

Координационная химия, 2019, T. 45, № 3, стр. 131-157
Управляемый молекулярный магнетизм би- и полиядерных комплексов переходных металлов на основе гидразонов, азометинов и их аналогов
В. В. Луков 1, *, И. Н. Щербаков 1, С. И. Левченков 1, 2, Ю. П. Туполова 1, Л. Д. Попов 1, И. В. Панков 1, С. В. Посохова 3
1 Южный федеральный университет
Ростов-на-Дону, Россия
2 Южный научный центр РАН
Ростов-на-Дону, Россия
3 Азово-Черноморский инженерный институт,
Донской государственный аграрный университет
Зерноград, Россия
* E-mail: vlukov@mail.ru
Поступила в редакцию 13.04.2018
После доработки 05.09.2018
Принята к публикации 12.07.2018
Аннотация
В настоящем обзоре предпринята попытка изложить возможности магнетохимического метода при описании строения и свойств би- и тетраядерных металлхелатов и супрамолекулярных архитектур на основе координационных соединений с ограниченным по типам числом лигандных систем, главным образом гидразонов, азометинов и их аналогов. Систематизированы известные литературные данные для би- и полиядерных комплексов, парамагнитные центры которых связаны как внутри-, так и межмолекулярным обменным взаимодействием. Рассмотрена взаимосвязь между особенностями электронного и геометрического строения комплексов и характером обменных эффектов, проведен систематический анализ магнетоструктурных корреляций в обсуждаемых соединениях, представляющих собой важные модельные объекты при разработке стратегии направленного дизайна одно-, двух- и трехмерных магнитно-упорядоченных структур.
ВВЕДЕНИЕ
Получение и анализ физико-химических свойств полиядерных комплексов металлов с магнитными и электронными взаимодействиями между металлическими центрами представляют собой одно из центральных направлений исследований современной координационной химии, молекулярной электроники и молекулярной биологии [1–17]. C фундаментальных позиций очевидно, что обменное взаимодействие и электронный перенос представляют собой тесно взаимосвязанные процессы, в основе которых лежат эффекты взаимодействия спина и электростатического заряда электронов. Транспорт электронов в рамках электронных устройств или в металлопротеинах представляет собой замечательный пример межэлектронного взаимодействия в искусственных и природных системах соответственно. Необходимо также отметить, что молекулы-магниты (SMM’s) и многоядерные координационные соединения, проявляющие эффект спин-кроссовера, могут рассматриваться как весьма удобные системы, моделирующие уникальные магнитные свойства, характерные для обычных неорганических магнитных материалов (металлические сплавы и их оксиды), и дополняющие их технологически значимыми оптическими, электронными, хемосенсорными и адсорбционными свойствами [18–22]. Не вызывает сомнений то обстоятельство, что детальный экспериментально-теоретический анализ подобных систем и явлений приведет исследователей молекулярного магнетизма к выделению принципиально новых классов управляемых извне многофункциональных молекулярных магнитных материалов [23]. Пористые и люминесцентные магниты, объекты нелинейной оптики, мультиферроики, протонные и электронные магнитные проводники, с одной стороны, а также термо-, пьезо-, фото- или хемопереключаемые магниты с другой, представляют яркие иллюстративные примеры нанотехнологических систем ближайшего будущего [24–35]. Считается общепринятым, что теоретической основой бурного развития основных положений молекулярного магнетизма послужили классические работы Р. Хоффмана и О. Кана [36–41]. В последующие годы работы данных ученых и возглавляемых ими научных коллективов определили пути рационального дизайна и синтеза обменно-связанных гомо- и гетеробиядерных комплексов переходных металлов с прогнозируемыми магнитными свойствами, иллюстрируя таким образом способность исследователей преобразовывать данные природой материалы в соответствии с задачами научно-технического прогресса [42–45]. Тем не менее к настоящему моменту изучение электронного строения и свойств полиядерных комплексов, в которых два или больше парамагнитных центров связаны магнитным обменным взаимодействием, остается одной из актуальных задач координационной химии [17, 46, 47]. При этом выявление взаимосвязи параметров обменного взаимодействия с особенностями строения полиядерных комплексов играет ключевую роль для понимания магнитных свойств подобных систем и составляет теоретическую основу для синтеза обменно-связанных систем с заданными магнитными характеристиками и в конечном счете для направленной модификации магнитоактивных материалов [48–50]. В настоящем обзоре предпринята попытка систематизировать известные литературные данные для би- и полиядерных комплексов, парамагнитные центры которых связаны как внутри-, так и межмолекулярным обменным взаимодействием с лигандами на основе органических производных гидразина, оснований Шиффа и родственных им систем. Очевидно, что потенциальные магнетоструктурные корреляции, полученные для данных классов соединений, могут быть весьма полезными для решения более широкого круга обозначенных выше проблем. В отдельной главе рассмотрены особенности строения и магнитных свойств тетраядерных комплексов, олигомерная структура которых является важным промежуточным звеном между “классическими” биядерными производными и полимерными супрамолекулярными архитектурами.
I. МАГНЕТОСТРУКТУРНЫЕ КОРРЕЛЯЦИИ В БИЯДЕРНЫХ КОМПЛЕКСАХ, СОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИЕ ФРАГМЕНТЫ
Гетарилгидразоны карбонильных соединений – популярные объекты современной органической и координационной химии благодаря их биологической активности. В литературе имеются многочисленные данные об их антибактериальной, противовирусной, противоопухолевой и противосудорожной способности [51–58]. Многие гетарилгидразоны являются хромогенными агентами, экстрагентами, аналитическими сенсорами на ионы металлов [59, 60]. Высокая комплексообразующая активность гетарилгидразонов в сочетании с относительной легкостью синтетического варьирования деталей структуры таких систем позволяет получать биядерные комплексы, представляющие собой, как отмечено выше, удобные модели для изучения основных факторов, определяющих характер и силу обменных взаимодействий между парамагнитными центрами вследствие возможности варьирования природы донорных центров, центральных ионов и экзогенных мостиковых групп; обмен может иметь как антиферро-, так и ферромагнитный характер. Так, например, в [61] представлены результаты синтеза, физико-химического исследования и квантово-химического моделирования биядерных комплексов меди(II) с 1'-фталазинилгидразонами замещенных салициловых альдегидов.
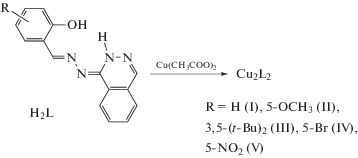
Биядерное строение I–V подтверждено наличием сильного антиферромагнитного обменного взаимодействия между ионами меди(II), причем присутствие в гидразонах H2L фталазинового фрагмента существенно расширяет потенциальные возможности данных лигандных систем в образовании биядерных комплексов. Описанные ранее гетарилгидразоны салицилового альдегида способны образовывать димерные комплексы только в том случае, когда мостиковую функцию выполняют феноксидные атомы кислорода [62–64]. Строение гидразонов H2L предопределяет два возможных способа димеризации: через феноксидные атомы альдегидного фрагмента с образованием четырехчленного металлохелатного цикла Cu–O(O')–Cu либо через атомы фталазинового фрагмента с образованием шестичленного цикла Cu–N–N'(N''–N''')–Cu. В этом отношении гидразоны H2L в известной степени сходны с ацил- и ароилгидразонами салицилового альдегида, для которых также возможны два способа димеризации: через феноксидные либо α-оксиазинные атомы кислорода альдегидного или гидразонного фрагментов соответственно [65]. Таким образом, для биядерных комплексов I–V возможна реализация димерных структур типа A или B:
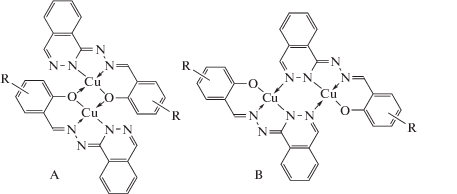
В случае комплекса III наличие трет-бутильной группы в орто-положении к фенольной группировке исключает возможность реализации димерной структуры A [66]. То обстоятельство, что комплекс III, безусловно, биядерный, поскольку в нем имеет место сильный антиферромагнитный обмен (2J = –416 см–1), однозначно указывает на реализацию в данном случае структуры типа B с димеризацией через диазинные мостики. В остальных случаях теоретически возможна реализация любой из изомерных структур.
Следует отметить, что существенное отличие биядерных комплексов меди(II) с 1'-фталазинилгидразонами производных салицилового альдегида от димерных комплексов меди(II) с ацилгидразонами заключается в характере магнитного обменного взаимодействия в координационных изомерах. Как показано ранее [66–71], в случае ацилгидразонов салицилового альдегида разные способы димеризации приводят к существенному различию магнитных свойств изомеров. В обоих случаях обмен антиферромагнитный, однако при димеризации через феноксидные атомы в комплексах имеет место более сильное обменное взаимодействие; величины параметров обмена составляют в этом случае –300…–500 против –50…–150 см–1 в случае димеризации через α-оксиазинные атомы кислорода [8, 24]. Это различие в магнитных свойствах позволяет на основании данных магнетохимического исследования определить структуру изомера. Однако для биядерных комплексов меди(II) с 1'-фталазинилгидразонами производных салицилового альдегида так называемый “магнетохимический критерий определения структуры изомера” [72] неприменим, поскольку в комплексах различных фталазинилгидразонов через диазинные мостики реализуется сильный антиферромагнитный обмен [73–75].
С целью теоретической интерпретации влияния способа димеризации в биядерных комплексах I–V на обменное взаимодействие авторами был выполнен квантово-химический расчет параметров обмена методом теории функционала плотности. Молекулярная структура димеров A и B (R = H) в вакууме по данным расчетов показана на рис. 1.
Рис. 1.
Молекулярная структура димеров A и B по данным расчета, R = H (справа вверху – вид в другой проекции) [61].
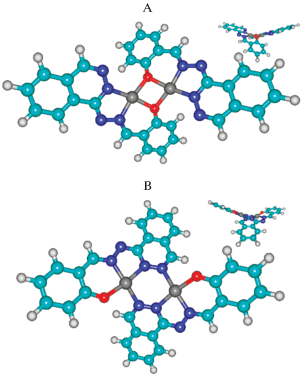
Во всех случаях оптимизация геометрии приводит к структурам, обменный фрагмент в которых неплоский. В димерах A четырехчленный цикл Cu–O(O')–Cu слегка искажен в результате перегиба по линии, соединяющей мостиковые атомы кислорода; диэдральный угол между среднеквадратичными плоскостями координационных полиэдров атомов меди составляет ~30° (рис. 1). Расстояния медь–медь в димерах A не зависят от природы заместителя R и составляют 2.97–2.98 Å.
Следует отметить, что расстояние медь–медь в комплексах меди(II) с гидразонами салицилового альдегида, в которых димеризация осуществляется через феноксидный атом кислорода, по данным РСА, равно 3.01–3.05 Å [76–80], что весьма близко к результатам расчета геометрии димеров A. Некоторое занижение расстояния обусловлено вышеупомянутым искажением обменного фрагмента, который, по данным РСА, обычно является совершенно плоским. В димерах B шестичленный обменный фрагмент Cu–N–N'(N''–N''')–Cu имеет, согласно результатам расчета геометрии, конформацию ванны с атомами меди в апикальных позициях (рис. 1б). Как и в предыдущем случае, природа заместителя R слабо влияет на степень искажения обменного фрагмента, и расстояния медь–медь достаточно близки (3.70–3.73 Å). Сравнение полных энергий триплетных состояний димеров A и B показало, что димеризация через атомы азота фталазиновых фрагментов оказывается выгоднее. Полная энергия димеров B на 8.8–12.9 ккал/моль ниже, чем димеров A, что составляет весьма существенную величину. Это позволяет предположить, что в комплексах I–V димеризация осуществляется именно через фталазинные мостики. Хорошим аргументом в пользу этого предположения является димерный характер комплекса III, для которого, как было сказано выше, димеризация через феноксидные атомы (структура A) исключена по стерическим причинам.
Рассчитанные и экспериментальные значения параметров обмена в димерных структурах A и B представлены в табл. 1. Для димеров A (с димеризацией через феноксидный атом кислорода) абсолютная величина параметра обмена для любых заместителей R заметно больше, чем для B, однако в целом результаты квантово-химического моделирования обменного взаимодействия в димерах A–B не позволяют сделать однозначный выбор между координационными изомерами комплексов I–V, хотя с экспериментом лучше согласуются рассчитанные величины 2J для димеров B с димеризацией через фталазиновые фрагменты.
Таблица 1.
Рассчитанные величины параметров обмена (см–1) в димерных структурах A–B и экспериментально полученные значения 2J в комплексах I–V
| Соединение | R | 2Jрасч | 2Jэксп | |
|---|---|---|---|---|
| димер A | димер B | |||
| I | H | –712 | –595 | –485 |
| II | 5-OCH3 | –754 | –593 | –445 |
| III | 3,5-(t-Bu)2 | – | –609 | –416 |
| IV | 5-Br | –699 | –606 | –416 |
| V | 5-NO2 | –642 | –629 | –402 |
Таким образом, решающим аргументом в проблеме выбора между координационными изомерами комплексов I–V может быть только экспериментальное определение структуры комплексов. К сожалению, авторам не удалось получить пригодные для РСА монокристаллы биядерных комплексов, поэтому был использован другой экспериментальный метод, способный дать информацию о структуре комплексов – рентгеновская спектроскопия поглощения (XAS), позволяющая определять локальную структуру координационной сферы ионов переходных металлов. Близость структурных параметров координационных узлов в комплексах I–V, полученных в рамках данного метода, доказывает реализацию во всех соединениях одного и того же способа димеризации. Величина расстояния медь–медь позволяет сделать экспериментально обоснованный вывод о структуре димеров. Как отмечалось выше, расстояние медь–медь в комплексах с димеризацией через феноксидный атом салицилальдегидного фрагмента не может превышать 3.1 Å. Поскольку погрешность определения расстояние металл–металл методом EXAFS составляет около 0.02–0.05 Å [81, 82], полученные данные позволяют однозначно исключить реализацию в данном случае димерных структур A. Меньшее по сравнению с рассчитанным расстояние Cu–Cu, по данным EXAFS, может быть обусловлено искажением структуры биядерной молекулы за счет межмолекулярных взаимодействий в кристаллической упаковке.
Таким образом, на основании совокупности данных магнетохимии, квантово-химических расчетов и ХАS можно с высокой степенью вероятности утверждать, что в комплексах меди(II) с 1'-фталазинилгидразонами замещенных производных салицилового альдегида димеризация реализуется через атомы азота фталазиновых фрагментов. Однозначное подтверждение предположению о том, что косвенное обменное взаимодействие между ионами меди(II) c участием фталазиновых фрагментов носит выраженный антиферромагнитный характер, приведено в [83], посвященной физико-химическому исследованию 1'-фталазинилгидразона 2-(N-тозиламино)бензальдегида (H2L) и комплексов переходных металлов на его основе.
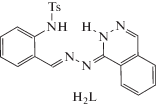
Синтез комплексов осуществлен в соответствии со схемой:
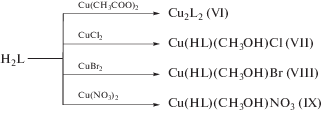
Взаимодействием гидразона H2L с ацетатом, хлоридом и бромидом меди(II) получили комплексы состава Cu2L2 и Cu(HL)(СН3ОН)X (L2– и HL– – дважды- и монодепротонированная форма гидразона, X = Cl, Br) (VI–VIII). Взаимодействие гидразона H2L с нитратом меди(II) приводит к выделению комплекса состава Cu(HL)(СН3ОН)NO3 (тип IX).
Измерение электропроводности 0.001 М растворов комплексов в ДМФА показало, что комплекс IX можно отнести к двойным электролитам [84], что указывает на внешнесферный характер координации входящего в состав комплекса нитрат-аниона. Данные ИК-спектроскопии указывают на координацию лиганда с ацетатом меди(II) в дважды депротонированной, с галогенидами и нитратом меди(II) – в монодепротонированной форме соответственно. Эффективный магнитный момент (μэф) комплекса VI при комнатной температуре (1.23 М.Б.) заметно ниже чисто спинового значения (1.73 М.Б.) и понижается до 0.18 М.Б. при охлаждении до точки кипения жидкого азота, величина параметра обмена 2J составляет –416 см–1. Эти данные однозначно указывают на то, что комплекс имеет димерное строение. В данном случае возможен только один способ димеризации комплекса – через атомы азота фталазинового фрагмента, поскольку выполнение мостиковой функции атомом азота альдегидного фрагмента практически исключено из-за стерических препятствий, создаваемых тозильной группой. Величина обменного параметра в комплексе VI укладывается в интервал, характерный для биядерных комплексов меди(II) с диазиновыми мостиками (для подобных соединений в литературе приводятся значения 2J от –300 до –600 см–1) [85–87].
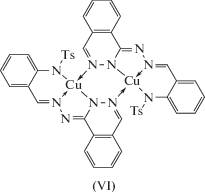
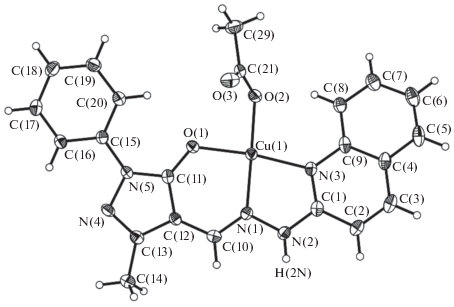
Строение комплекса VI было установлено также и методом РСА (рис. 2), результаты которого подтвердили выводы, сделанные на основе спектральных и магнетохимических исследований. Ион меди(II) в молекуле биядерного комплекса пентакоординирован. Три положения в координационной сфере иона Cu2+ занимают атомы азота одного лиганда, расположенные на примерно одинаковом расстоянии от иона меди: Cu(1)–N(1) 1.934(7), Cu(1)–N(2) 1.944(8) и Cu(1)–N(5) 1.937(8) Å, четвертое – атом N(4#) второго лиганда на заметно большем расстоянии (Cu(1)–N(4#) 2.014(8) Å), через который и осуществляется димеризация моноядерных фрагментов с образованием шестичленного обменного цикла Cu(1)–N(5)–N(4)–Cu(1#)–N(5#)–N(4#). Завершает координационную сферу иона меди атом кислорода O(2) тозильного фрагмента. Пятичленный хелатный цикл имеет конформацию конверта, “клапан” которого занимает атом меди, отклоняясь от средней плоскости остальных четырех атомов на 0.171(1) Å. Шестизвенный хелатный цикл искажен значительно сильнее за счет перегиба по линиям N(1)–С(7) и N(1)–N(2). Строение мономерных комплексных фрагментов близко к плоскому (за исключением тозильных фрагментов, находящихся во “флагштоковой” конформации по отношению к плоскости лиганда). Плоскости двух мономерных фрагментов составляют двугранный угол 64.4°. В результате, в отличие от описанных в литературе комплексов с фталазиновыми мостиковыми группировками [85], плоскость шестичленного обменного фрагмента в VI значительно искажена и по конформации близка к ванне с атомами меди в апикальных позициях. Атомы меди располагаются на расстоянии 3.367(1) Å друг от друга. Авторы отмечают, что для описанного в [88] биядерного комплекса меди(II) с 1'-фталазинилидразоном 2-ацетилбензимидазола, в котором стерические затруднения менее существенны, квантово-химический расчет приводит к похожей структуре обменного фрагмента. За счет стерического отталкивания плоскости фенильных колец тозильных фрагментов в структуре VI практически параллельны, угол между ними составляет ~9°.
Рис. 2.
Структура комплекса VI (атомы водорода не показаны, эллипсоиды тепловых колебаний приведены с вероятностью 50%); проекции структуры комплекса вдоль линии Cu–Cu (а) и перпендикулярно ей (б) [83].
Было проведено квантово-химическое моделирование обменного взаимодействия в комплексе VI в рамках метода “нарушенной симметрии”. Рассчитанное в этом приближении значение параметра антиферромагнитного обмена 2J = –457 см–1 удовлетворительно согласуется с определенной из экспериментальных данных величиной (2J = = –416 см–1).
Эффективные магнитные моменты комплексов VII–IX близки к чисто спиновым значениям для иона Cu2+ и практически не изменяются при охлаждении. Отсутствие обменного взаимодействия между ионами Cu2+ в совокупности с другими результатами физико-химического исследования позволило авторам с высокой долей вероятности приписать им следующее мономерное строение:
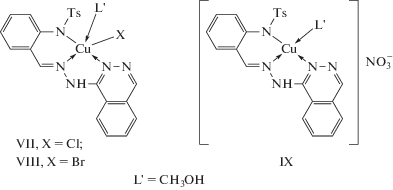
Вместе с тем для некоторых обменно-связанных комплексов с фталазинилгидразонами только данные магнетохимического анализа позволяют осуществить достаточно надежное отнесение структуры. Об этом свидетельствуют результаты [89], посвященной физико-химическому исследованию гетарилгидразонов глиоксалевой кислоты X и комплексов переходных металлов на их основе.
Как отмечалось выше, согласно данным РСА, электронной, ИК- и ЯМР-спектроскопии, известные гидразоны на основе 1-гидразинофталазина в свободном состоянии существуют во фталазоновой таутомерной форме (в форме смешанных азинов) [90]. При взаимодействии гидразонов X с ацетатом меди(II) были получены металлохелаты состава Cu2L2, где L – дважды депротонированная форма гидразонов. Измерение магнитной восприимчивости комплексов показало, что для всех соединений значения µэф в расчете на один ион меди при комнатной температуре существенно меньше чисто спиновой величины и понижаются при охлаждении до температуры кипения жидкого азота, что свидетельствует о наличии обменного взаимодействия антиферромагнитного типа между двумя ионами меди и позволяет предположить димерную структуру металлохелатов типа XI–XIII.
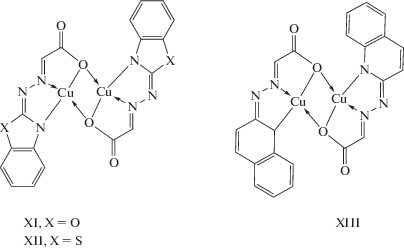
Способ димеризации комплексов на основе данных гидразонов однозначно задается структурой лиганда, поскольку выполнение мостиковой функции гетероциклическими атомами азота в данном случае невозможно. В случае комплекса на основе фталазинилгидразона типа X димеризация моноядерных фрагментов комплекса может реализоваться как через атомы кислорода карбоксильной группы, так и через диазиновую цепочку атомов азота фталазинового фрагмента с реализацией структур типа XIV и XV соответственно.
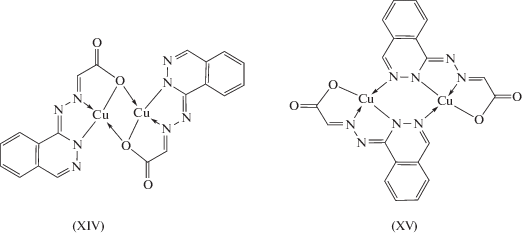
Как показали результаты магнетохимического анализа, данный комплекс, как и комплексы XI–XIII, характеризуется не очень значительным антиферромагнитным обменным взаимодействием (2J = –136 см–1). Такая величина параметра обмена, согласно развитой в [65, 72] концепции геометрического моделирования обменных фрагментов биядерных комплексов переходных металлов с гидразонами, свидетельствует о реализации димерной структуры типа XIV. В случае реализации биядерной структуры типа XV значение параметра обменного взаимодействия, как уже обсуждалось выше, должно быть существенно больше по абсолютной величине [91–93].
Сочетание магнетохимического эксперимента, квантово-химических расчетов и данных EXAFS спектроскопии позволяет, даже в отсутствие результатов РСА, делать практически однозначные выводы в пользу реализации определенной структуры обменно-связанных комплексов с фталазинилгидразоновыми лигандами, о чем свидетельствуют результаты работы [94]. В данной работе приводятся результаты изучения строения комплексов 1'-фталазинилгидразона диацетилмонооксима, полученных реакцией с нитратом меди(II) (комплексы XVI–XVIа),
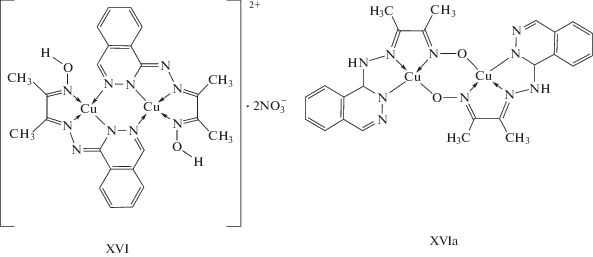
структурные модели которых были предложены на основе геометрического моделирования с использованием стандартных длин связей и валентных углов. Пространственное строение моделей структуры комплекса типа XVI изображено на рис. 3.
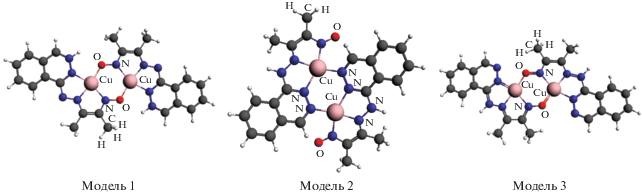
Оптимизированные структурные модели комплекса XVI были использованы в дальнейшем для расчета спектров рентгеновского поглощения (XANES). Как оказалось, наилучшее согласие с экспериментальным XANES дает спектр, вычисленный для структурной модели 2. Для уточнения наиболее вероятных моделей строения комплекса XVI данное исследование было дополнено встречным квантово-химическим моделированием обменного взаимодействия. В этом комплексе антиферромагнитное обменное взаимодействие достаточно велико (2J = –295 см–1). Такая величина обмена и данные физико-химического исследования говорят о том, что в соединении мостиковую функцию могут выполнять либо атомы азота фталазиновых фрагментов, и комплекс имеет строение XVI, либо атомы нитрозогруппы, что соответствует структуре XVIа. В обоих случаях обменный фрагмент должен быть близок к плоскому, что будет способствовать эффективному перекрыванию “магнитных” атомных dx2–y2-орбиталей ионов меди(II), реализуемому с участием орбиталей мостиковых атомов.
Для оценки величин параметра магнитного обмена для возможных изомеров и их сопоставления с экспериментально измеренными величинами, в рамках подхода “нарушенной симметрии” авторами были рассчитаны значения 2J для альтернативных структур комплексов (табл. 2).
Таблица 2.
Полные энергии (а.е.) высокоспинового состояния E(HS), состояния с нарушенной симметриeй E(BS), рассчитанные значения 2J (см–1) для комплексов типа XVI
| Тип | E(HS) | E(BS) | 2Jрасч, см–1 |
|---|---|---|---|
| Модель 1 | –4908.232104 | –4908.239578 | –1640 |
| Модель 2 | –4908.222266 | –4908.223993 | –379 |
| Модель 3 | –4908.220893 | –4908.227890 | –1536 |
В случае дикатионных комплексов типа XVI, образующихся за счет внешнесферной координации нитрат-анионов взаимодействие носит антиферромагнитный характер для всех рассмотренных изомерных структур, различается только его величина (табл. 2), причем довольно резко. Так, координация через нитрозогруппу (цепочка N=O) определяет очень сильное взаимодействие, 2J = –1640 или –1536 см–1 для моделей 1 и 3 соответственно. В случае модели 2 (димеризация через диазиновые цепочки фталазиновых фрагментов) величина синглет-триплетного расщепления составляет 379 см–1, что соответствует экспериментально полученной величине (2J = –295 см–1) и согласуется с приведенными выше результатами исследования XANES спектроскопии.
Резюмируя сказанное выше, можно констатировать, что гетероциклические, в первую очередь, фталазинильные мостики, – весьма удобные объекты для экспериментально-теоретического анализа особенностей проявления обменных эффектов в биядерных металлохелатах на основе гидразоновых лигандов. Подобные системы могут быть использованы также в качестве удобных линкеров и для получения цепочечных полимерных обменно-связанных структур [95–100]. Как отмечают авторы данных исследований, электронные и геометрические факторы: состав мостика, валентные и диэдральные углы в обменно-связанных хелатных циклах и межъядерное расстояние между парамагнитными центрами – основные факторы в формировании обменного взаимодействия определенной природы и силы. Эти системы представляют собой важные модельные объекты при разработке стратегии направленного дизайна одно-, двух- и трехмерных магнитно-упорядоченных структур, открывающих, по мнению авторов вышеуказанных работ, новую главу в физико-химии управляемого молекулярного магнетизма, причем подобное упорядочение во многих случаях реализуется за счет эффектов межмолекулярного магнитного взаимодействия, некоторые важные детали которого рассмотрены в следующем разделе.
II. КОМПЛЕКСЫ С МЕЖМОЛЕКУЛЯРНЫМ ОБМЕННЫМ ВЗАИМОДЕЙСТВИЕМ, ТРАНСЛИРУЕМЫМ ПО СИСТЕМЕ ВОДОРОДНЫХ СВЯЗЕЙ
Межмолекулярные нековалентные взаимодействия, определяющие молекулярную упаковку функциональных наноразмерных материалов, могут рассматриваться в качестве основной “движущей силы” создания или средства управления магнитным обменным взаимодействием в супрамолекулярных архитектурах [101]. В частности, реализация водородных связей, катион-анионные, π–π-взаимодействия и взаимодействия ван-дер-ваальса между отдельными молекулами могут быть направленно использованы для формирования каналов обменного взаимодействия, величина которого может быть предопределена как по знаку, так и по абсолютному значению [102]. В цитируемых выше работах отмечается, что указанные взаимодействия в ряде случаев могут быть столь же и даже более эффективными в формировании ферро-, антиферро- и ферримагнетиков, как и традиционные ковалентные взаимодействия, определяющие внутримолекулярный характер обмена.
Вместе с тем направленный дизайн магнитных материалов с использованием нековалентных взаимодействий достаточно ограничен, вследствие того что подобные эффекты, с одной стороны, могут действовать как организаторы кристаллической архитектуры и в некоторых случаях как посредники магнитного обмена между металлическими центрами; с другой стороны, характер и проявление магнитных параметров не столь однозначно предсказуем, как в случае внутримолекулярного обмена [103–108]. В этой связи разработка относительно новых подходов к теоретическому моделированию обмена в таких объектах, учитывающих разнообразие структурных факторов, вовлеченных в межмолекулярные взаимодействия, природу акцепторных и донорных атомов, электронную природу взаимодействующих металлических центров и т.д., является актуальной и важной. Справедливость последнего положения подтверждается приведенными ниже примерами.
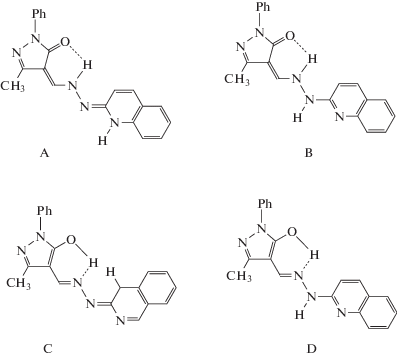
Так, работа [109] посвящена исследованию структуры, магнитных свойств и квантово-химическому моделированию комплекса меди(II) типа XVII c хинолилгидразоном 1-фенил-3-метил-4-пиразола-5, в котором зарегистрирован межмолекулярный обмен по каналу водородной связи NH···O. Для указанной лигандной системы методом ЯМР спектроскопии и квантово-химических расчетов из четырех возможных таутомерных форм A–D структура A выбрана в качестве наиболее стабильной.
Подобная структура лиганда предполагала получение моноядерного металлохелата меди(II), однако данные ЭПР спектроскопии и магнетохимических измерений показали наличие слабого обменного взаимодействия антиферромагнитного типа между ионами меди. В ЭПР спектре был зарегистрирован сигнал с g = 4.121, отвечающий запрещенному переходу в димерных комплексах меди(II) [110], что подтверждается магнитными свойствами данного соединения, интерпретированными с помощью выражения (1), учитывающего как парный, так и межмолекулярный об-мен,
(1)
${{\chi }_{m}} = \frac{{\chi _{m}^{'}}}{{{{1 - 7.6853zj{\text{'}}x_{m}^{'}} \mathord{\left/ {\vphantom {{1 - 7.6853zj{\text{'}}x_{m}^{'}} {{{g}^{2}}}}} \right. \kern-0em} {{{g}^{2}}}}}}$с параметрами наилучшего приближения, составляющими 2J = –3.54 cм–1, zJ' = –0.08 cм–1. Как и ожидалось, молекула комплекса XVII имеет моноядерное строение (рис. 4).
В этой связи наличие обменного взаимодействия может быть объяснено межмолекулярной ассоциацией (рис. 5).
С помощью квантово-химического моделирования обменного взаимодействия авторы цитируемой работы показали, что из двух возможных способов ассоциации (рис. 6, 7 соответственно) в данном случае реализуется строение, представленное на рис. 6, с необычно протяженным каналом обмена по цепочке атомов Cu–N–N···H–O–C–O–Cu.
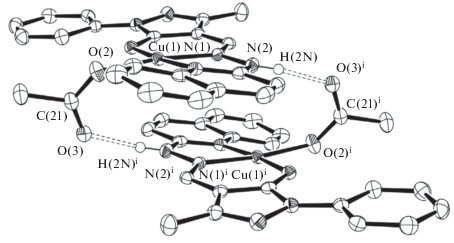
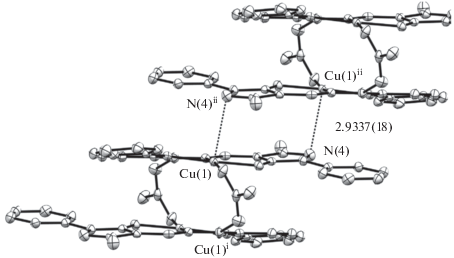
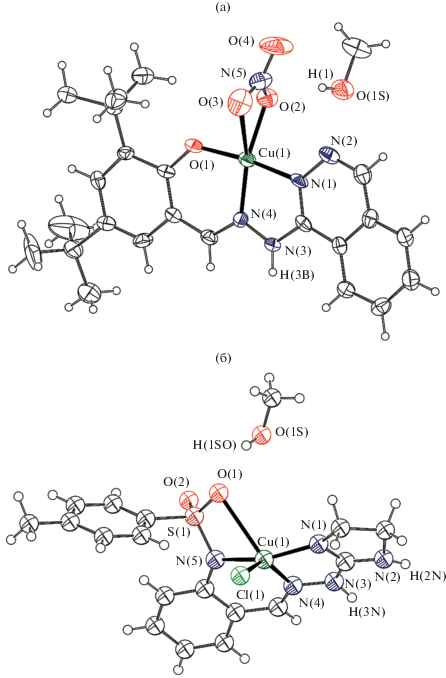
Фталазинилгидразоновые производные, представляющие собой, как показано в предыдущем разделе, удобные лигандные системы для получения обменно-связанных комплексов с предсказуемым и управляемым характером обмена, также можно использовать для получения супрамолекулярных архитектур, в первую очередь благодаря наличию в молекулах комплексов дополнительных донорных центров и NН-групп [111–114]. Так, например, в [114] синтезированы комплексы меди(II) [CuL1(NO3)]CH3OH (XVIII) и [CuL2Cl]CH3OH (XIX) (L1, L2 = монодепротонированные остатки 1'-фталазинилгидразона 3,5-ди-трет-бутилсалицилового альдегида и 2-имидазолинилгидразона 2-N-тозиламинобензальдегида соответственно), строение комплексов изображено на рис. 8.
Рис. 8.
Структура комплексов XVIII (а) и XIX (б) в представлении атомов эллипсоидами тепловых смещений с 30%-ной вероятностью [114].
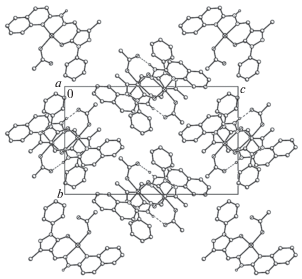
Молекула метанола, входящая в кристаллическую структуру комплекса XVIII, образует две водородные связи O(1S)–H(1)···O(2) и N(3)–H(3B)···O(1S), что приводит к образованию водородно-связанных димеров, упакованных в бесконечные стопки, вытянутые вдоль кристаллографической оси а (рис. 9а).
Рис. 9.
Упаковка водородно-связанных димеров комплексов XVIII (а) и XIX (б) в кристалле (вид вдоль кристаллографической оси b) [114].
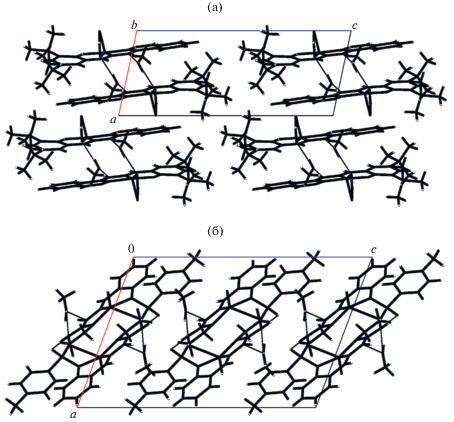
В случае комплекса XIX каждая молекула метанола образует две водородные связи с двумя ближайшими молекулами комплекса; NH-группы имидазолиновых фрагментов и атомы O(1S) тозильных групп этих молекул образуют еще две водородные связи. Это приводит к формированию в кристалле комплекса XIX водородно-связанных димеров, упакованных в бесконечные стопки, вытянутые вдоль кристаллографической оси с (рис. 9б).
Структуры комплексов XVIII и XIX предполагают возможность магнитного обменного взаимодействия между ионами меди(II). В обоих случаях возможный канал обмена весьма протяженный, причем в трансляции обмена должна участвовать водородная связь N–H···O [109]. В димере комплекса XVIII имеется лишь один парный канал обмена, в котором ионы меди разделены линкером, включающим шесть атомов и две водородных связи: Cu–N–N–H···O–H···O–Cu; расстояние медь–медь составляет 5.208 Å. В димере комплекса XIX имеются два альтернативных парных обменных канала: цепочки из семи атомов с одной водородной связью Cu–N=C–N–H···O=S–N–Cu и цепочки из восьми атомов с двумя водородными связями Cu–N–N–H···O–H···O=S–N–Cu; расстояние медь–медь составляет 5.756 Å.
Комплексы XVIII и XIX изучены методом ЭПР спектроскопии в поликристаллическом состоянии в интервале 5–300 K. В ЭПР спектре комплекса XVIII при 5 K при значительном увеличении чувствительности спектрометра в “половинном” поле авторы наблюдали линию запрещенного перехода (рис. 10). Спектр описывается ромбически искаженным спиновым гамильтонианом (2) димера со спином S = 1 с зеемановским, сверхтонким и тонким взаимодействием:
(2)
$\begin{gathered} \hat {H} = {{g}_{z}}\beta {{H}_{z}}{{S}_{z}} + {{g}_{x}}\beta {{H}_{x}}{{S}_{x}} + {{g}_{y}}\beta {{H}_{y}}{{S}_{y}} + \\ + \,\,A{{I}_{z}}{{S}_{z}} + B{{I}_{x}}{{S}_{x}} + C{{I}_{y}}{{S}_{y}} + \\ + \,\,D\left( {S_{z}^{2} - \frac{1}{3}S(S + 1)} \right) + E\left( {S_{x}^{2} - S_{y}^{2}} \right). \\ \end{gathered} $Рис. 10.
Спектр ЭПР поликристаллического образца комплекса XVIII при 5 К: эксперимент (1), теория (2). На вставке слева вверху – увеличенный участок спектра в “половинном” поле [114].
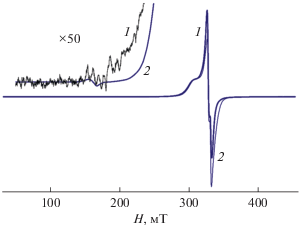
При моделировании спектра получены следующие параметры: gz = 2.196; gx = 2.041; gy = 2.009; A = 3.35 × 10–3 см–1; B = 2.59 × 10–5 см–1; C = 2.65 × × 10–3 см–1; D = 2.77 × 10–3 см–1; E = 2.20 × 10–4 см–1.
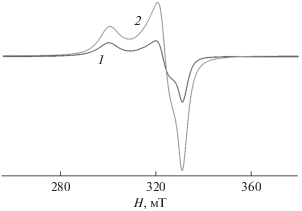
В случае комплекса XIX сигнал в “половинном” поле практически отсутствует даже при 5 K. Однако ряд особенностей спектра позволил авторам цитируемой работы предположить, что в данном случае также имеет место магнитное обменное взаимодействие между ионами меди(II). На рис. 11 показана температурная эволюция спектров ЭПР комплекса XIX. Обращает на себя внимание то, что интенсивность спектра при 20 К выше, чем при 5 К, в то время как из теории ЭПР известно, что с понижением температуры интенсивность сигнала ЭПР возрастает. Наблюдаемую аномалию можно объяснить наличием в образце комплекса межмолекулярных обменных взаимодействий антиферромагнитного типа с величиной изотропного обменного параметра |J| ≈ 1–5 см–1 [115].
Изучение температурной зависимости магнитной восприимчивости комплексов XVIII и XIX подтвердило, что между ионами меди(II) имеет место очень слабое антиферромагнитное обменное взаимодействие. Эффективный магнитный момент комплексов XVIII и XIX в расчете на один ион меди(II) составляет при комнатной температуре 1.88 и 1.91 М.Б., а при 2 K – 1.25 и 1.35 М.Б. соответственно. Величины обменных параметров, рассчитанные с помощью выражения (1), составили для комплекса XVIII 2J = –3.0 см–1, zJ ' = = 0.55 см–1, для XIX – 2J = –2.6 см–1, zJ' = = 0.60 см–1. Эти значения весьма близки к величине 2J (–3.54 см–1) в вышеописанном водородно-связанном димере типа XVII, в котором обменное взаимодействие также транслируется через водородные связи N–H···O, причем между взаимодействующими ионами меди находятся шесть атомов (цепочки связей Cu–N–N···H–O–C–N–Cu, расстояние медь–медь 4.431 Å).
Отмеченная выше роль нековалентных взаимодействий при формировании магнитных свойств супрамолекулярных систем иллюстрируется результатами работы [116], посвященной синтезу и исследованию спектральных и магнитных свойств полиядерных комплексов меди(II) с изоникотиновой кислотой, представляющую собой удобную N,O-донорную систему и подходящую для формирования супрамолекулярных архитектур комплексов за счет водородных связей. Варьируя условия синтеза, авторам [116] удалось выделить два типа таких структур (XX, XXI; рис. 12, 13), отличающихся характером координации лиганда и кристаллохимическими особенностями образования водородных связей.
Рис. 12.
Фрагмент кристаллической структуры комплекса XX (a); 3D супрамолекулярная структура XX, отражающая образование водородных связей с участием молекул воды и некоординированных карбоксилатных групп молекулы лиганда (б) [116].
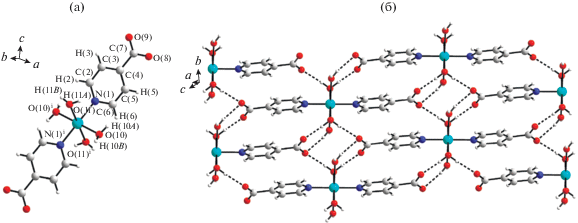
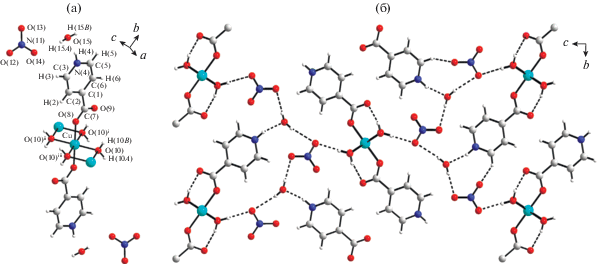
Рис. 13.
Фрагмент кристаллической структуры комплекса XXI (a); показаны водородные связи между отдельными фрагментами XXI, приводящие к образованию 3D супрамолекулярной архитектуры (б) [116].
Изучение магнитных свойств выделенных супрамолекулярных систем показало, что соединение XX представляет собой, по определению авторов [16], “quasi-2D magnet” с обменным взаимодействием между парамагнитными центрами по системе водородных связей, в то время как для соединения XXI характерно внутримолекулярное обменное взаимодействие ферромагнитного типа по цепи Cu–O–Cu вследствие, как утверждают авторы работы [16], уменьшения расстояния между ионами Cu2+. Этому же авторскому коллективу удалось также препаративно выделить два типа супрамолекулярных комплексов Cu2+ с лигандной системой транс-4,4´-азо-бис(пиридином) [116], первый из которых соответствует 1D координационному полимеру c антиферромагнитным характером обмена, а второй образует 2D полимер, стабилизация которого обуславливается за счет межмолекулярных водородных связей и для которого экспериментально доказано сохранение остаточной намагниченности образца.
Ряд экспериментальных доказательств положения о том, что относительно слабые межмолекулярные взаимодействия являются прекрасным инструментом для формирования супрамолекулярных структур, в том числе с управляемым характером обменного взаимодействия, представлен в [117–121]. Так, например, в [118] описан синтез и строение семи супрамолекулярных комплексов Mn(II), Co(II), Fe(II), Cu(II), Zn(II) и Cd(II) на основе тетратиафульвален-тетракарбоксилата (TC–TTF) и бипиридина (Bipy) состава [M(Вipy)2(H2O)4] [M(TC–TTF)(H2O)2]. На рис. 14 приведены примеры формирования катионных и анионных 1D-цепочек в случае ионов Mn(II) (XXII), которые далее самоорганизуются в 3D-структуру (рис. 15) за счет водородных связей O–H···O и O–H···N, а также π···π-взаимодействия.
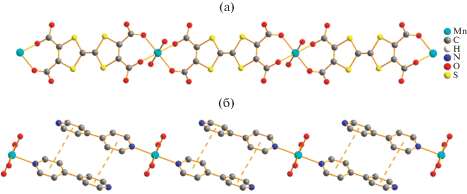
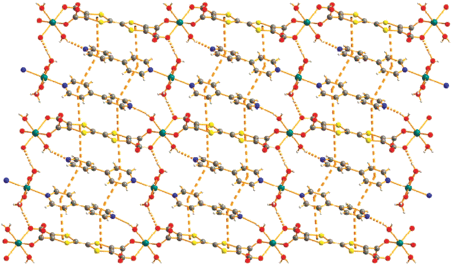
Комплекс XXII обладает свойствами тривиального парамагнетика, однако совершенно иная ситуация отмечена в [122], где описаны тетраядерные и синтезированные на их основе супрамолекулярные комплексы. В состав этих комплексов, помимо ионов переходных металлов, входят ионы редкоземельных элементов, магнетохимическая значимость которых обуславливается выраженной магнитной анизотропией, что является одним из необходимых элементов для направленного дизайна SMM’s [123]. В качестве лигандных систем авторы работы [122] выбрали 4-трифторметил-бензойную кислоту и 1,10-фенантролин, что обусловлено известной способностью карбоксилатсодержащих лигандов выступать как в роли хелатирующих агентов, так и связующих мостиков. Отмечается, что структурные фрагменты (a), (б) (рис. 16) достаточно легко объединяются в 1D‑цепочечные супрамолекулы (в) (XXIII) за счет водородных связей.
Анализ динамической магнитной восприимчивости XXIII однозначно подтвердил наличие медленной релаксации намагниченности, что свидетельствует в пользу SMM’s-характера полученного соединения.
III. КОМПЛЕКСЫ ПЕРЕХОДНЫХ МЕТАЛЛОВ, СОДЕРЖАЩИХ КУБАНОВЫЙ ФРАГМЕНТ С РАЗЛИЧНЫМИ КАНАЛАМИ ОБМЕНА
Как отмечалось выше, полиядерные комплексы меди(II) в течение последних десятилетий привлекают большое внимание исследователей вследствие образования многообразных координационных архитектур и потенциального применения в таких областях, как координационные полимеры [124–126], магнетохимия и бионеорганическая химия [127–131], катализ [132–134]. Среди них кубаноподобные Cu4O4 комплексы, содержащие гидроксо-, алкоксо- или феноксомостиковые фрагменты, заслужили особое внимание с точки зрения экспериментальной и теоретической магнетохимии [135–139].
На основе структурных особенностей кубаноподобных комплексов Cu4O4 были предложены две классификации их строения. Хронологически первая отмечена в [140], в которой классификация осуществлена на основе длин связей Cu–O в кубановом фрагменте. Второй тип классификации предложен в [141–143], подход был основан на длинах связей Cu···Cu в пределах кубанового фрагмента и позволил разделить данные комплексы на три типа (рис. 17).
Рис. 17.
Структурные типы комплексов с Cu4O4 кубановым фрагментом, классифицируемые в соответствии с расстоянием Cu···Cu (сплошные линии – короткие расстояния Cu···Cu и Cu–O, пунктирные – более длинные связи Cu···Cu и Cu–O) [141].
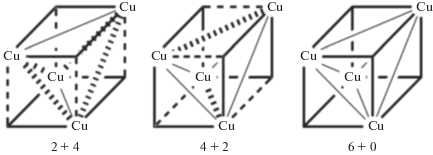
Несмотря на то что анализ магнетоструктурных данных для таких комплексов позволил выявить некоторые параметры, влияющие на магнитный обмен в подобных системах, определяющие магнетоструктурные корреляции установлены не были. Кроме того, были получены противоречивые результаты для комплексов типа 4 + 2, суть которых заключалась в противоположных знаках обменных параметров, полученных из теоретических расчетов и экспериментальных данных. К тому же, как будет показано ниже, широкое разнообразие состава и строения тетраядерных комплексов обуславливает необходимость детального подхода к анализу магнитных свойств данных систем с целью получения обобщающих магнетохимических закономерностей. Примером подобного подхода являются результаты работы [144], в которой представлены данные по получению супрамолекулярных структур, детально проанализированных в предыдущем разделе. Используя 2,6-пиридиндиметанол (H2L2, XXIV), а также ацетаты кобальта(II) и никеля(II), авторы путем молекулярной самосборки получили металл-кубаны состава [${\text{M}}_{4}^{{{\text{II}}}}$(HL2)4(OAc)4] типа XXV, строение которых изображено на рис. 18.
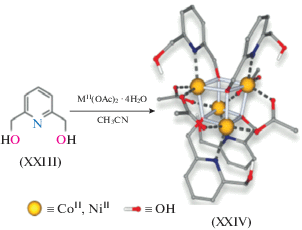
Отмечено, что образование монокристаллов XXV · 2CH3OH обусловлено π–π-взаимодействием и образованием как внутри-, так и межмолекулярных O–H···O водородных связей, что приводит к реализации 3D-архитектуры. Магнитные измерения доказали “смешанный” характер внутримолекулярного обмена с ферро- и антиферромагнитными параметрами, схема которого изображена ниже на рисунке к табл. 3.
Таблица 3.
Величины обменных параметров и факторов для кубанов XXVa, XXVb
| Соединение | 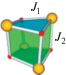 |
J1 [K] | J2 [K] | G |
| XXVa | 10.8 | –14.7 | 2.0 | |
| XXVb | 52.3 | –4.2 | 2.2 |
Эффекты кристаллической упаковки, оказывающие влияние на детали строения и магнитные свойства кубанов, отмечаются в [145–149]. Так, например, в [149] представлены результаты РСА и магнетохимического исследования тетраядерного комплекса меди(II) с азометиновым лигандом – продуктом конденсации 1-фенил-3-метил-4-формилпиразолона-5 и 1,3-диаминопропанола-2 (H3L).
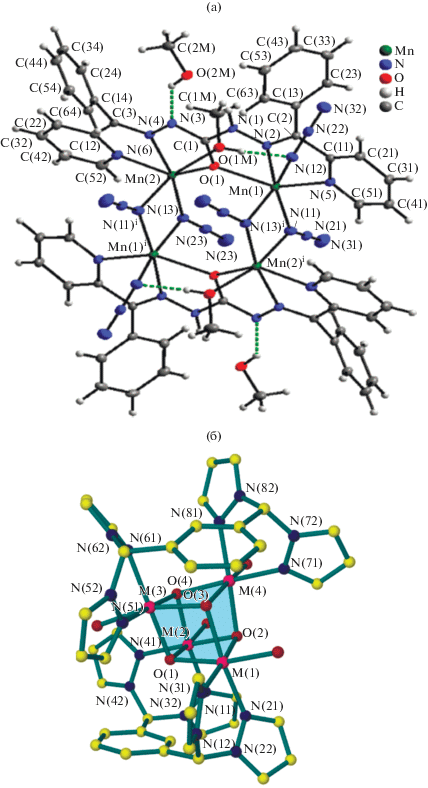
Взаимодействием бис-азометина H3L с перхлоратом меди(II) в присутствии триэтиламина и азида натрия с последующей перекристаллизацией из ацетонитрила авторы получили комплекс XXVI состава [Cu12L6(N3)6] · 9C2H3N · H2O. Симметрично-независимая часть элементарной ячейки соединения XXVI содержит 1.5 молекулы комплекса (A и B; B находится в частном положении на двойной оси симметрии), 4.5 молекул ацетонитрила и 0.5 молекулы воды. Соотношение молекул A и B в элементарной ячейке составляет 2 : 1. Строение обоих комплексов A и B можно формально описать как два биядерных фрагмента, связанных мостиковыми азидными группами (рис. 19). Это приводит к образованию искаженного центрального кубического фрагмента (КФ) Cu4N2O2, в котором атомы меди лежат в вершинах вписанного в этот куб тетраэдра. Координация атомов меди в комплексе различная, так же как различным является и строение этих симметрично-независимых молекул. Наиболее существенное различие комплексов A и B проявляется в строении их центральных КФ Cu4N2O2 (рис. 20).
Рис. 19.
Общий вид молекул A и B комплекса XXVI в представлении атомов эллипсоидами атомных смещений с 50%-ной вероятностью (атомы водорода не показаны, фенильные группы молекулы A заменены обозначением Ph; для молекулы B показана лишь симметрично-независимая часть за исключением координационных узлов атомов Cu, показанных полностью) [149].
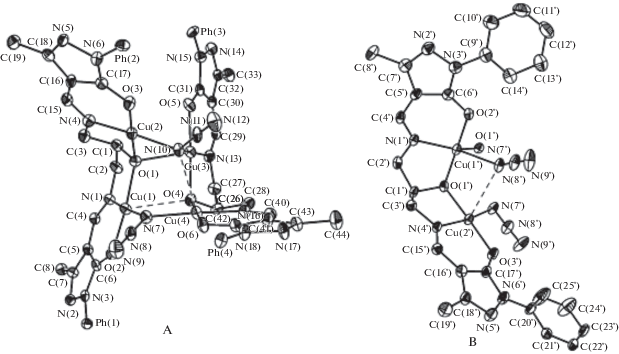
Рис. 20.
Структура обменных фрагментов в молекулах A и B комплекса XXVI (показаны только донорные атомы лигандов и мостиковые группы ${\text{N}}_{3}^{ - }$) [149].
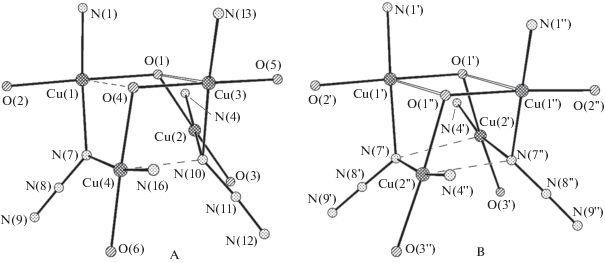
В комплексе A наиболее короткая дополнительная координация реализуется для атома Cu(3) (Cu(3)–O(1) 2.289(3) Å), что не вызывает сомнения в интерпретации координационного полиэдра атома Cu(3) как 4 + 1 (тетрагональная пирамида). Молекула B имеет симметрию С2, и КФ искажен в значительно меньшей степени. Оба симметрично-независимых атома меди имеют координацию 4 + 1. Искажения КФ проявляются и в контактах Cu···Cu. В комплексе A расстояние между атомами меди Cu(2)···Cu(3) и Cu(3)···Cu(4) достаточно короткое (2.9743(8) и 3.0187(9) Å соответственно), а диапазон расстояний составляет 2.97–4.03 Å. В КФ молекулы B диапазон расстояний Cu···Cu несколько меньше: 3.17–3.92 Å. Наиболее интересным представляется тот факт, что две заметно различающиеся строением не только лигандов, но и КФ конформации реализуются в одной кристаллической решетке соединения XXVI. Сравнение структур комплексов позволяет предположить, что при образовании тетраядерного комплекса с кубической геометрией центрального фрагмента Cu4N2O2 атомы меди стремятся к образованию устойчивой и характерной для Cu(II) координации 4 + 1. На геометрию КФ, очевидно, влияет объем лигандов. Однако проявление этого влияния показывает, что симметрию КФ можно легко нарушить межмолекулярным взаимодействием. Реализующаяся в кристаллической структуре водородная связь между молекулой воды и комплексом A достаточно слабая (O(1w)–H(1w)···N(14): H···N 2.240, O···N 3.028(5) Å, угол OHN 154°). Проведенное авторами квантово-химическое моделирование позволило сделать вывод о том, что наличие двух конформеров комплекса в кристаллической решетке определяется исключительно межмолекулярным взаимодействием в твердом теле и эффектами упаковки, а не особенностями строения молекул, так как теоретически наиболее устойчивой является единственная конформация. Спин-гамильтониан для кластера A авторами работы представлен в виде выражения (3),
(3)
$\begin{gathered} {{{\hat {H}}}_{A}} = - {{J}_{1}}({{{\hat {S}}}_{1}}{{{\hat {S}}}_{2}} + {{{\hat {S}}}_{1}}{{{\hat {S}}}_{3}}) - \\ - \,\,{{J}_{2}}({{{\hat {S}}}_{2}}{{{\hat {S}}}_{3}}) - {{J}_{3}}({{{\hat {S}}}_{1}}{{{\hat {S}}}_{4}} + {{{\hat {S}}}_{3}}{{{\hat {S}}}_{4}}), \\ \end{gathered} $а для кластера B – в виде (4)
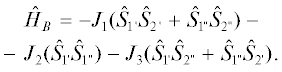
Принимая молярную магнитную восприимчивость комплекса равной сумме восприимчивостей “симметричного” и “несимметричного” кластеров и предполагая, что спин-гамильтониан для обоих кластеров включает три обменных параметра, температурную зависимость магнитной восприимчивости комплекса удалось удовлетворительно описать при следующих параметрах модели: J1A = –178, J2A = 80, J3A = 18, J1B = –26, J2B = = –74, J3B = 46 см–1, gA = gB = 2.05.
Сочетание ферро- и антиферромагнитных каналов обмена – довольно общее явление для обменно-связанных кубаноподобных систем [150, 151], причем возможны ситуации, при которых ферромагнитные вклады имеют доминирующий характер. Так, например, в [152] получены два комплекса меди(II) составов [{Cu(H2L1)}4] (XXVII) (H2L1 = N-(2-гидроксиэтил)-3,5-ди-трет-бутилсалицилальдимин) и {Cu(H2L2)}4 (XXVIII) (H2L2 = = N-(2- гидроксиэтил)-4-метоксисалицилальдимин), методом РСА показано, что соединения XXVII, XXVIII представляют собой тетраядерные алкоксомостиковые комплексы, содержащие существенно искаженные кубановые фрагменты Cu4O4 типа 4 + 2 (рис. 21).
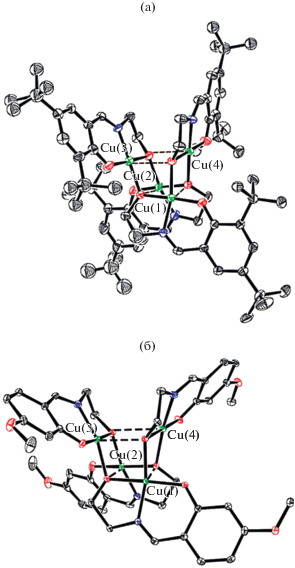
Магнитные свойства обоих комплексов отвечают сильному внутримолекулярному обменному взаимодействию ферромагнитного типа, приводящему к основному состоянию со спином S = 2. В данных соединениях зарегистрированы четыре коротких и два более длинных расстояния между атомами меди (тип 4 + 2), что, по мнению авторов, должно подразумевать шесть различных каналов и параметров обмена (рис. 22).
Рис. 22.
Структура обменных фрагментов в молекулах XXVII–XXVIII (a) и потенциальные каналы обмена (б) [152].
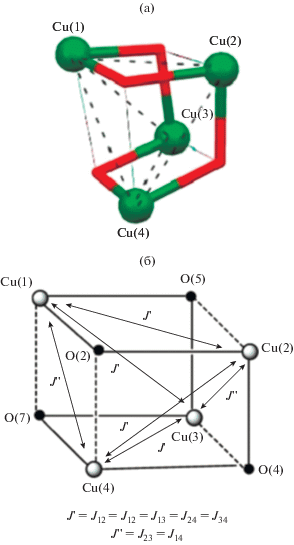
Для уменьшения числа параметров авторы предположили, что обменное взаимодействие по “короткому пути” между атомами меди носит идентичный характер и описывается параметром J ', а каналы обмена, соответствующие большему межатомному расстоянию, – параметром J '', что отвечает изотропному гамильтониану (5)
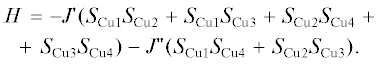
Величины обменных параметров составили J ' = +28.7 см–1, J '' = +7.8 см–1, g = 2.036 для комплекса XXVII и J ' = +39.8 см–1, J '' = +10.2 см–1, g = = 2.025 для XXVIII.
Авторы отмечают, что DFT расчеты дают хорошее соответствие теоретических и экспериментальных значений обменных параметров как по знаку, так и по величине, и комплексы типа XXVII, XXVIII представляют собой один из редких примеров кубаноподобных структур, в которых обменное взаимодействие между атомами меди носит исключительно ферромагнитный характер.
Как уже отмечалось выше в случае тетраядерного комплекса XXVI канал обмена с участием азидного мостика при μ-1,1-характере координации последнего отвечает ферромагнитному взаимодействию. Аналогичная ситуация реализуется в случае тетраядерного координационного соединения состава [Mn4(L)2(CH3OH)2(μ-N3)4(N3)2] · 2(CH3OH) (XXIX), описанного в [153], структура которого приведена на рис. 23а, где L – депротонированная форма бис-[(E)-N'-(фенил(пиридин-2-ил)метилен)]карбогидразида.
Авторы отмечают, что тетраядерный фрагмент в XXIX включает два различных по характеру трансляции обменных эффектов мостика – ферромагнитный азидный и превалирующий по абсолютной величине обменного параметра антиферромагнитный енолатный, что приводит к общему антиферромагнитному характеру обмена. Спин-гамильтониан системы имеет вид (6)
(6)
$\begin{gathered} H = - 2{{J}_{1}}({{{\hat {S}}}_{1}}{{{\hat {S}}}_{2}} + {{{\hat {S}}}_{3}}{{{\hat {S}}}_{4}}) - \\ - \,\,2{{J}_{2}}({{{\hat {S}}}_{1}}{{{\hat {S}}}_{{\text{4}}}} + {{{\hat {S}}}_{2}}{{{\hat {S}}}_{3}}) \\ \end{gathered} $с параметрами g = 1.98, J1 = +1.1 см−1, J2 = −1.3 см−1.
Авторы отмечают, что теоретические расчеты также предсказывают ферромагнитный характер обмена по каналу Mn-(μ-1,1-N3)2-Mn, если валентный угол αN фрагмента Mn–N–Mn составляет ~100°. В соответствии с данным положением параметр обмена J1 = +1.1 cm−1, без сомнений, отвечает каналу с мостиковым фрагментом (μ-1,1-N3)2(αсредн = 100.85°) в комплексе XXIX, параметр J2 = −1.3 см−1 описывает обменное взаимодействие через енольный атом кислорода.
Аналогичная зависимость параметров обмена от валентного угла МXМ в обменном фрагменте отмечена и в [154], посвященной физико-химическому исследованию комплекса никеля(II) типа XXX состава [Ni4(μ3-OH)4(μ-Lm)2(DMF)4]-(ClO4)4 · · DMF · EtOH, где Lm – м-бис[бис(1-пиразолил)метил]бензол, строение которого изображено на рис. 23б.
В указанной работе отмечается, что, если ядро Ni4 имеет тетраэдрическую симметрию, шесть обменных параметров Jij должны быть одинаковыми, однако детальный анализ рентгеноструктурных данных выявил два фрагмента с различными валентными углами NiONi: в первом валентный угол лежит в пределах 97.9°–99.5°, во втором – 100.3°–101.5°. Следовательно, можно ожидать реализацию двух ферромагнитных обменных каналов, причем первый будет превалирующим. Обменный гамильтониан в данном случае имеет вид (7)
(7)
$\begin{gathered} {{{\hat {H}}}_{{HDVV}}} = - {{J}_{1}}({{{\hat {S}}}_{1}}{{{\hat {S}}}_{2}} + {{{\hat {S}}}_{2}}{{{\hat {S}}}_{4}} + {{{\hat {S}}}_{3}}{{{\hat {S}}}_{4}}) - \\ - \,\,{{J}_{2}}({{{\hat {S}}}_{1}}{{{\hat {S}}}_{{\text{3}}}} + {{{\hat {S}}}_{1}}{{{\hat {S}}}_{4}} + {{{\hat {S}}}_{2}}{{{\hat {S}}}_{3}}) \\ \end{gathered} $с параметрами J1 = 9.1 см−1, J2 = 2.1 см−1; полученные результаты подтверждаются DFT расчетами.
В заключение отметим, что в настоящем обзоре авторы попытались изложить возможности магнетохимического метода, используемого для описания строения и свойств би- и тетраядерных металлхелатов и супрамолекулярных архитектур на основе координационных соединений с ограниченным по типам числом лигандных систем, главным образом гидразонов, азометинов и их аналогов. При этом мы старались очертить круг наиболее важных задач, которые были решены с помощью магнетохимического метода преимущественно для обменно-связанных систем с указанными лигандами. Совершенно очевидно, что дальнейшее теоретическое и экспериментальное развитие магнетохимического метода в плане адекватной интерпретации свойств подобных систем – весьма актуальная задача, особенно при направленном дизайне супрамолекулярных структур с заданными или управляемыми физико-химическими характеристиками, а также для выявления основополагающих закономерностей взаимосвязи “структура–свойство” с целью получения новых материалов, например, молекулярных магнетиков для инновационных технологий ближайшего будущего. Значительные возможности координационной химии в плане получения мультифункциональных молекул-магнитов, обладающих уникальными магнитными, электронными, оптическими, окислительно-восстановительными и фотохимическими свойствами составляют важный потенциал для научного сообщества. Без сомнения, систематизация имеющегося к настоящему времени обширного материала по данным проблемам, начало которой положено в цитированных выше обзорах и монографиях, будет продолжено в обобщающих исследованиях творческих коллективов, работающих в различных областях современной координационной химии.
Работа выполнена в рамках Программы развития ЮФУ (внутренний грант № ВнГр-07/2017-29).
Список литературы
Itoh K., Kinoshita M. Molecular Magnetism: New Magnetic Materials. Tokio: Kodansha and Gordon & Breach Science Pub., 2000.
Magnetism: Molecules to Materials: Models and Experiments V. 1–3 / Eds. Miller J.S., Drillon M. Weimheim: Wiley-VCH, 2001.
Launay J.P., Verdaguer M. Electrons in Molecules: From Basic Principles to Molecular Electronics. Oxford: Oxford University Press, 2014.
Joachim C., Gimzewski J.K., Aviram A. // Nature. 2000. V. 408. P. 541.
Park J., Pasupathy A.N., Goldsmith J.I. et al. // Nature. 2002. V. 417. P. 722.
Smith R.H.M., Noat Y., Untiedt C. et al. // Nature. 2002. V. 419. P. 906.
Ng M.K., Lee D.C., Yu L. // J. Am. Chem. Soc. 2002. V. 124. P. 11862.
Carrol R.L., Gorman C.B. // Angew. Chem. Int. Ed. 2002. V. 41. P. 4378.
Lippard S.J., Berg J.M. Principles of Bioinorganic Chemistry. California: Univ. Sci. Books, Mill Valley, 1994.
Que L. Jr., Dong Y. // Acc. Chem. Res. 1996. V. 29. P. 190.
Law N.A., Caudle M.T., Pecoraro V.L. // Adv. Inorg. Chem. 1998. V. 46. P. 305.
Wu A.J., Penner-Hahn J.E., Pecoraro V.L. // Chem. Rev. 2004. V. 104. P. 903.
Herrero C., Lassalle-Kaiser B., Leibl W. et al. // Coord. Chem. Rev. 2008. № 252. P. 456.
Kärkäs M.D., Verho O., Johnston E.V. et al. // Chem. Rev. 2014. V. 114. P. 11863.
Gerey B., Gouré E., Fortage J. et al. // Coord. Chem. Rev. 2016. № 319. P. 1.
Engelmann X., Monte-Pérez I., Ray K. // Angew. Chem. Int. Ed. 2016. V. 55. P. 7632.
Ferrando-Soria J., Vallejo J., Castellano M. et al. // Coord. Chem. Rev. 2017. V. 339. P. 17.
Clemente-León M., Coronado E., Martí-Gastaldo C. et al. // Chem. Soc. Rev. 2011. V. 40. P. 473.
Muñoz M.C., Real J.A. // Coord. Chem. Rev. 2011. V. 255. P. 2068.
Guo J.F., Yeng W.F., Gao S. // Eur. J. Inorg. Chem. 2008. P. 158.
Coronado E., Mínguez-Espallargas G. // Chem. Soc. Rev. 2013. V. 42. P. 1525.
Grancha T., Ferrando-Soria J., Castellano M. et al. // Chem. Commun. 2014. V. 50. P. 7569.
Ouahab L. Multifunctional Molecular Materials. Singapore: Pan Standford Publishing, 2013.
Ferrando-Soria J., Ruiz-García R., Cano J. et al. // Chem. Eur. J. 2012. V. 18. P. 1608.
Sessoli R., Boulon M.-E., Caneschi A. et al. // Nat. Phys. 2015. V. 10. № 1. P. 69.
Chorazy S., Nakabayashi K., Ohkoshi S. et al. // Chem. Mater. 2014. V. 26. P. 4072.
Pardo E., Train C., Gontard G. et al. // J. Am. Chem. Soc. 2011. V. 133. P. 15328.
Risset O.N., Quintero P.A., Brinzari T.V. et al. // J. Am. Chem. Soc. 2014. V. 136. P. 15660.
Darago L.E., Aubrey M.L., Yu C.J. et al. // J. Am. Chem. Soc. 2015. V. 137. P. 15703.
Jeon I.-R., Sun L., Negru B. et al. // J. Am. Chem. Soc. 2016. V. 138. P. 6583.
Pinkowicz D., Rams M., Misek M. et al. // J. Am. Chem. Soc. 2015. V. 137. P. 8795.
Romero-Morcillo T., Dela Pinta D., Callejo L.M. et al. // Chem. Eur. J. 2015. № 21. P. 12112.
Trzop E., Zhang D., Piñeiro-Lopez L. et al. // Angew. Chem. Int. Ed. 2016. V. 55. P. 8675.
Clements J.E., Price J.R., Neville S.M. et al. // Angew. Chem. Int. Ed. 2016. V. 55. P. 15105.
Reed D.A., Xiao D.J., Gonzalez M.I. et al. // J. Am. Chem. Soc. 2016. V. 138. P. 5594.
Hay J.P., Thibeault J.C., Hoffmann R. // J. Am. Chem. Soc. 1975. V. 97. P. 4884.
Kahn O., Briat B. // Faraday Trans. II. 1976. V. 268. P. 79.
Girerd J.J., Joumaux Y., Kahn O. // Chem. Phys. Lett. 1981. V. 82. P. 534.
Kahn O., Galy J., Journaux Y. et al. // J. Am. Chem. Soc. 1982. V. 104. P. 2165.
Julve M., Verdaguer M., Gleizes A. et al. // Inorg. Chem. 1984. V. 23. P. 3808.
Julve M., Faus J., Verdaguer M. et al. // J. Am. Chem. Soc. 1984. V. 106. P. 8306.
Willet R., Gatteschi D., Kahn O. Magneto-Structural Correlations in Exchange-Coupled Systems (NATO ASI Series C). Dordrecht: D. Reidel., 1985.
Kahn O. // Angew. Chem. Int. Ed. 1985. V. 24. P. 834.
Kahn O. // Z. Anorg. Allgem. Chem. 1987. V. 68. P. 89.
Blondin G., Girerd J.J. // Chem. Rev. 1990. V. 90. P. 1359.
Коган В.А., Луков В.В., Щербаков И.Н. // Коорд. химия. 2010. Т. 36. № 6. С. 403 (Kogan V.A., Lukov V.V., Shcherbakov I.N. // Russ. J. Coord. Chem. 2010. V.36. № 6. P. 401. doi 10.1134/S1070328410060011).
Луков В.В., Щербаков И.Н., Левченков С.И. и др. // Коорд. химия. 2017. Т. 43. № 1. С. 3 (Lukov V.V. Shcherbakov I.N., Levchenkov S.I. et al. // Russ. J. Coord. Chem. 2017. V. 43. № 1. P. 1. doi 10.1134/ S1070328417010055).
Калинников В.Т., Ракитин Ю.В. Введение в магнетохимию. Метод статической магнитной восприимчивости. М.: Наука, 1980. 301 с.
Ракитин Ю.В., Калинников В.Т. Современная магнетохимия. СПб.: Наука, 1994. 272 с.
Калинников В.Т., Ракитин Ю.В., Новоторцев В.М. // Успехи химии. 2003. Т. 72. № 12. С. 1123 (Kalinnikov V.T., Rakitin Yu.V., Novotortsev V.M. // Russ. Chem. Rev. 2003. V. 72. № 12. Р. 995).
Щегольков Е.В., Бургарт Я.В., Худина О.Г. и др. // Успехи химии 2010. Т. 79. № 1. С. 33 (Shchegolkov E.V., Burgart Ya. V., Khudina O.G. et al. // Russ. Chem. Rev. 2010. V. 79. № 1. P. 31).
Hakki E., Safak C., Mevlut E. et al. // J. Indian. Chem. Soc. 1989. V. 66. P. 45.
Mishra P., Gupta P.N., Shakya A.K. // J. Indian. Chem. Soc. 1991. V. 68. P. 618.
Likhate M.A., Fernandes P.S. // J. Indian. Chem. Soc. 1990. V. 67. P. 862.
Badr M.Z.A., Mahmoud M.A., Mahgoub S.A. et al. // Bull. Pol. Acad. Sci. Chem. 1989. V. 37. P. 185.
Revankar V.K., Arali V.H., Mehale V.B. // Indian. J. Chem. A. 1990. V. 29. P. 889.
Bartolucci C., Cellai L., Patrizia F. et al. // Farmaco. 1992. V. 41. P. 945.
Vicini P., Incerti M., Doychinova I.A. et al. // Eur. J. Med. Chem. 2006. V. 41. P. 624.
Odashima T., Yamaguchi M., Ishii H. // Microchim. Acta. 1991. V. 1. P. 267.
Sakamoto H., Ishikawa J., Nakagamo H. et al. // Chem. Lett. 1992. V. 21. P. 481.
Левченков С.И., Попов Л.Д., Щербаков И.Н. и др. // Журн. общ. химии. 2014. Т. 84. № 10. С. 1693 (Levchenkov S.I., Popov L.D., Shcherbakov I.N. et al. // Russ. J. Gen. Chem. 2014. V. 84. № 10. P. 1970).
Абраменко В.Л., Гарновский А.Д., Абраменко Ю.В. // Коорд. химия. 1994. Т. 20. № 1. С. 39.
Луков В.В., Коган В.А., Богатырева Е.В. и др. // Журн. неорган. химии. 1989. Т. 34. № 10. С. 2554.
Богатырева Е.В., Коган В.А., Луков В.В. и др. // Журн. неорган. химии. 1990. Т. 35. № 8. С. 2010.
Коган В.А., Луков В.В. // Коорд. химия. 1993. Т. 19. № 6. С. 476.
Луков В.В., Туполова Ю.П., Коган В.А. и др. // Коорд. химия. 2003. Т. 29. № 5. С. 359 (Lukov V.V., Tupolova Yu.P., Kogan V.A. et al. // Russ. J. Coord. Chem. 2003. V. 29. № 5. P. 335).
Левченков С.И., Коган В.А., Луков В.В. // Журн. неорган. химии. 1993. Т. 38. № 12. С. 1992.
Луков В.В., Левченков С.И., Коган В.А. // Журн. неорган. химии. 1997. Т. 42. № 4. С. 606.
Луков В.В., Левченков С.И., Коган В.А. // Коорд. химия. 1998. Т. 24. № 12. С. 946.
Kogan V.A., Lukov V.V., Levchenkov S.I. et al. // Mendeleev Commun. 1998. № 4. P. 145.
Iskander M.F., El-Sayed L., Salem N.M.H. et al. // Polyhedron. 2004. V. 23. № 1. P. 23.
Стариков А.Г., Коган В.А., Луков В.В. и др. // Коорд. химия. 2009. Т. 35. № 8. С. 625 (Starikov A.G., Kogan V.A., Lukov V.V. et al. // Russ. J. Coord. Chem. 2009. V. 35. № 8. P. 616. doi 10.1134/S1070328409080090).
Tandon S.S., Thompson L.K., Hynes R.C. // Inorg. Chem. 1992. V. 31. P. 2210.
Brooker S., Davidson T.C., Hay S.J. et al. // Coord. Chem. Rev. 2001. V. 216. P. 3.
Попов Л.Д., Левченков С.И., Щербаков И.Н. и др. // Журн. общ. химии. 2010. Т. 80. № 3. С. 471 (Popov L.D., Levchenkov S.I., Shcherbakov I.N. et al. // Russ. J. Gen. Chem. 2010. V. 80. № 3. P. 493).
Sangeetha N.R., Baradi R., Gupta R. et al. // Polyhedron. 1999. V. 18. P. 1425.
Haba P.M., Diouf O., Sy A. et al. // Z. Kristallogr. Cryst. Mater. 2005. V. 220. P. 479.
Chan S.C., Koh L.L., Leung P.-H. et al. // Inorg. Chim. Acta. 1995. V. 236. P. 101.
Roth A., Buchholz A., Gärtner V. et al. // Z. Anorg. Allgem Chem. 2007. V. 633. P. 2009.
Симонов Ю.А., Боурош П.Н., Ямпольская М.А. и др. // Коорд. химия. 1990. Т. 16. № 8. С. 1072.
Yamamoto T. // X-ray Spectrom. 2008. V. 37. P. 572.
Кочубей Д.И., Бабанов Ю.А., Замараев К.И. и др. // Рентгеноспектральный метод изучения структуры аморфных тел: EXAFS-спектроскопия. Новосибирск: Наука. Сиб. отд-ние, 1988. 306 с.
Попов Л.Д., Щербаков И.Н., Левченков С.И. и др. // Коорд. химия. 2011. T 37. № 7. С. 483 (Popov L.D., Levchenkov S.I., Shcherbakov I.N. et al. // Russ. J. Coord. Chem. 2011. V. 37. № 7. P. 483. doi 10.1134/ S1070328411060078).
Geary W.J. // Coord. Chem. Rev. 1971. V. 7. № 1. P. 81.
Коган В.А., Левченков С.И., Попов Л.Д. и др. // Рос. хим. журн. 2009. Т. 53. № 1. С. 86.
Tandon S.S., Thompson L.K., Hynes R.C. // Inorg. Chem. 1992. V. 31. P. 2210.
Abraham F., Lagrenee M., Sueur S. et al. // Dalton Trans. 1991. P. 1443.
Попов Л.Д., Левченков С.И., Щербаков И.Н. и др. // Журн. общ. химии. 2010. Т. 80. № 12. С. 2040 (Popov L.D., Levchenkov S.I., Shcherbakov i.N., et al. // Russ. J. Gen. Chem. 2010. V. 80. № 12. P. 2501).
Попов Л.Д., Мищенко А.В., Туполова Ю.П. и др. // Журн. общ. химии. 2011. Т. 81. № 8. С. 1346 (Popov L.D., Mishchenko A.V., Tupolova Yu. P. et al. // Russ. J. Gen. Chem. 2011. Т. 81. № 8. P. 1691).
Попов Л.Д., Левченков С.И., Щербаков И.Н. и др. // Журн. общ. химии. 2012. Т. 82. № 3. С. 472 (Popov L.D., Levchenkov S.I., Shcherbakov I.N. et al. // Russ. J. Gen. Chem. 2012. V. 82. № 3. P. 465).
Popov L.D., Shcherbakov I.N., Levchenkov S.I. et al. // J. Coord. Chem. 2008. V. 61. № 3. P. 392.
Popov L.D., Tupolova Yu.P., Lukov V.V. et al. // Inorg. Chim. Acta. 2009. V. 362. № 6. P. 1673.
Tupolova Yu.P., Popov L.D., Lukov V.V. et al. // Z. Anorg. Allgem. Chem. 2009. V. 635. № 3. P. 530.
Брылева М.А., Кравцова А.Н., Щербаков И.Н. и др. // Журн. структур. химии. 2012. Т. 53. № 2. С. 297 (Bryleva M.A., Kravtsova A.N., Shcherbakov I.N. et al. // Russ. J. Struct. Chem. 2012. V. 53. № 2. P. 295).
Castro I., Calatayud M.L., Barros W.P. et al. // Inorg. Chem. 2014. V. 53. P. 5759.
Щербаков И.Н., Левченков С.И., Попов Л.Д. и др. // Коорд. химия. 2015. Т. 41. № 2. С. 67 (Shcherbakov I.N., Levchenkov S.I., Popov L.D. et al. // Russ. J. Coord. Chem. 2015. V. 41. № 2. P. 69. doi 10.1134/ S1070328415020098).
Pavlishchuk A.V., Satska Yu. A., Kolotilov S.V. et al. // Cur. Inorg. Chem. 2015. V. 5. P. 5.
Hazra S., Karmakar A., Silva M. et al. // Inorg. Chem. Commun. 2014. V. 46. P. 113.
Liu X., Cen P., Li H. et al. // Inorg. Chem. 2014. V. 53. P. 8088.
Ogawa H., Mori K., Murashima K. et al. // Inorg. Chem. 2016. V. 55. P. 717.
Atzori M., Serpe A., Deplano P. et al. // Inorg. Chem. Front. 2015. V. 2. P. 108.
Biswas R., Mukherjee S., Ghosh S. et al. // Inorg. Chem. Commun. 2015. V. 56. P. 108.
Desplanches C., Ruiz E., Rodríguez-Fortea A. et al. // J. Am. Chem. Soc. 2002. V. 124. P. 5197.
Desplanches C., Ruiz E., Alvarez S. // Chem. Commun. 2002. P. 2614.
Bandeira N.A.G., Guennic B.L. // J. Phys. Chem. A. 2012. V. 116. P. 3465.
Bandeira N.A.G., Maynau D., Robert V. et al. // Inorg. Chem. 2013. V. 52. P. 7980.
Perić M., Zlatar M., Grubišić S. et al. // Polyhedron. 2012. V. 42. P. 89.
Ракитин Ю.В., Калинников В.Т., Ходасевич С.Г., Новоторцев В.М. // Коорд. химия. 2007. Т. 33. № 8. С. 563 (Rakitin Yu.V., Kalinnikov V.T., Khodasevich S.G., Novotortsev V.M. // Russ. J. Coord. Chem. 2007. V. 33. № 8. Р. 551. doi 10.1134/S1070328407080015).
Levchenkov S.I., Shcherbakov I.N., Popov L.D. et al. // Inorg. Chim. Acta. 2013. V. 405. P. 169.
Ларин Г.М., Шульгин В.Ф., Мельникова Е.Д., Зуб В.Ю., Ракитин Ю.В. // Изв. АН. Сер. хим. 2002. Т. 51. № 4. С. 592 (Larin G.M., Shul’gin, V.F., Mel’nikova E.D., Zub V.Y., Rakitin Yu.V. // Russ. Chem. Bull. Int. Ed. 2002. V. 51. № 4. P. 632).
Попов Л.Д., Левченков С.И., Щербаков И.Н. и др. // Коорд. химия. 2013. Т. 39. № 12. С. 743 (Popov L.D., Levchenkov S.I., Shcherbakov I.N. et al. // Russ. J. Coord. Chem. 2013. V. 39. № 12. P. 849. doi 10.1134/ S1070328413110079).
Попов Л.Д., Левченков С.И., Щербаков И.Н. и др. // Журн. структур. химии. 2015. V. 56. № 1. P. 120 (Popov L.D., Levchenkov S.I., Shcherbakov I.N. et al. // Russ. J. Struct. Chem. 2015. V. 56. № 1. P. 102).
Левченков С.И., Щербаков И.Н., Попов Л.Д. и др. // Журн. общ. химии. 2013. V. 83. № 10. С. 1722 (Levchenkov S.I., Shcherbakov I.N., Popov L.D. et al. // Russ. J. Gen. Chem. 2013. V. 83. № 10. P. 1928).
Левченков С.И., Попов Л.Д., Ефимов Н.Н. и др. // Журн. неорган. химии. 2015. V. 60. № 9. P. 1238 (Levchenkov S.I., Popov L.D., Efimov N.N. et al. // Russ. J. Inorg. Chem. 2015. V. 60. № 9. P. 1129. doi 10.1134/S0036023615040129).
Ларин Г.М., Минин В.В., Шульгин В.Ф. // Успехи химии. 2008. Т. 77. № 5. С. 476 (Larin G.M., Minin V.V., Shul’gin V.F. // Russ. Chem. Rev. 2008. V. 77. № 5. P. 451).
Almáši M., Vargová Z., Gyepes R. et al. // Inorg. Chem. Commun. 2014. V. 46. P. 118.
Qian J., Hu J., Yoshikawa H. et al. // Eur. J. Inorg. Chem. 2015. P. 2110.
Qin J.-H., Chang X-H., Ma L.-F. et al. // Inorg. Chem. Commun. 2015. V. 41. P. 92.
Song J. H., Lim K.S., Ryu D.W. et al. // Inorg. Chem. 2014. V. 53. P. 7936.
Tan X., Ji X., Zheng J.-M. // Inorg. Chem. Commun. 2015. V. 60. P. 27.
Sessoli R., Powell A. // Coord. Chem. Rev. 2009. V. 253. P. 2328.
Tan X., Ji X., Zheng J.-M. // Inorg. Chem. Commun. 2015. V. 60. P. 27
Sessoli R., Powell A. // Coord. Chem. Rev. 2009. V. 253. P. 232.
Adams C.J., Kurawa M.A., Lusi M. et al. // CrystEngComm. 2008. V. 10. P. 1790.
Chang M., Chung M., Lee B.S. et al. // Nanosci. Nanotechnol. 2006. V. 6. P. 3338.
Kang S.G., Kim H., Bang S. // Inorg. Chim. Acta. 2013. V. 396. P. 10.
Coronado E., Day P. // Chem. Rev. 2004. V. 104. P. 5419.
Liu C.M., Xiong R.G., Zhang D.Q. et al. // J. Am. Chem. Soc. 2010. V. 132. P. 4044.
Lu Z., Fan T., Guo W. et al. // Inorg. Chim. Acta. 2013. V. 400. P. 191.
Henkel G., Krebs B. // Chem. Rev. 2004. V. 104. P. 801.
Tjioe L., Meininger A., Joshi T. et al. // Inorg. Chem. 2011. V. 50. P. 4327.
Wegner R., Gottschaldt M., Görls H. et al. // Chem. Eur. J. 2001. V. 7. P. 2143.
Dıaz-Requejo M.M., Pérez P.J. // Chem. Rev. 2008. V. 108. P. 3379.
Safaei E., Kabir M.M., Wojtczak A. et al. // Inorg. Chim. Acta. 2011. V. 366. P. 275.
Lu J.W., Huang Y.H., Lo S. et al. // Inorg. Chem. Commun. 2007. V.10. P. 1210.
Monfared H.H., Sanchiz J., Kalantari Z. et al. // Inorg. Chim. Acta. 2009. V. 362. P. 3791.
Ruiz E., Alvarez S., Rodríguez-Fortea A. et al. // J. Mater. Chem. 2006. V. 16. P. 2729.
Ruiz E., Alvarez S., Rodríguez-Fortea A. et al. Electronic Structure and Magnetic Behavior in Polynuclear Transition-Metal Compounds. V. 1. Weinheim (Germany): Wiley-VCH, 2001.
Ruiz E., Rodríguez-Fortea A., Alemany P. et al. // Polyhedron. 2001. V. 20. P. 1323.
Mergehenn R., Haase W. // Acta Crystallogr. B. 1977. V. 33. P. 1877.
Tercero J., Ruiz E., Alvarez S., Rodríguez-Fortea A., Alemany P. // J. Mater. Chem. 2006. V. 16. P. 2729.
Ruiz E., Alvarez S., Rodríguez-Fortea A. et al. // Electronic Structure and Magnetic Behavior in Polynuclear Transition-Metal Compounds. V. 2. Weinheim (Germany): Wiley-VCH, 2001. P. 227.
Ruiz E., Rodríguez-Fortea A., Alemany P. et al. // Polyhedron. 2001. V. 20. P. 1323.
Scheurer A., Korzekwa J., Nakajima T. et al. // Eur. J. Inorg. Chem. 2015. V. 2015. P. 1892.
Carter K.P., Thomas K.E., Pope S.J.A. et al. // Inorg. Chem. 2016. V. 55. P. 6902.
Costes J.-P., Duhayon C., Vendier L. // Inorg. Chem. 2014. V. 53. P. 2181.
Li Y., Guo Y., Tian H. et al. // Inorg. Chem. Commun. 2014. V. 43. P. 135.
Tian C.-B., He C., Han Y.-H. et al. // Inorg. Chem. 2015. V. 54. P. 2560.
Левченков С.И., Щербаков И.Н., Попов Л.Д. и др. // Коорд. химия. 2014. Т. 40. № 2. С. 67 (Levchenkov S.I., Shcherbakov I.N., Popov L.D. et al. // Russ. J. Coord. Chem. 2014. V. 40. № 2. P. 69. doi 10.1134/S1070328414020055).
Gao Y.-Z., Zhang Y.-A., Zhang J. // Inorg. Chem. Commun. 2015. V. 54. P. 85.
Dias S.S.P., Kirillova M.V., André V. et al. // Inorg. Chem. 2015. V. 54. P. 5204.
Gungor E., Kara H., Colacio E. et al. // Eur. J. Inorg. Chem. 2014. P. 1552.
Bikas R., Hosseini-Monfared H., Siczek M. et al. // Inorg. Chem. Commun. 2015. V. 62. P. 60.
Reger D.L., Pascui A.E., Pellechia P.J. et al. // Inorg. Chem. 2014. V. 53. № 9. P. 4325.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия