

Координационная химия, 2019, T. 45, № 11, стр. 654-662
Новые гетерометаллические пивалатные комплексы кобальта(III) с 1,3-(CH2)3(NH2)2
И. А. Луценко 1, *, Ю. В. Нелюбина 1, 2, П. В. Примаков 2, 3, Т. М. Иванова 1, К. И. Маслаков 3, А. В. Хорошилов 1, М. А. Кискин 1, А. А. Сидоров 1, И. Л. Еременко 1
1 Институт общей и неорганической химии им. Н.С. Курнакова PAH
Москва, Россия
2 Институт элементорганических соединений им. А.Н. Несмеянова PAH
Москва, Россия
3 Московский государственный университет им. М.В. Ломоносова
Москва, Россия
* E-mail: irinalu05@rambler.ru
Поступила в редакцию 30.04.2019
После доработки 15.05.2019
Принята к публикации 03.06.2019
Аннотация
Взаимодействие комплекса [Co(DAP)2(Piv)2] · Piv (Ia), где Piv – анион пивалиновой кислоты, DAP – 1,3-диаминопропан, с пивалатами Cd(II) и Li(I) приводит к формированию гетерометаллических соединений: молекулярного [CdCo(DAP)2(Piv)5] (I) и полимерного [LiCо(DAP)2(Piv)4]n (II) строения (CIF files CCDC № 1908223 и 1908224 соответственно). По данным рентгено-фотоэлектронной спектроскопии электронное строение ионов кобальта в комплексах Ia и I соответствует низкоспиновому состоянию Со(III) в окружении лигандов сильного поля. Термическая деструкция I и II, изученная методом синхронного термического анализа, включает стадии десорбции сольватных молекул, разрушение молекулы 1,3-диаминопропана с выделением азота и деструкцию карбоксилатных анионов. Финальный продукт термолиза I представляет собой сложный оксид CdCoO2.
Многочисленные результаты по синтезу и исследованию устойчивых молекулярных гетерометаллических карбоксилатных комплексов [1–11] и установление факта сохранения исходного металлоостова при синтезе из них каркасных координационных полимеров [12–14] показали несомненную перспективность поиска и исследования новых гетерометаллических систем. В качестве стартового объекта исследования привлекают внимание карбоксилатные комплексы кобальта(III), которые легко получаются при взаимодействии дипивалата кобальта(II) c первичными аминами в присутствии кислорода воздуха [15]. В отличие от карбоксилатных комплексов Fe(III) и Mn(III) [10, 16, 17] достаточно просто получить комплексы Со(III), в которых отсутствуют мостиковые анионы О2, что, в свою очередь, предполагает возможность получения гетерометаллических соединений по тем же методикам, что и комплексы 3d-металлов в степени окисления +2.
Цель настоящей работы – установление состава и строения продуктов взаимодействия ранее полученного комплекса [Co(DAP)2(Piv)2] · Piv (Ia), где Piv – анион пивалиновой кислоты, DAP – 1,3-диаминопропан [15], с пивалатами Cd(II) и Li(I). Выбор кадмия обусловлен тем, что большой ионный радиус Cd(II) позволяет металлоцентру координировать большее количество донорных атомов (до 8), чем ионы 3d-металлов, и образование гетерометаллического соединения в результате этого более вероятно. Различие в ионных радиусах металлов также упрощает идентификацию атомов в структуре, определенную методом РСА. Перспективность получения Li–Co комплексов обусловлена существованием устойчивых тетраядерных соединений [Li2M2(Piv)6L2] (M = Co, Zn) [12, 18–20] и металлоорганических каркасных полимеров (МОКП) на их основе [12, 13, 20]. Поиск исходных комплексов для синтеза Li(I)–Co(III) МОКП представляет собой актуальную задачу для расширения круга новых типов каркасов с определенными функциональными свойствами.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез комплексов проводили с использованием коммерческих реагентов и растворителей без дополнительной очистки: пивалиновая кислота (HPiv) (Merck), CoCl2 · 6Н2О (Химмед, “ч. д. а.”), Cd(NO3)2 · 4Н2О (Acros), бензол (“ч. д. а.”), ацетонитрил (Химмед, “ос. ч.”), DAP (Acros). Пивалат лития, Li(Piv), синтезирован взаимодействием эквимолярных водных растворов LiOH и HPiv с последующим испарением; образовавшаяся твердая фаза промыта гексаном. Комплекс Ia получали в рамках методики, предложенной в [15]. Анаэробный термолиз проводили в потоке аргона (Ar, >99.998%; O2, 0.0002%; N2, <0.001%; H2O, <0.0003%; CH4, <0.0001%). Исходный полимерный комплекс [Co(OH)n(O)(Piv)2 –n]x синтезирован по методике [21] и охарактеризован ИК-спектроскопией и данными химического анализа.
ИК-спектры соединений регистрировали на ИК-спектрофотометре с Фурье преобразованием Perkin-Elmer Spectrum 65 методом нарушенного полного внутреннего отражения (НПВО) в интервале частот 400–4000 см–1.
Элементный анализ выполняли на автоматическом С,H,N,S-анализаторе Carlo Erba EA 1108.
Рентгенофотоэлектронные спектры (РФЭС) снимали на спектрометре Kratos Axis Ultra (монохроматическое AlKα-излучение с мощностью, не превышающей 180 Вт). Для компенсации заряда на поверхности образцов использовали пушку низкоэнергетических электронов. Разложение спектров на компоненты проведено по программе Kratos Analytical. Каждую линию спектра аппроксимировали гауссовым профилем или их суммой. Измерения проводили не менее 2 раз при давлении ~10–9 Tорр. Спектры снимали как при температуре жидкого азота, так и при комнатной. Калибровку спектров осуществляли по линии С1s, принимая энергию связи компоненты, отвечающей связям C–(C,H), равной 285.0 эВ. Полученные результаты находятся в согласии с данными РСА.
Синтез [CdCo(DAP)2(Piv)5] ∙ 1/2С6Н6 (I). К суспензии [Co(OH)n(O)(Piv)2 –n]x 0.2 г (0.8 ммоль) в 50 мл ацетонитрила добавляли 0.3 г (0.7 ммоль) CdPiv2 и раствор 0.13 мл (1.5 ммоль) диаминопропана в 10 мл бензола. Полученный вишневый раствор выдерживали при нагревании (70°С) 40 мин. Далее отфильтровывали и концентрировали. Выпавшие через сутки бордовые кристаллы I отделяли от маточного раствора декантацией, промывали холодным эфиром и высушивали в токе аргона. Выход I 120 мг (60% для высушенного до постоянного веса десольватированного продукта).
ИК (ν, сm–1): 3262 у.ср, 3224 у.ср, 3154 сл, 2960 с, 2930 ср, 2870 ср, 2300 о.сл, 2253 сл, 1620 ср, 1583 сл, 1540 о.с, 1480 о.с, 1458 сл, 1410 о.с, 1356 о.с, 1336 о.с, 1306 сл, 1211 у.с, 1136 сл, 1112 ср, 1050 у.ср, 924 ср, 898 о.с, 807 ср, 792 ср, 752 у.сл, 682 сл, 640 сл, 604 ср, 571 сл, 521 ср, 460 ср, 425 сл.
Синтез [LiCо(DAP)2(Piv)4]n (II). К суспензии полимера [Co(OH)n(O)(Piv)2 –n]x 0.2 г (0.8 ммоль) в 50 мл ацетонитрила добавляли 0.08 г (0.7 ммоль) LiPiv и раствор 0.13 мл (1.5 ммоль) диаминопропана в 10 мл бензола. Образовавшуюся розовую суспензию выдерживали при нагревании (70°С) 40 мин. Выпавшие через сутки розовые кристаллы II отделяли от маточного раствора декантацией, промывали холодным эфиром и высушивали в токе аргона. Выход II 70 мг (35% для высушенного до постоянного веса десольвати-рованного продукта).
ИК (ν, сm–1): 3297 сл, 3200 у.сл, 3153 сл, 3089 сл, 2953 ср, 2930 ср, 2866 ср, 1638 ср, 1595 о.с, 1564 о.с, 1479 о.с, 1454 сл, 1421 сл, 1346 о.с, 1302 ср, 1261 сл, 1200 у.сл, 1111 ср, 1047 ср, 1036 ср, 931 ср, 907 сл, 895 сл, 881 ср, 789 ср, 726 у.сл, 679 о.с, 648 ср, 592 ср, 577 сл, 547 сл, 528 сл, 446 о.с, 423 сл.
РСА кристаллов I и II выполнен на дифрактометрах Bruker Apex II и Bruker Apex II DUO, оборудованных CCD-детекторами (MoKα-излучение, λ = 0.71073 Å, графитовый монохроматор), при 120 К. Структуры расшифрованы прямым методом и уточнены в полноматричном МНК в анизотропном приближении с использованием комплекса программ SHELXL-2014/7 [22] и Olex2 [23]. Атомы водорода групп NH2 локализовали из разностного синтеза Фурье, положения остальных атомов водорода рассчитаны геометрически. Все атомы водорода уточнены в изотропном приближении в модели “наездника”. Кристаллографические параметры и детали уточнения комплексов I и II приведены в табл. 1.
Таблица 1.
Кристаллографические параметры и детали уточнения структур для I и II
| Параметр | Значение | |
|---|---|---|
| I | II | |
| Брутто-формула | C37H71N4O10CoCd | C32H62N4O8LiCo |
| M | 903.34 | 696.74 |
| Т, K | 120 | |
| Сингония | Моноклинная | |
| Пр. гр. | Р21/с | |
| Размер кристалла, мм | 0.3 × 0.15 × 0.158 | 0.3 × 0.1 × 0.1 |
| a, Å | 14.808(4) | 15.1439(14) |
| b, Å | 23.691(6) | 9.4332(9) |
| c, Å | 18.113(5) | 17.4604(19) |
| β, град | 134.204(7) | 130.171(2) |
| V, Å3 | 4555(2) | 1906.0(3) |
| Z | 4 | 2 |
| ρ(выч.), г/см3 | 1.3171 | 1.2140 |
| μ, мм−1 | 0.884 | 4.99 |
| F(000) | 1904 | 753 |
| Область сбора данных по θ, град | 1.64–28.00 | 2.16–27.99 |
| Интервалы индексов отражений | –18 ≤ h ≤ 21,
–33 ≤ k ≤ 31, –25 ≤ l ≤ 25 |
–19 ≤ h ≤ 15,
0 ≤ k ≤ 12, 0 ≤ l ≤ 22 |
| Число измерено отражений | 27 135 | 33 228 |
| Число независимых отражений (Rint) | 10 856 (0.0632) | 4603 (0.1117) |
| Число отражений с I > 2σ(I) | 7365 | 3235 |
| Переменных уточнения | 555 | 217 |
| GООF | 0.9628 | 1.0061 |
| R-факторы по F 2 > 2σ(F 2) | R1 = 0.0438, wR2 = 0.0753 |
R1 = 0.0445, wR2 = 0.0942 |
| R-факторы по всем отражениям | R1 = 0.0785, wR2 = 0.0889 |
R1 = 0.0777, wR2 = 0.1086 |
| Остаточная электронная плотность (min/max), e/Å3 | –0.844/0.971 | –0.563/0.607 |
Полный набор рентгеноструктурных данных депонирован в Кембриджском банке структурных данных (ССDC № 1908223 (I), 1908224 (II); deposit@ccdc.cam.ac.uk или http://www.ccdc.cam.ac.uk/ data_request/cif).
РФА анализ для обоих образцов проведен на приборе Bruker D8 Advance (CuKα, λ = 1.54 Å, Ni-filter, LYNXEYE-детектор).
Термическое поведение I и II изучали методом синхронного термического анализа (СТА) в атмосфере аргона с одновременной регистрацией кривых термогравиметрии (ТГ) и дифференциальной сканирующей калориметрии (ДСК). Исследование проводили на приборе CTA 449 F1 J-upiter (фирмы NETZSCH) в алюминиевых тиглях под крышкой с отверстием, обеспечивающим давление паров при термическом разложении образцов в 1 атм. Скорость нагрева составляла 10°С/мин до 450°С. Масса навески 5.57 мг (I)/ 1.13 мг (II). Точность измерения температуры ±0.7°С, изменения массы ±1 × 10−2 мг. При съемке кривых ТГ и ДСК использовали файл коррекции, а также калибровки по температуре и чувствительности для заданной температурной программы и скорости нагрева. После проведения термического анализа качественное определение химического состава и микроморфологию остаточного вещества анализировали с помощью растрового электронного микроскопа CarlZeissNVision 40, оснащенного рентгеноспектральным детектором Oxford X-Max, при ускоряющем напряжении 1 и 20 кВ соответственно. Увеличение составляло от ×3000 до ×300 000.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Продукт реакции [Co(DAP)2(Piv)2] · Piv (Ia) c дипивалатом кадмия [24] в растворе бензола представляет собой молекулярный биядерный гетерометаллический комплекс [CdCo(DAP)2(Piv)5] или [CdII(κ2-Piv)3(µ2-Piv)CoIII(κ2-DAP)2(κ1-Piv)] (I, рис. 1). По данным РСА для его кристаллосольвата с бензолом в соотношении 1 : 0.5 ион кобальта(III) имеет такое же искаженное октаэдрическое окружение, как в исходном соединении Ia. Оно образовано двумя хелатными пропандиаминами и двумя пивалат-анионами (с близкими расстояниями Сo–N и Co–O(4), табл. 2), один из которых становится мостиковым, связывая ионы Co3+ и Cd2+. Внешнесферный пивалатный анион исходного комплекса Ia достраивает координационное окружение иона кадмия(II), связываясь с ним хелатно, до {Cd(κ2-OOC-)3(κ1-OOC-)} и формирует вокруг него искаженную одношапочную тригональную призму. При этом две из четырех групп NH2 пропандиаминовых лигандов образуют внутримолекулярные водородные связи с терминальным, мостиковым и двумя хелатными пивалат-анионами (N···O 2.848(4)–3.057(5) Å, NHO 123.0(4)°–152.5(3)°). Другие две группы NH2 связаны с пивалат-анионами, координированными к иону кадмия(II) в соседней молекуле комплекса I (N···O 2.874(5)–2.964(7) Å, NHO 139.5(6)°–156.9(3)°), с образованием водородно-связанной бесконечной цепочки.
Рис. 1.
Строение комплекса [CdCo(DAP)2Piv5] (I) в представлении атомов эллипсоидами тепловых колебаний (p = 50%). Атомы водорода за исключением принадлежащих группам NH2 лигандов и сольватная молекула бензола не показаны для ясности; t-Bu отвечают трет-бутильным группам пивалат-анионов.
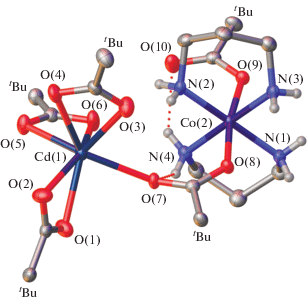
Таблица 2.
Основные длины связей (d) в структурах Ia, I и II
| I | II | ||
|---|---|---|---|
| Связь | d, Å | Связь | d, Å |
| Cd(1)–O(1) | 2.336(3) | Li(1)–O(2) | 1.903(4) |
| Cd(2)–O(2) | 2.354(3) | Li(1)–O(3) | 1.967(3) |
| Cd(3)–O(3) | 2.342(3) | Co(1)–O(1) | 1.906(1) |
| Cd(1)–O(4) | 2.510(2) | Co(1)–N(1) | 1.975(2) |
| Cd(1)–O(5) | 2.378(3) | Co(1)–N(2) | 1.984(3) |
| Cd(1)–O(6) | 2.348(2) | ||
| Cd(2)–O(7) | 2.382(3) | Ia | |
| Co(2)–O(8) | 1.930(2) | Co–O | 1.922(1)–1.937(1) |
| Co(2)–O(9) | 1.896(2) | Со–N | 1.969(2)–1.977(2) |
| Co(2)–N(1) | 1.964(3) | ||
| Co(2)–N(2) | 1.962(3) | ||
| Co(2)–N(3) | 1.972(5) | ||
| Co(2)–N(5) | 1.977(5) | ||
Поскольку при образовании I один Piv–, связанный в исходном соединении с ионом кобальта(III) монодентатно, становится мостиковым, была сделана попытка получить гетерометаллический координационный полимер [Cd2Co(DAP)2Piv7] за счет введения в реакцию двух эквивалентов [Cd(H2O)2(Piv)2]. Однако при этом никаких гетерометаллических соединений кроме I из раствора выделено не было.
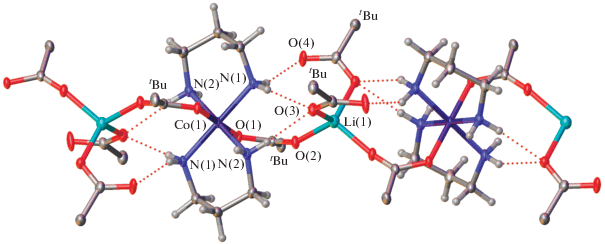
Реакция Iа с LiPiv приводит к образованию 1D-координационного полимера [LiCо(DAP)2(Piv)4]n или [Li(κ1-Piv)2(µ2-Piv)2Cо(DAP)2]n (II, рис. 2). Координационное окружение иона кобальта(III) также остается неизменным (табл. 2), но оба Piv–, монодентатно координированные к иону кобальта(III) в исходном соединении, становятся мостиковыми, связывая его с Li+. За счет такого связывания формируется бесконечная 1D-цепочка, в которой чередующиеся ионы лития и кобальта связаны мостиковым карбоксилатным анионом. Ион лития(I), в свою очередь, окружен еще двумя пивалат-анионами, образующими вокруг него искаженное тетраэдрическое окружение, наиболее типичное для иона Li+. Некоординированные к иону лития атомы кислорода связаны с группами NH2 диаминопропана водородными связями (N···O 2.848(2)–3.027(3) Å, NHO 148.1(2)°–175.5(3)°), дополнительно стабилизирующими полученный 1D-координационный полимер. В кристалле такие цепочки связаны только слабыми взаимодействиями, а не водородными связями с участием группы N(2)H2, которые не образуются, по-видимому, из-за экранирования соответствующих атомов водорода объемными трет-бутильными заместителями Piv–.
Рис. 2.
Строение 1D-координационного полимера [LiCо(DAP)2(Рiv)4]n (II) в представлении атомов эллипсоидами тепловых колебаний (p = 50%). Атомы водорода за исключением принадлежащих группам NH2 лигандов не показаны для ясности; t-Bu отвечают трет-бутильным группам пивалат-анионов. Ион кобальта(III) в кристалле занимает частное положение, центр инверсии.
Степень окисления кобальта была подтверждена методом РФЭС в исходном соединении Ia и гетерометаллическом комплексе I. Как правило, при переходе от состояния с меньшей валентностью к состоянию с большей энергия связи (Есв) основного уровня возрастает. Но в ряде соединений кобальта высокоспиновые состояния Со(II) имеют большие значения Есв, чем низкоспиновые Со(III) [25]. В случае кобальта спин-орбитальное расщепление (ΔE) линии Со2р более информативно [25, 26]. Величина ΔE представляет собой разницу в Есв между энергиями связи линий Со2р1/2 и Со2р3/2. Результаты исследования приведены в табл. 3.
Таблица 3.
Характеристики рентгено-фотоэлектронных спектров I и Ia
| I | Ia | |
|---|---|---|
| Т, К | –77 | 273 |
| Линии | Энергия связи, эВ | |
| Co2p3|2 | 781.4 (ΔE* =14.9) | 781.4 (ΔE =14.8) |
| C1s | 285.0 | 285.0 (С–H) |
| 286.0 | 286.0 (С–N) | |
| 288.0 | 288.3 (–ОСО–) | |
| O1s | 531.4 | 531.5 (–ОСО–) |
| 532.6 (cледы) | ||
| N1s | 400.0 | 400.0 (–NH2) |
| Cd3d5|2 | 405.5 | |
Спектры РФЭ-линии кобальта Со2р для образцов I и Ia, полученные при температуре жидкого азота (–77 K), представлены на рис. 3а.
Рис. 3.
Рентгеновские фотоэлектронные спектры линии Со2р: а – I и II (–77 К); б – I (273 К); а – в начале эксперимента, б – в конце.
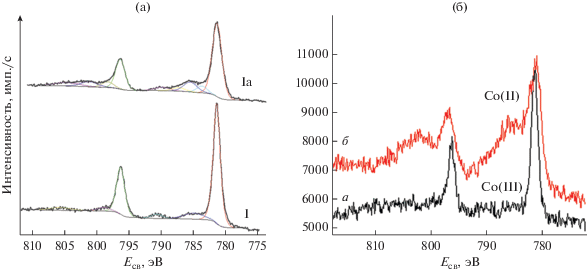
Спектр линии Co2p3/2 этих комплексов представляет собой довольно узкую линию со слабой сателлитной составляющей и с ΔE, равными 14.9 и 14.8 эВ (табл. 3), что соответствует Со(III). Согласно теоретическим расчетам [27], спин-орбитальное расщепление увеличивается в зависимости от числа неспаренных 3d-электронов. Ранее проведенные эксперименты [25, 26, 28] подтверждают теоретический результат: в диамагнитных соединениях Со(III) и парамагнитных Со(II) это расщепление близко к 15 и 16 эВ соответственно. Следует отметить тот факт, что для образца Ia при комнатной температуре первые сканы зафиксировали одно состояние кобальта в степени окисления +3, спектр которого полностью идентичен спектру комплекса I, полученного при –77 K. В конце съемки под действием рентгеновского излучения произошло частичное восстановление кобальта до состояния Со(II) (рис. 3б).
Неустойчивость I к рентгеновскому излучению может быть связана со структурными параметрами этого комплекса. Известно, что переход в низкоспиновое состояние сопровождается подавлением сателлитов в спектрах 3d-металлов, что указывает на уменьшение ионности связи металл–лиганд [29]. Структура РФЭ-спектра линии Со2р комплексов I и Ia (рис. 3a) имеет слабую сателлитную составляющую, что свидетельствует о диамагнитном состоянии кобальта. Это означает, что заселенность 3d-подоболочки равна 6. Единственным объяснением отсутствия спина на атомах кобальта в такой конфигурации является отнесение данных соединений к комплексам с лигандами сильного поля. В этом случае шесть 3d‑электронов занимают трехкратно вырожденный уровень 3d(t2g), при этом проигрыш в энергии за счет антипараллельной ориентации спинов компенсируется понижением энергии t2g-уровня в поле лигандов. Известно [30], что в ряду 3d-металлов именно атомы кобальта Со(III) с наибольшей вероятностью образуют диамагнитные соединения типа комплексов с лигандами сильного поля. Отнесение линий кислорода O1s и азота N1s (табл. 3) соответствует структурным формулам изученных образцов. Таким образом, по результатам анализа РФЭ-спектров линии Со2р комплексов Ia и I идентифицированы как комплексы сильного поля с Co(III) в диамагнитном состоянии.
Термическое поведение комплексов I и II было исследовано методом СТА в атмосфере аргона (с одновременной регистрацией кривых ТГ и ДСК) до 450°С. Различная природа гетероатомов, входящих в состав комплексов, а также разный способ структурной организации (I – молекулярного строения; II – полимер) обусловила принципиально различный характер протекания термического разложения (рис. 4). Комплекс I устойчив до 125°С. На кривой ТГ можно выделить следующие стадии термолиза (рис. 4а, табл. 4): первая протекает в диапазоне 125–206°С и включает два небольших эндоэффекта на кривой ДСК (рис. 4б) с экстремумами 167 и 196°С. Два близких эндоэффекта соответствуют десорбции двух молекул DAP с выделением двух молекул N2 (mтеор/эксп = = 6.2/6.3%).
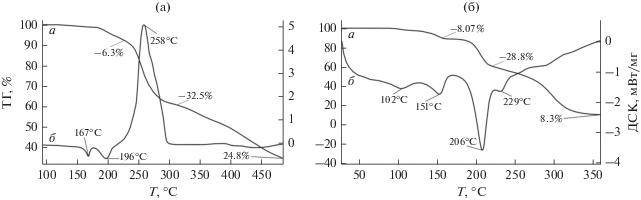
Таблица 4.
Данные СТА для комплексов I и II*
| Комплекс | Этап/ΔT, °С | Δm (TГ), % | Tэндо/экзо, °С | mфин, % |
|---|---|---|---|---|
| I | 1 (125–206) | 6.3 | 167/196 ± 0.7 | 24.8 |
| 2 (206–295) | 32.3 | 258 ± 0.7 | ||
| 3 (295–500) | 36.6 | |||
| II | 1 (89–117) | 3.5 | 102 ± 0.7 | 8.3 |
| 2 (117–161) | 8.1 | 151 ± 0.7 | ||
| 3 (161–216) | 28.8 | 206 ± 0.7 | ||
| 4 (216–400) | 50.8 | 229 ± 0.7 | ||
| [Сd(Piv)2]n | 1 (55–104) | 8.0 | 95 ± 0 .7 | 19.8 |
| 2 (104–172) | 6.9 | 162 ± 0.7 | ||
| 3 (172–415) | 64.95 | 348 ± 0.7 (плав.) | ||
| 412 ± 0.7 |
За разрушением азотсодержащего фрагмента следует этап деструкции {Cd(Piv)3} (о том, что первым протекает процесс разрушения азотсодержащего фрагмента свидетельствует Eдисс связейN–C 70; О–С 84 ккал/моль [31]). Данная стадия на кривой ДСК (рис. 4б) характеризуется высокоинтенсивным экзотермическим пиком (экстремум 258°С), соответствующим возгонке газообразных продуктов термолиза фрагмента {Cd(Piv)3} (mтеор/эксп = 33.5/32.3%). (Термолиз индивидуального комплекса [Cd(Piv)2]n (табл. 4) также показывает, что термическое разложение сопровождается высокоинтенсивным экзотермическим пиком с экстремумом при 412°С.) Потеря массы на кривой ТГ для I (рис. 4а) в интервале 206–589°С соответствует постепенной деструкции пивалатной части (mтеор/эксп = 61.0/58.8%). Это согласуется с данными ранее проведенных экспериментов для пивалатных комплексов [29–35]).
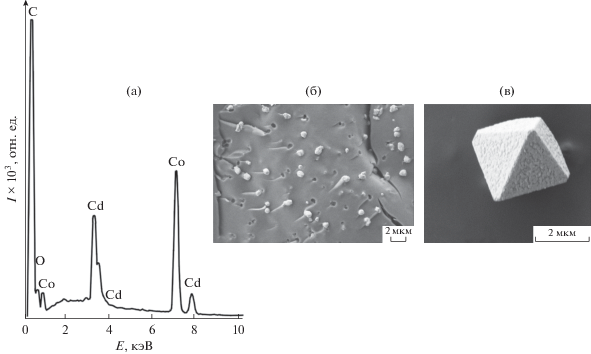
В отличие от I комплекс II менее термостабилен, и деструкционные процессы начинаются уже с 89°С (рис. 4б, табл. 4). Первый этап соответствует разрушению диаминопропана с выделением двух молекул азота (mтеор/эксп = 8.04/8.07%). Температурные данные этого процесса в комплексе II несколько сдвинуты в низкотемпературную область по сравнению с I (табл. 4). Четко выраженный на кривой ТГ этап потери массы (28.8%) соответствует удалению двух групп Piv–, связанных водородными связями с молекулами DAP (mтеор = 28.9%) (рис. 2, 4б). Заключительная стадия соответствует деструкции оставшихся органических частей (суммарная потеря массы составила 79.6%; mтеор = 79.4%). Финальная масса (8%) свидетельствует о летучести исследованного комплекса (на крышке тигля после эксперимента наблюдаются следы “побежалости” металла). В отличие от II остаточная масса для комплекса I составляет 24.8% и соответствует CdCoO2 (mтеор = 22.5%; небольшое завышение массы связано с зауглероженностью остатка после термолиза в инертной атмосфере). Высокоинтенсивный пик от углерода на энергодисперсионном спектре соответствует материалу подложки (рис. 5).
Рис. 5.
Энергодисперсионный спектр (а) и фотографии микроморфологии (б – ×10 000; в – ×30 000 раз) остатка I.
Таким образом показано, что моноядерный комплекс Ia реагирует с пивалатами кадмия и лития с образованием биядерного гетерометаллического молекулярного комплекса I и гетерометаллического 1D-координационного полимера II соответственно. Координационное окружение атома кобальта и степень окисления +3 в этих соединениях сохраняются. При замещении анионов триметилуксусной кислоты на анионы дикарбоновой кислоты (например, терефталиевой) существует реальная возможность получения новых гетерометаллических каркасных полимеров с атомами Co(III). Сохранение исходного координационного окружения позволяет надеяться на вероятность его сохранения и в каркасных структурах на основе мостиковых анионов дикарбоновых (и поликарбоновых кислот).
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Cидоров А.А., Фомина И.Г., Талисманов С.С. и др. // Коорд. химия. 2001. Т. 27. № 8. С. 584.
Михайлова Т.Б., Фомина И.Г., Сидоров А.А. и др. // Журн. неорган. химии. 2003. Т. 48. № 10. С. 1648.
Малков А.Е., Фомина И.Г., Сидоров А.А. и др. // Изв. АН. Сер. хим. 2003. № 2. С. 489.
Михайлова Т.В., Пахмутова Е.В., Малков А.Е. и др. // Изв. АН. Сер. хим. 2003. № 10. С. 1994.
Пахмутова Е.В., Малков А.Е., Михайлова Т.В. // Изв. АН. Сер. хим. 2003. №. 1. С. 131.
Malkov A.E., Fomina I.G., Sidorov A.A. et al. // J. Mol. Struct. 2003. V. 656. № 1–3. P. 207.
Фомина И.Г., Сидоров А.А., Александров Г.Г. и др. // Изв. АН. Сер. хим. 2004. № 7. С. 1422.
Александров Г.Г., Фомина И.Г., Сидоров А.А. и др. // Изв. АН. Сер. хим. 2004. № 6. С. 1153.
Сидоров А.А., Никифирова М.Е., Пахмутова Е.В. и др. // Изв. РАН. Сер. хим. 2006. № 11. С. 1851.
Kiskin M.A., Fomina I.G., Aleksandrov G.G. et al. // In-org. Chem. Commun. 2004. V. 7. № 6. P. 734.
Eremenko I.L., Kiskin M.A., Fomina I.G. et al. // J. Cluster Sci. 2005. V. 16. № 3. P. 331.
Sapianik A.A., Zorina-Tikhonova E.N., Kiskin M.A. et al. // Inorg. Chem. 2017. V. 56. P. 1599.
Sapianik A.A., Kiskin M.A., Samsonenko D.G. et al. // Polyhedron. 2018. V. 145. P. 147.
Сапьяник А.А., Луценко И.А., Кискин М.А. и др. // Изв. АН. Cер. хим. 2016. № 11. С. 2106.
Сидоров А.А., Талисманова М.О., Фомина И.Г. и др. // Изв. АН. Сер. хим. 2001. №. 11. С. 2106.
Kiskin M.A., Sidorov A.A., Fomina I.G. et al. // Inorg. Chem. Commun. 2005. V. 8. № 6. P. 524.
Kiskin M.A., Fomina I.G., Aleksandrov G.G. et al. // In-org. Chem. Commun. 2005. V. 8. № 1. P. 89.
Dobrokhotova Z., Emelina A., Sidorov A. et al. // Polyhedron. 2011. V. 30. P. 132.
Dobrohotova Z.V., Sidorov A.A., Kiskin M.A. et al. // J. Solid State Chem. 2010. V. 183. P. 2475.
Sapianik A.A., Kiskin M.A., Kovalenko K.A. et al. // Dalton Trans. 2019. V. 48. P. 3676.
Фомина И.Г., Александров Г.Г., Доброхотова Ж.В. и др. // Изв. РН. Сер. хим. 2006. С. 1841.
Sheldrick G.M. // Acta Crystallogr. A. 2008. V. 64. P. 112.
Bourhis L.J., Dolomanov O.V., Gildea-Bourhis R.J. et al. // Acta Crystallogr. A. 2015. V. 71. P. 59.
Гоголева Н.В., Сидоров А.А., Нелюбина Ю.В. и др. // Коорд. химия. 2018. Т. 44. № 8. С. 473 (Gogoleva N.V., Sidorov A.A., Nelyubina Yu.V. et al. // J. Cood. Chem. 2018. V. 44. № 4. P. 219.).https://doi.org/10.1134/S107032841808002X
Frost D.C., McDowell C.F., Woolsey I.S. // Mol. Phys. 1974. V. 27. P. 1473.
Okamoto Y., Nakano H., Imanaka T. // Bull. Chem. Soc. Jpn. 1975. V. 48. P. 1163.
Баринский Р.Л., Нефедов В.И. Рентгено-спектральное определение заряда атомов в молекулах. М.: Наука, 1966. 247 с.
Ivanova T., Naumkin A., Sidorov A. et al. // J. Electron Spectrosc. Relft. Phenom. 2007. V. 156–158. P. 200.
Burger K., Furlani C., Mattogno G. // J. Electron Spectrosc. 1980. V. 21. P. 249.
Cкворцова Л.И. Магнетометрический метод анализа. Новосибирск: Гео, 2006. 157 с.
Лидин Р.А., Андреева Л.Л., Молочко В.А. Константы неорганических веществ. М.: Дрофа, 2008. 686 с.
Lutsenko I.A., Kiskin M.A., Efimov N.N. et al. // Polyhedron. 2017. V. 137. P. 165.
Луценко И.А., Кискин М.А., Александров Г.Г. и др. // Изв. АН. 2018. Cер. хим. № 3. С. 449.
Lutsenko Irina A., Kiskin Mikhail A., Nelyubina Yulia V. et al. // Polyhedron. 2019. V. 159. P. 426.
Фомина И.Г., Александров Г.Г., Доброхотова Ж.В. и др. // Изв. АН. Cер. хим. 2006. № 11. С. 1841.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия