в растворе бензола-d6 при 293 K, присутствуют сигналы, относящиеся к амидинатному лиганду и фенилтригидроборатному аниону. Гидридные атомы [PhBH3]− лиганда проявляются в виде уширенного мультиплета, состоящего из четырех сигналов, в области 1.60–2.23 м.д. Эти химические сдвиги сопоставимы со значениями, наблюдаемыми в спектрах родственных комплексов Ti и Zr моно- и бис(циклопентадиенильных) рядов (δH(BH3) = 1.39 м.д. для Cp2Zr(PhBH3)2; 2.28 м.д. для CpZr(PhBH3)3; 2.37 м.д. для Cp2ZrCl(PhBH3)) [57, 59]. С целью более надежной идентификации наблюдаемых в спектрах ЯМР 1Н и 11B сигналов был записан двумерный спектр ЯМР 11B–1Hge-HSQC, в котором наблюдается кросс-пик (−21.0; 1.90). При помощи двумерного спектра ЯМР 11B–1Hge-HSQC без широкополосной развязки была уточнена мультиплетность гидридного сигнала. Наличие в мультиплете четырех линий хорошо согласуется с ожидаемым для расщепления трех протонов на одном 11B (спин протонов: 1/2, спин основного изотопа бора: 3/2).
Таким образом, было установлено, что взаимодействие гидридного комплекса двухвалентного иттербия I, стабилизированного объемным амидинатным лигандом, с кислотой Льюиса BPh3 сопровождается разрывом связей C−B и восстановлением последнего до фенилтригидроборатного аниона [PhBH3]−. Продуктом реакции является амидинатный комплекс Yb(II) II, в котором два фрагмента (Amd)Yb связаны между собой двумя мостиковыми фенилтригидроборатными анионами.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Ringelberg S.N., Meetsma A., Hessen B. et al. // J. Am. Chem. Soc. 1999. V. 121. P. 6082.
Werkema E.L., Messines E., Perrin L. et al. // J. Am. Chem. Soc. 2005. V. 127. P. 7781.
Maron L., Werkema E.L., Perrin L. et al. // J. Am. Chem. Soc. 2005. V. 127. P. 279.
Jeske G., Lauke H., Mauermann H. et al. // J. Am. Chem. Soc. 1985. V. 107. P. 8111.
Conticello V.P., Brard L., Giardello M.A. et al. // J. Am. Chem. Soc. 1992. V. 114. P. 2761.
Molander G.A., Romero J.A.C. // Chem. Rev. 2002. V. 102. P. 2161.
Oyamada J., Nishiura M., Hou Z. // Angew. Chem. Int. Ed. 2011. V. 50. P. 10720.
Evans W.J., Meadows J.H., Wayda A.L. et al. // J. Am. Chem. Soc. 1982. V. 104. P. 2008.
Evans W.J., Meadows J.H., Wayda A.L. // J. Am. Chem. Soc. 1982. V. 104. P. 2015.
Evans W.J., Bloom I., Hunter W.E. et al. // J. Am. Chem. Soc. 1983. V. 105. P. 1403.
Hou Z., Zhang Y., Tardif O. et al. // J. Am. Chem. Soc. 2001. V. 123. P. 9216.
Tardif O., Nishiura M., Hou Z. // Organometallics. 2003. V. 22. P. 1171.
Cui D., Tardif O., Hou Z. // J. Am. Chem. Soc. 2004. V. 126. P. 1312.
Hou Z., Nishiura M., Shima T. // Eur. J. Inorg. Chem. 2007. P. 2535.
Nishiura M., Hou Z. // Nature Chem. 2010. V. 2. P. 257.
Hagadorn J.R., Arnold J. // Organometallics. 1996. V. 15. P. 984.
Duchateau R., van Wee C.T., Meetsma A. et al. // Organometallics. 1996. V. 15. P. 2279.
Trifonov A.A., Skvortsov G.G., Lyubov D.M. et al. // Chem. Eur. J. 2006. V. 12. P. 5320.
Kissel A.A., Mahrova T.V., Lyubov D.M. et al. // Dalton Trans. 2015. V. 44. P. 12137.
Skvortsov G.G., Tolpyguin A.O., Fukin G.K. et al. // Eur. J. Inorg. Chem. 2010. P. 1655.
Rong W., He D., Wang M., et al. // Chem. Commun. 2015. V. 51. P. 5063.
Cheng J., Saliu K., Kiel G.Y. et al. // Angew. Chem. Int. Ed. 2008. V. 47. P. 4910.
Cheng J., Ferguson M.F., Takats J. // J. Am. Chem. Soc. 2010. V. 132. P. 2.
Cheng J., Saliu K., Ferguson M.J. et al. // J. Organomet. Chem. 2010. V. 695. P. 2696.
Cheng J., Shima T., Hou Z. // Angew. Chem. Int. Ed. 2011. V. 50. P. 1857.
Johnson K.R.D., Kamenz B.L., Hayes P.G. // Organometallics. 2014. V. 33. P. 3005.
Ohashi M., Konkol M., Del Rosal I. et al. // J. Am. Chem. Soc. 2008. V. 130. P. 6920.
Zhou J., Chu J., Zhang Y. et al. // Angew. Chem. Int. Ed. 2013. V. 52. P. 4243.
Martin D., Kleemann J., Abinet E. et al. // Eur. J. Inorg. Chem. 2013. P. 3987.
Lyubov D.M., Döring C., Ketkov S. Yu. et al. // Chem. Eur. J. 2011. V. 17. P. 3824.
Fegler W., Venugopal A., Spaniol T.P. et al. // Angew. Chem. Int. Ed. 2013. V. 52. P. 7976
Venugopal A., Fegler W., Spaniol T.P. et al. // J. Am. Chem. Soc. 2011. V. 133. P. 17574.
Arndt S., Kramer M.U., Fegler W. et al. // Organometallics. 2015. V. 34. P. 3739.
Abinet E., Martin D., Standfuss S. et al. // Chem. Eur. J. 2011. V. 11. P. 15014.
Li X., Baldamus J., Hou Z. // Angew. Chem. Int. Ed. 2005. V. 44. P. 962.
Lyubov D.M., Döring C., Fukin G.K. et al. // Organometallics. 2008. V. 27. P. 2905.
Lyubov D.M., Fukin G.K., Cherkasov A.V. et al. // Organometallics. 2009. V. 28. P. 1227.
Luconi L., Lyubov D.M., Bianchini C. et al. // Eur. J. Inorg. Chem. 2010. P. 608.
Lyubov D.M., Cherkasov A.V., Fukin G.K. et al. // Dalton Trans. 2014. V. 43. P. 14450.
Lyubov D.M., Cherkasov A.V., Fukin G.K. et al. // Organometallics. 2016. V. 35. P. 126.
Trifonov A.A. // Russ. Chem. Rev. 2007. V. 76. P. 1049.
Trifonov A.A., Lyubov D.M. // Coord. Chem. Rev. 2017. V. 340. P. 10.
Abakumov G.A. et al. // Russ. Chem. Rev. 2018. V. 87. P. 393.
Ferrence G.M., McDonald R., Takats J. // Angew. Chem. Int. Ed. 1999. V. 38. P. 2233.
Ruspic C., Spielman J., Harder S. // Inorg. Chem. 2007. V. 46. P. 5320.
Basalov I.V., Lyubov D.M., Fukin G.K. et al. // Angew. Chem. Int. Ed. 2012. V. 52. P. 3444.
Basalov I.V., Lyubov D.M., Fukin G.K. et al. // Organometallics. 2013. V. 32. P. 1507.
Schuhknecht D., Truong K.-N., Spaniol T.P. et al. // Chem. Commun. 2018. V. 54. P. 11280.
Smart. Madison (WI, USA): Bruker AXS Inc., 2012.
Krause L., Herbst-Irmer R., Sheldrick G.M. et al. // J. Appl. Cryst. 2015. V. 48. P. 30.
Sheldrick G.M. // Acta Crystallogr. C. 2015. V. 71. P. 3.
Werkema E.L., Andersen R.A., Yahia A. et al. // Organometallics. 2009. V. 28. P. 3173.
Summerscales O.T., Batista E.R., Scott R.L. et al. // Eur. J. Inorg. Chem. 2016. P. 4551.
Takenaka Y., Hou Z. // Organometallics. 2009. V. 28. P. 5196.
Lyubov D.M., Bubnov A.M., Fukin G.K. et al. // Eur. J. Inorg. Chem. 2008. P. 2090.
Cheng J., Hou Z. // Chem. Commun. 2012. V. 48. P. 814.
Liu F.-C., Chen J.-H., Chen S.-C. et al. // J. Organomet. Chem. 2005. V. 690. P. 291.
Liu F.-C., Chen S.-C., Lee G.-H. et al. // J. Organomet. Chem. 2007. V. 692. P. 2375.
Liu F.-C., Yang C.-C., Chen S.-C. et al. // Dalton Trans. 2008. № 27. P. 3599.
Hesp K.D., Kannemann F.O., Rankin M.A. et al. // Inorg. Chem. 2011. V. 50. P. 2431.
Arnold N., Mozo S., Paul U. et al. // Organometallics. 2015. V. 34. P. 5709.
Samigullin K., Bolte M., Lerner H.-W. et al. // Organometallics. 2014. V. 33. P. 3564.
Murosaki T., Kaneda S., Maruhashi R. et al. // Organometallics. 2016. V. 35. P. 3397.
Schmid M., Guillaume S.M., Roesky P.W. // J. Organomet. Chem. 2013. V. 744. P. 68.
Saliu K.O., Maunder G., Ferguson M.J. et al. // Inorg. Chim. Acta. 2009. V. 362. P. 4616.
Schmid M. Guillaume S.M., Roesky P.W. // Organometallics. 2014. V. 33. P. 5392.
Schmid M., Ona-Burgos P., Guillaume S.M. // Dalton Trans. 2015. V. 44. P. 12338.
Chen X., Lim S., Plečnik C.E. et al. // Inorg. Chem. 2005. V. 44. P. 6052.
Tolpygin A.O., Cherkasov A.V., Fukin G.K. et al. // Inorg. Chem. 2014. V. 53. P. 1537.
Basalov I.V., Rosca S.C., Lyubov D.M. et al. // Inorg. Chem. 2014. V. 53. P 1654.
Trifonov A.A., Lyubov D.M., Basalov I.V. et al. //Russ. Chem. Bull. Int. Ed. 2018. V. 67. P. 455.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия