

Координационная химия, 2019, T. 45, № 11, стр. 663-671
Внутримолекулярные невалентные взаимодействия в комплексе ${\text{Eu}}_{{\text{2}}}^{{{\text{II}}}}$EuIII(μ-ORF)2(μ2-ORF)3(μ3-ORF)2(ДМЭ)2
Р. В. Румянцев 1, Г. К. Фукин 1, *
1 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
* E-mail: gera@iomc.ras.ru
Поступила в редакцию 10.04.2019
После доработки 17.05.2019
Принята к публикации 03.06.2019
Аннотация
Проведено квантово-химическое исследование фторированного алкоголята европия ${\text{Eu}}_{{\text{2}}}^{{{\text{II}}}}$EuIII(μ-ORF)2(μ2-ORF)3(μ3-ORF)2(DME)2 (I), где DME – 1,2-диметоксиэтан. В структуре комплекса обнаружены многочисленные F…F, F…O, F⋅⋅⋅H и C–F → Eu взаимодействия. Исследована природа и энергия этих невалентных взаимодействий в рамках квантовой теории “атомы в молекулах” Р. Бейдера. Показано, что реализация невалентных взаимодействий между отрицательно заряженными атомами фтора в структуре I возможна за счет донирования электронной плотности из областей относительной ее концентрации в области относительного разрежения. Дополнительное исследование с использованием комплекса структурно-топологических программ Topos Pro позволило также обнаружить более слабые C–F → Eu взаимодействия. Их наличие подтверждено исследованием индексов делокализации электронов в области потенциальных Eu⋅⋅⋅F взаимодействий.
В последнее время активно исследуются галогенные связи, в которых “тяжелые” атомы галогенов I, Br и Cl участвуют в роли акцепторов электронной плотности [1–3]. Такие взаимодействия получили свое применение в молекулярном дизайне. Их можно использовать для создания надмолекулярных цепочек или разветвленных сетей [4–8]. Однако Fδ–···Fδ– взаимодействия по-прежнему представляют немалый интерес для исследования. Имеются многочисленные свидетельства увеличения летучести соединений при замене атомов водорода в структуре на фторы [9–12]. Следует также отметить, что в ряде случаев фторирование приводит к увеличению энтальпии сублимации и, соответственно, уменьшению способности к летучести соединения по сравнению с нефторированным аналогом. Так, в случае C10H2F12O4Pb энтальпия сублимации составляет 111.7 ± 1.3 кДж/моль, а для нефторированного аналога C10H14O4Pb – 102.4 ± 5.0 кДж/моль [13]. Также известно, что введение F-заместителя может оказывать влияние на биологическую активность аминокислот [14].
Существует несколько работ, посвященных непосредственно исследованию природы и энергии взаимодействий F⋅⋅⋅F [15–17]. Для любых взаимодействий галоген···галоген (Hal⋅⋅⋅Hal) показано, что возможность реализации взаимодействия существенно зависит от взаимного расположения атомов. Существуют две геометрические возможности для реализации таких взаимодействий [18]. Первая возможность возникает, когда θ1 ≈ θ2 (тип I), где θ1 и θ2 – углы C–Hal1⋅⋅⋅Hal2 и Hal1⋅⋅⋅Hal2–C соответственно. Вторая возможность реализации Hal···Hal взаимодействия при θ1 ≈ 180° и θ2 ≈ 90° (тип II). Классификация взаимодействий галоген···галоген представлена на схеме 1 :
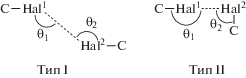
Схема 1 .
Следует отметить, что подавляющее число таких работ посвящено межмолекулярным взаимодействиям в органических или элементоорганических соединениях с относительно “легкими” атомами (P, Cl). В то же время наибольший интерес представляет исследование такого рода взаимодействий именно в металлоорганических соединениях, поскольку замена атомов водорода существенным образом влияет на физико-химические свойства комплексов. В частности, можно ожидать существенное изменение физико-химических свойств в координационных комплексах лантанидов при фторировании. Комплексы Ln(III)/Ln(II) за счет возможных f–f- и f–d-электронных переходов могут использоваться в роли эмиссионных материалов [19, 20]. Большое количество H–C-фрагментов в структуре вызывают тушение люминесценции, и фторирование лигандов используется как один из способов предотвращения этого [21]. Кроме того, в структурах с фторированными лигандами возможна реализация внутримолекулярных взаимодействий C–F → Ln. За счет реализации таких взаимодействий значительно расширяются области применения данных комплексов. Предположительно, именно с реализации взаимодействий C–F → Ln начинаются некоторые каталитические процессы [22, 23], а также процессы разложения [24, 25] органолантанидных комплексов.
Таким образом, исследование нековалентных взаимодействий в структуре металлоорганических комплексов – важная задача на пути понимания связи их строения и физико-химических свойств.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Полная оптимизация геометрии комплекса ${\text{Eu}}_{{\text{2}}}^{{{\text{II}}}}$EuIII(μ-ORF)2(μ2-ORF)3(μ3-ORF)2(DME)2 (I) была проведена методом функционала плотности (DFT) с использованием гибридного функционала B3LYP, реализованном в программном пакете Gaussian 09 [26]. Для органической части использовали базис 6-31+G*, для атомов европия – псевдопотенциал ECP28MWB. Для описания внешних оболочек европия, участвующих в образовании химической связи, использовали валентные базисные наборы ((12s11p9d8f)/[9s8p6d5f]) с диффузными и поляризационными орбиталями [27]. Функцию электронной плотности (EDF) для ECP рассчитывали с использованием программы Molden2aim.
Топологический анализ теоретической функции распределения электронной плотности проводили с помощью программы AIMALL [28].
Расчеты индексов делокализации (DI) и анализ распределения деформационной электронной плотности (ДЭП) для нековалентных взаимодействий в комплексе I проводили с использованием программы Multiwfn v. 3.3.8 [29]. Визуализацию 3d‑распределения ДЭП выполняли при помощи программы GPView [30].
Геометрический анализ наличия внутримолекулярных взаимодействий Eu···F проводили при помощи комплекса структурно-топологических программ ToposPro 5.0.2.1 [31] с использованием модуля AutoCN (метод сферических секторов, [32] MinOm = 0.50).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Основным объектом исследования выбран комплекс европия I, методика синтеза и подробное исследование кристаллического строения которого обсуждалось ранее [33]. Молекулярное строение комплекса представлено на рис. 1.
Рис. 1.
Молекулярное строение комплекса I. Тепловые эллипсоиды приведены с 30%-ной вероятностью. Атомы водорода не приведены для ясности.
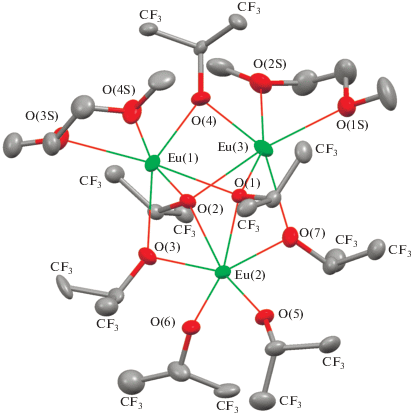
Большое число близко расположенных атомов фтора в молекуле I позволило предположить наличие внутримолекулярных взаимодействий F⋅⋅⋅F в структуре. Для проверки этой гипотезы мы провели квантово-химическое исследование комплекса I.
Проведенная оптимизация удовлетворительно воспроизводит основные геометрические характеристики комплекса и может быть использована для дальнейших расчетов (табл. 1). Среднее отклонение связей Eu–X (X = O, F, Eu) составляет 0.045 Å. Наибольшие изменения наблюдаются для связей Eu–O(ДМЭ). Очевидно, это связано с координационным характером такого взаимодействия, а также с влиянием на это расстояние в кристалле эффекта упаковки.
Таблица 1.
Сравнение экспериментальных и расчетных Eu–X (X = O, F, Eu) длин связей в комплексе I
| Связь | Eu–XX-ray, Å | Eu–Xcalc, Å | Δ | Связь | Eu–XX-ray, Å | Eu–Xcalc, Å | Δ |
|---|---|---|---|---|---|---|---|
| Eu(1)–O(1) | 2.520(4) | 2.5313 | 0.011 | Eu(2)–O(7) | 2.333(3) | 2.3401 | 0.007 |
| Eu(1)–O(2) | 2.740(4) | 2.6667 | 0.073 | Eu(3)–O(1) | 2.738(4) | 2.6820 | 0.056 |
| Eu(1)–O(3) | 2.532(3) | 2.4760 | 0.056 | Eu(3)–O(2) | 2.513(4) | 2.5390 | 0.026 |
| Eu(1)–O(4) | 2.469(4) | 2.4683 | 0.001 | Eu(3)–O(4) | 2.472(3) | 2.4799 | 0.008 |
| Eu(1)–O(3S) | 2.640(3) | 2.6793 | 0.039 | Eu(3)–O(7) | 2.519(3) | 2.4817 | 0.037 |
| Eu(1)–O(4S) | 2.640(5) | 2.7213 | 0.081 | Eu(3)–O(1S) | 2.630(5) | 2.7909 | 0.161 |
| Eu(1)–F(1) | 2.920(4) | 2.8690 | 0.051 | Eu(3)–O(2S) | 2.659(5) | 2.7631 | 0.104 |
| Eu(1)–F(10) | 2.699(4) | 2.7873 | 0.088 | Eu(3)–F(4) | 2.675(4) | 2.7730 | 0.098 |
| Eu(2)–O(1) | 2.446(4) | 2.4681 | 0.022 | Eu(3)–F(7) | 2.893(4) | 2.8158 | 0.077 |
| Eu(2)–O(2) | 2.437(4) | 2.4690 | 0.032 | Eu(1)–Eu(2) | 3.7172(4) | 3.6991 | 0.018 |
| Eu(2)–O(3) | 2.331(4) | 2.3500 | 0.019 | Eu(1)–Eu(3) | 3.8202(5) | 3.7927 | 0.028 |
| Eu(2)–O(5) | 2.167(4) | 2.1649 | 0.002 | Eu(2)–Eu(3) | 3.7096(5) | 3.7061 | 0.004 |
| Eu(2)–O(6) | 2.160(4) | 2.1472 | 0.013 | Δср | 0.045 | ||
Атомы Eu(1) и Eu(3) в I двухвалентны, тогда как Eu(2) имеет формальный заряд +3. Такое представление хорошо согласуется с атомными зарядами, рассчитанными с использованием программы AIMALL [28]. Все атомы фтора и кислорода в комплексе I имеют отрицательный заряд, тогда как все углеродные атомы и водороды заряжены электроположительно (табл. 2).
Таблица 2.
Атомные заряды в комплексе I
| Атом | Заряд, e |
|---|---|
| Eu(1) | 1.68 |
| Eu(2) | 2.16 |
| Eu(3) | 1.67 |
| q(F) | –0.65…–0.63 |
| q(C) | 0.43–1.74 |
| q(O) | –1.24…–1.09 |
| q(H) | 0.02–0.12 |
Исследование топологии электронной плотности (ЭП) в комплексе I с использованием программы AIMALL [28] показало, что все взаимодействия Eu(II)–O и Eu(II)···F соответствуют типу закрытых оболочек (∇2ρ(r)) > 0, he(r) > 0. В свою очередь, все связи Eu3+ с терминальными и μ-2-мостиковыми O-изо-PrF-лигандами относятся к типу промежуточных взаимодействий (∇2ρ(r)) > 0, he(r) < 0 (табл. 3).
Таблица 3.
Основные топологические характеристики связей Eu–O и Eu···F в комплексе I
| Связь | ν(r), a.e. | ρ(r), a.e. | ∇2ρ(r), a.e. | he(r), a.e. | EEML [35], ккал/моль |
|---|---|---|---|---|---|
| Eu(1)–O(1) | –0.036 | 0.037 | 0.155 | 0.001 | 11.28 |
| Eu(1)–O(2) | –0.024 | 0.028 | 0.108 | 0.001 | 7.67 |
| Eu(1)–O(3) | –0.042 | 0.041 | 0.179 | 0.002 | 13.07 |
| Eu(1)–O(4) | –0.042 | 0.041 | 0.183 | 0.002 | 13.15 |
| Eu(1)–O(3S) | –0.023 | 0.026 | 0.106 | 0.002 | 7.28 |
| Eu(1)–O(4S) | –0.021 | 0.024 | 0.095 | 0.002 | 6.44 |
| Eu(1)–F(1) | –0.012 | 0.013 | 0.064 | 0.002 | 3.92 |
| Eu(1)–F(10) | –0.015 | 0.016 | 0.076 | 0.002 | 4.85 |
| Eu(2)–O(1) | –0.044 | 0.045 | 0.179 | 0.0004 | 13.74 |
| Eu(2)–O(2) | –0.044 | 0.045 | 0.178 | 0.0003 | 13.73 |
| Eu(2)–O(3) | –0.062 | 0.057 | 0.241 | –0.001 | 19.50 |
| Eu(2)–O(5) | –0.113 | 0.082 | 0.396 | –0.007 | 35.47 |
| Eu(2)–O(6) | –0.120 | 0.084 | 0.417 | –0.008 | 37.56 |
| Eu(2)–O(7) | –0.063 | 0.058 | 0.249 | –0.001 | 19.91 |
| Eu(3)–O(1) | –0.023 | 0.027 | 0.103 | 0.001 | 7.33 |
| Eu(3)–O(2) | –0.035 | 0.037 | 0.151 | 0.001 | 10.94 |
| Eu(3)–O(4) | –0.042 | 0.042 | 0.177 | 0.001 | 13.25 |
| Eu(3)–O(7) | –0.042 | 0.042 | 0.176 | 0.001 | 13.14 |
| Eu(3)–O(1S) | –0.017 | 0.021 | 0.080 | 0.001 | 5.44 |
| Eu(3)–O(2S) | –0.019 | 0.022 | 0.087 | 0.002 | 5.81 |
| Eu(3)–F(4) | –0.016 | 0.017 | 0.079 | 0.002 | 5.07 |
| Eu(3)–F(7) | –0.015 | 0.015 | 0.072 | 0.002 | 4.59 |
Интересной особенностью структуры является наличие многочисленных внутримолекулярных невалентных взаимодействий. К важным структурным особенностям следует отнести дативные взаимодействия C–F → Eu (рис. 2а). В рамках квантовой теории “Атомы в молекулах” (AIM) Р. Бейдера [34] мы обнаружили в структуре комплекса I четыре внутримолекулярных взаимодействия F⋅⋅⋅Eu (табл. 4).
Рис. 2.
Молекулярный граф комплекса I (на рисунках отдельно приведены только КТ(3, –1) для взаимодействий F···Eu (а), F···F (б), F···H (в) и F···O (г). Атомы F и H, не принимающие участие в соответствующих взаимодействиях не приведены для ясности).
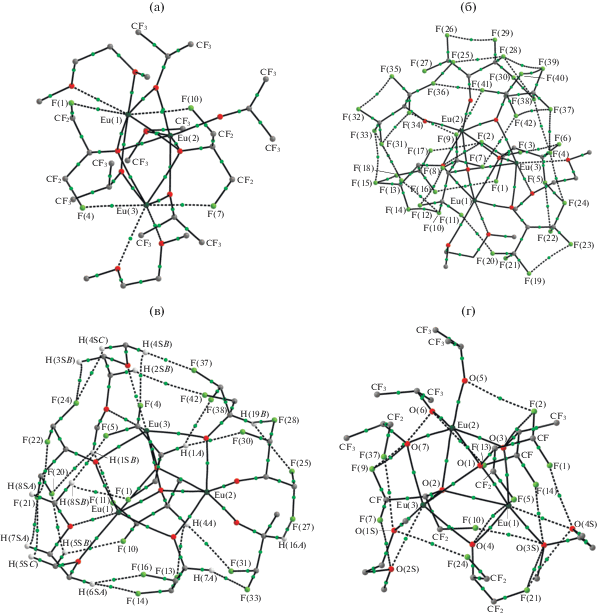
Согласно корреляции Эспинозы–Моллинса–Лекомта [35] энергия этих взаимодействий лежит в интервале 3.92–5.07 ккал/моль, что лишь незначительно меньше соответствующих значений для координационных связей Eu–O(DME) (5.44–7.28 ккал/моль). Для сравнения, энергия Eu–O(изо-PrF) варьирует в диапазоне от 7.33 до 37.56 ккал/моль при переходе от μ3-мостиковых лигандов к терминальным. Таким образом, эти четыре взаимодействия Eu–F, безусловно, необходимо учитывать при определении координационного числа, а также координационного окружения атома лантанида.
Однако помимо четырех взаимодействий Eu–F, которые удалось локализовать при исследовании топологии ЭП, в структуре комплекса I наблюдаются пять укороченных расстояний Eu⋅⋅⋅F (3.305–3.691 Å). Такие геометрические параметры говорят о возможном взаимодействии между атомами европия и фтора [36]. Геометрически топологический анализ, проведенный с использованием комплекса структурно-топологических программ ToposPro [31], подтвердил наличие четырех сильных взаимодействий Eu–F, обнаруженных в рамках AIM-подхода. Кроме того, проведенный Topo-sPro расчет позволил обнаружить еще пять более слабых взаимодействий Eu⋅⋅⋅F (табл. 4).
Таблица 4.
Внутримолекулярные взаимодействия Eu···F в комплексе I
| Контакт | Eu···FX-ray, Å | Eu···Fcalc, Å | EEML [35], ккал/моль | SA*, % | DI, e |
|---|---|---|---|---|---|
| Eu(3)–F(4) | 2.675(4) | 2.7730 | 5.07 | 11.35 | 0.296 |
| Eu(1)–F(10) | 2.699(4) | 2.7873 | 4.84 | 11.11 | 0.294 |
| Eu(3)–F(7) | 2.893(4) | 2.8158 | 4.59 | 8.28 | 0.284 |
| Eu(1)–F(1) | 2.920(4) | 2.8690 | 3.92 | 8.39 | 0.269 |
| Eu(1)…F(21) | 3.305 | 3.4228 | 3.36 | 0.111 | |
| Eu(3)…F(24) | 3.343 | 3.4182 | 3.21 | 0.111 | |
| Eu(3)…F(42) | 3.447 | 3.2248 | 2.89 | 0.166 | |
| Eu(1)…F(13) | 3.543 | 3.4635 | 0.91 | 0.018 | |
| Eu(2)…F(9) | 3.691 | 3.6528 | 0.52 | 0.061 |
* SA (Solid Angle) – телесный угол, соответствующий взаимодействию, рассчитанный с использованием программы ToposPro [31].
Интересно отметить, что взаимодействиям, подтвержденным AIM-методом, соответствуют контакты с телесными углами (SA) 8.39–11.35%. В то же время оставшиеся пять взаимодействий Eu⋅⋅⋅F соответствуют гораздо более низким значениям телесных углов (0.52–3.36%). Для того чтобы выяснить реальное количество делокализованных между атомами европия и фтора электронов в комплексе I и подтвердить возможность реализации взаимодействий между этими атомами, мы произвели расчет DI [29]. Определено, что в области взаимодействий Eu···F, подтвержденных как геометрически, так и исследованием ЭП, индексы DI составляют 0.27–0.30 e. В области взаимодействий C–F → Eu, обнаруженных с использованием ToposPro и не локализованных в рамках AIM-подхода, индексы делокализации оказались значительно меньше (0.06–0.17 e). Однако следует отметить, что такое количество делокализованных электронов оказывается существенным (табл. 4) по сравнению с парами невзаимодействующих атомов (DI < 10–5 e) и подтверждает наличие взаимодействия между атомами европия и фтора во всех девяти случаях.
Анализ ДЭП показал, что наличие как сильных (рис. 3а), так и слабых (рис. 3б) взаимодействий Eu···F в комплексе I обусловлено донированием ЭП из области ее концентрации на атоме фтора на положительно заряженный атом европия. Таким образом, комплексное исследование SA, DI и ДЭП подтверждает наличие девяти взаимодействий C–F → Eu в комплексе I. Следует отметить, что величины SA и DI обнаруженных взаимодействий свидетельствуют о том, что для контактов Eu(1)–F(1), Eu(1)–F(10), Eu(3)–F(4) и Eu(3)–F(7) энергия взаимодействия должна быть существенно выше.
Рис. 3.
3d-Pаспределение ДЭП (0.05 а.е.) в комплексе I в областях, соответствующих взаимодействиям С(3)–F(4) → Eu(3) (а) и C(5)–F(9) → Eu(2) (б). Синий цвет соответствуют области концентрации ЭП, красный цвет – области разрежения ЭП.
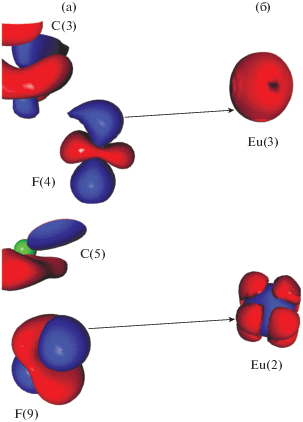
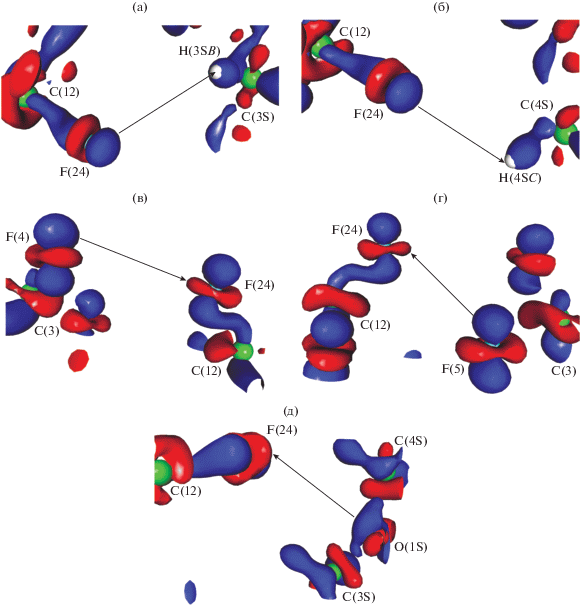
Кроме дативных взаимодействий C–F → Eu в структуре комплекса I обнаружены многочисленные внутримолекулярные невалентные взаимодействия F⋅⋅⋅F, F⋅⋅⋅H и F⋅⋅⋅O (рис. 2б–2г). Исследование топологии ЭП позволило обнаружить взаимодействия: 31 F⋅⋅⋅F, 24 F⋅⋅⋅H и 14 F⋅⋅⋅O. Интересно отметить, что каждый атом фтора в структуре I принимает участие, по меньшей мере, в одном дополнительном невалентном взаимодействии. Для структуры I максимальное число таких дополнительных взаимодействий равно пяти. При этом один и тот же атом фтора выступает как в роли донора (рис. 4а, 4б), так и в роли акцептора (рис. 4в–4д) ЭП.
Рис. 4.
3d-Распределение ДЭП (0.05 а.е.) в комплексе I в областях, соответствующих взаимодействиям F(24)···H(3SB) (а), F(24)···H(4SC) (б), F(4)···F(24) (в), F(5)···F(24) (г) и F(24)···O(1S) (д). Синий цвет соответствуют области концентрации ЭП, красный цвет – области разрежения ЭП.
Анализ геометрии галогенных взаимодействий показал, что подавляющее большинство взаимодействий F⋅⋅⋅F в комплексе I можно отнести к типу I (схема 1 ). Важно отметить, что все взаимодействия F⋅⋅⋅H в структуре I по геометрическим критериям также можно условно отнести либо к I (θ1 ≈ θ2), либо к II типу (θ1 ≈ 90°, θ2 ≈ 180°) взаимодействий. При этом подавляющее большинство взаимодействий F···H в структуре I также будут относиться именно к типу I.
Расстояния для взаимодействий F···F, F···H и F···O в комплексе I варьируют в достаточно широких пределах (2.654–3.196, 2.235–3.197 и 2.777–3.448 Å соответственно). Таким образом, значительным оказывается и разброс энергии соответствующих взаимодействий (0.67–4.10, 0.19–3.25 и 0.51–3.03 ккал/моль) (табл. 5). Суммарный вклад всех взаимодействий Fδ–···Fδ– в стабилизацию молекулярного строения комплекса I составляет 81.86 ккал/моль. Это значение превосходит суммарный вклад взаимодействий Fδ–···Hδ+ (43.26 ккал/моль) и Fδ–···Oδ– (25.19 ккал/моль).
Таблица 5.
Основные геометрические и топологические характеристики, соответствующие внутримолекулярным взаимодействиям F···X (X = F, H, O) в комплексе I
| Взаимодействие | Кол-во | F···Xcalc, Å | ρ(r), a.e. | ∇2ρ(r), a.e. | EEML [35], ккал/моль | DI, e | ∑E, ккал/моль |
|---|---|---|---|---|---|---|---|
| F···F | 31 | 2.6535–3.1962 | 0.003–0.013 | 0.019–0.061 | 0.66–4.11 | 0.003–0.030 | 81.86 |
| F···H | 24 | 2.2350–3.1968 | 0.001–0.012 | 0.007–0.049 | 0.19–3.25 | 0.003–0.018 | 43.26 |
| F···O | 14 | 2.7769–3.4483 | 0.003–0.010 | 0.015–0.043 | 0.51–3.03 | 0.001–0.019 | 25.19 |
Для всех невалентных взаимодействий F···X (X = O, F, H) в I, обнаруженных нами, также были проведены расчеты индексов делокализации. Значения DI составили 0.001–0.019, 0.003–0.018 и 0.003–0.030 e для взаимодействий F···O, F···H и F···F соответственно. Такие значения существенно превышают значения DI для невзаимодействующих атомов (<10–5 e) и свидетельствуют о наличии взаимодействия между этими атомами.
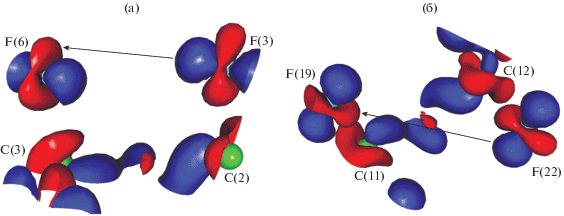
Сразу для нескольких взаимодействий F···X (X = O, F, H), которые удается локализовать, расстояния между атомами превосходят сумму ван-дер-ваальсовых радиусов элементов [37]. В то же время в структуре комплекса I наблюдается ряд пар F···F, для которых расстояние между атомами лежит внутри интервала локализованных взаимодействий, однако критической точки (КТ) (3, –1) обнаружить не удалось. Например, расстояние F(3)···F(5) составляет 3.0516 Å, а связевый путь и КТ(3, –1) отсутствуют. Также в структуре I наблюдается несколько примеров F···F, для которых расстояние между атомами лишь незначительно превышает интервал локализованных взаимодействий. Например, для пары атомов F(19)···F(22) это значение составляет 3.3838 Å. Интересно отметить, что для этих взаимодействий как геометрические (θ1 = 69.50°, θ2 = 65.28° для F(3)···F(5), θ1 = 60.09°, θ2 = 58.04° для F(19)···F(22)), так и электронные факторы (DI составляет 0.005 и 0.001 e для F(3)···F(5) и F(19)···F(22) соответственно) свидетельствуют о возможном наличии между этими атомами взаимодействия. Исследование фрагмента ДЭП также подтверждает этот факт (рис. 5). Таким образом, можно сделать вывод о том, что в структуре комплекса I есть также более слабые взаимодействия F···F, локализовать которые в рамках AIM-теории не удается.
Рис. 5.
3d-Распределение ДЭП (0.05 а.е.) в комплексе I в областях, соответствующих возможным взаимодействиям F(3)···F(6) (а) и F(19)···F(22) (б). Синий цвет соответствуют области концентрации ЭП, красный цвет – области разрежения ЭП.
Помимо взаимодействий F···F, F···O, F···H и C–F → Eu в структуре комплекса I наблюдаются также три внутримолекулярных взаимодействия O···H, относящиеся к промежуточному типу (3.66, 2.15 и 1.57 ккал/моль).
Таким образом, было проведено исследование электронного строения комплекса I, а также природы и энергии нековалентных взаимодействий Eu···F, F···F, F···H и F···O. В структуре I были обнаружены четыре “сильных” и пять “слабых” взаимодействий Eu···F. Наличие всех взаимодействий C–F → Eu в I подтверждено расчетами DI электронов. Показано, что такого рода взаимодействия обусловлены донированием ЭП из области ее концентрации на атоме фтора на положительно заряженный катион лантанида. Проведено исследование природы и энергии взаимодействий 31 F···F, 24 F···H и 14 F···O в структуре I. Вклад всех взаимодействий Fδ–···Fδ– в комплексе I в стабилизацию молекулярного строения комплекса (81.86 ккал/моль) превосходит суммарный вклад взаимодействий Fδ–···Hδ+ (43.26 ккал/моль) и Fδ–···Oδ– (25.19 ккал/моль). Показано, что для всех локализованных взаимодействий F···F области концентрации ЭП на одном атоме фтора соответствуют области разрежения ЭП на другом атоме.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Барташевич Е.В., Цирельсон В.Г. // Успехи химии. 2014. Т. 83. № 12. С. 1181.
Bartashevich E.V., Yushina I.D., Stash A.I., Tsirelson V.G. // Cryst. Growth Des. 2014. V. 14. № 11. P. 5674.
Duarte D.J.R., Peruchena N.M., Alkorta I. // J. Phys. Chem. A. 2015. V. 119. № 16. P 3746.
Rissanen K. // CrystEngComm. 2008. V. 10. P. 1107.
Gilday L.C., Robinson S.W., Barendt T.A. et al. // Chem. Rev. 2015. V. 115. P. 7118.
Priimagi A., Cavallo G., Metrangolo P., Resnati G. // Acc. Chem. Res. 2013. V. 46. № 11. P. 2686.
Berger G., Soubhye J., Meyer F. // Polym. Chem. 2015. V. 6. P. 3559.
Li B., Zang S.-Q., Wang L.-Y., Mak Th.C.W. // Coord. Chem. Rev. 2016. V. 308. P. 1.
Grushin V.V., Herron N., LeCloux D.D. et al. // Chem. Commun. 2001. P. 1494.
Mancino G., Ferguson A.J., Beeby A. et al. // J. Am. Chem. Soc. 2005. V. 127. P. 524.
Tiitta M., Niinistou L. // Chem. Vap. Deposition. 1997. V. 3. № 4. P. 167.
Eisentraut K.J., Sievers R.E. // J. Inorg. Nucl. Chem. 1967. V. 29. P. 1931.
Krisyuk V.V., Sysoev S.V., Fedotova N.E. et al. // Thermochim. Acta. 1997. V. 307. P. 107.
Smart B.E. // J. Fluor. Chem. 2001. V. 109. P. 3.
Osuna R.M., Hernández V., Navarrete J.T.L. et al. // Theor. Chem. Acc. 2011. V. 128. P. 541.
Hathwar V.R., Row T.N.G. // Cryst. Growth Des. 2011. V. 11. P. 1338.
Karnoukhova V.A., Fedyanin I.V., Lyssenko K.A. // Struct. Chem. 2016. V. 27. P. 17.
Ramasubbu N., Parthasarathy R., Murray-Rust P. // J. Am. Chem. Soc. 1986. V. 108. P. 4308.
Bunzli J.-C.G., Piguet C. // Chem. Soc. Rev. 2005. V. 34. P. 1048.
Shipley C.P., Capecchi S., Salata O.V. et al. // Adv. Mater. 1999. V. 11. P. 533.
Glover P.B., Bassett A.P., Nockemann P. et al. // Chem. Eur. J. 2007. V. 13. P. 6308.
Evans W.J., Forrestal K.J., Ansari M.A., Ziller J.W. // J. Am. Chem. Soc. 1998. V. 120. P. 2180.
Liu B., Roisnel T., Maron L. et al. // Chem. Eur. J. 2013. V. 19. P. 3986.
Maleev A.A., Fagin A.A., Ilichev V.A. et al. // J. Organomet. Chem. 2013. V. 747. P. 126.
Melman J.H., Rohde C., Emge T.J., d Brennan J.G. // Inorg. Chem. 2002. V. 41. P. 28.
Frisch M.J., Trucks G.W., Schlegel H.B. et al. Gaussian 09. Wallingford, 2009.
Dolg M., Stoll H., Preuss H. // J. Chem. Phys. 1989. V. 90. P. 1730.
Keith T.A. AIMAll. Version 17.11.14. TK Gristmill Software, Overland Park (KS, USA), 2017.
Lu T., Chen F. // J. Comp. Chem. 2012. V. 33. P. 580.
Shi T., Wang P. // J. Mol. Graph. Model. 2016. V. 70. P. 305.
Blatov V.A., Shevchenko A.P., Proserpio D.M. // Cryst. Growth Des. 2014. V. 14. P. 3576.
Peresypkina E.V., Blatov V.A. // Acta Crystallogr. B. 2000. V. 56. P. 1035.
Кузяев Д.М., Румянцев Р.В., Фукин Г.К., Бочкарев М.Н. // Изв. АН. Сер. хим. 2014. № 4. С. 848.
Bader R.F.W. Atoms in Molecules – A Quantum Theory. Oxford: Oxford Univ. Press, 1990.
Espinosa E., Molins E., Lecomte C. // Chem. Phys. Lett. 1998. V. 285. P. 170.
Румянцев Р.В., Фукин Г.К. // Изв. АН. Сер. хим. 2017. № 9. С. 1557.
Бацанов С.С. // Неорган. материалы. 2001. Т. 37. № 9. С. 1031.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия