

Координационная химия, 2019, T. 45, № 4, стр. 211-218
Гидрирование связей C=C И C=N амидо-иминного лиганда в координационной сфере металла в реакции бис(алкильного) комплекса иттрия [2,6-изо-Pr2C6H3NC(=CH2)C(Me)=NC6H3-изо-Pr2-2,6]Y(CH2SiMe3)2(THF) с молекулярным водородом
А. А. Кисель 1, 2, Д. М. Любов 1, 2, А. В. Черкасов 1, А. А. Трифонов 1, 2, *
1 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
2 Институт элементоорганических соединений им. А.Н. Несмеянова РАН
Москва, Россия
* E-mail: trif@iomc.ras.ru
Поступила в редакцию 09.11.2018
После доработки 15.11.2018
Принята к публикации 27.11.2018
Аннотация
Установлено, что при действии молекулярного водородана бис(алкильный) комплекс иттрия [ArNC(=CH2)C(Me)=NAr]Y(CH2SiMe3)2(THF) (I, Ar = C6H3-изо-Pr2-2.6), содержащий амидо-иминный лиганд, наряду с гидрогенолизом связей Y−C протекает гидрирование кратных связей C=N и C=C последнего. Реакция также сопровождается перераспределением азотсодержащих лигандов и приво-дит к образованию комплексов иттрия [ArNCH(Me)CH(Me)NAr]Y[ArN(=CH2)CH(Me)N(H)Ar](THF) (IIMeMe) и [ArNCH(Me)C(=CH2)NAr]Y[ArN(=CH2)CH(Me)N(H)Ar](THF) $({\text{I}}{{{\text{I}}}^{{{\text{MeС }}{{{\text{H}}}_{{\text{2}}}}}}}),$ содержащих наряду с дианионным диамидным [ArNCH(Me)CH(Me)NAr]2− или [ArNCH(Me)C(=CH2)NAr]2− лигандом, моноанионный ен-амидо-аминный [ArNC(=CH2)CH(Me)N(H)Ar]− фрагмент (СIF file CCDC № 1873206).
1,4-Дизамещенные диазабута-1,3-диены и лигандые системы на их основе в настоящее время нашли широкое использование в химии органических и координационных соединений редкоземельных металлов (РЗМ) [1–9]. Интерес к этим лигандам обусловлен многообразием их координационных возможностей и редокс-активной природой, а также простотой модификации их стерических и электронных свойств [10]. Наличие в молекуле диазадиена неподеленных электронных пар атомов азота, а также π-электронов кратных связей C=N позволяет ей выступать в роли как n-, так и π-донора, координируясь на атом металла в виде нейтрального лиганда [11]. Восстановление диазабутадиенов при действии одного либо двух мольных эквивалентов щелочных металлов позволяет получать анион-радикальные [RNC(R')C(R')NR]•− (A, cхема 1) [12] и ен-диамидные [RN−C(R')=C(R')−NR]2− (B, cхема 1) [13–17] лиганды соответственно. С другой стороны, в случае диазабутадиенов, содержащих при иминных атомах углерода метильные группы, возможен и второй путь трансформации лиганда за счет активации одной или двух связей СН этих заместителей с образованием моноанионного амидо-иминного [ArN=C(Me)−C(=CH2)−NAr]− (C, cхема 1) [18–20] либо диен-диамидного [ArN−C(=CH2)−C(=CH2)−NAr]2− (D, cхема 1) лигандов [21]. Производные РЗМ на их основе оказались эффективными катализаторами полимеризации с раскрытием цикла лактида и β-бутиролактона [22], полимеризации 2-винилпиридина [23], стереоселективной полимеризации изопрена [19, 24], межмолекулярных реакций гидроаминирования, гидрофосфинирования [25] и гидросилилирования [26] алкенов и ацетиленов. Недавно опубликованы первые примеры комплексов лантанидов (Yb, Dy), содержащих лиганды на основе диазабутадиена и проявляющих свойства мономолекулярных магнетиков (Single Molecular Magnet, SMM) [27, 28]. Типы лигандных систем на основе диазабутадиенов (A, B, C, D) представлены на схеме 1 :

Схема 1 .
Недавно было также показано, что дианионная ен-диамидная, а также моноанионная амидо-иминная лигандые системы на основе диазабутадиенов формируют подходящее лигандное окружение для стабилизации моно- и бис(алкильных) производных РЗМ [9, 23, 25]. Более того, на основе дианионной ен-диамидной лигндной систем получен редкий пример металлациклического гидридного комплекса иттрия [25].
В настоящей работе мы сообщаем о взаимодействии бис(алкильного) комплекса иттрия, стабилизированного амидо-иминным лигандом, с молекулярным водородом H2 и фенилсиланом PhSiH3.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез комплексов проводили в условиях, исключающих контакт с кислородом и влагой воздуха, по стандартной технике Шленка. Tетрагидрофуран (THF) и толуол сушили над бензофенонкетилом натрия, гексан – над металлическим натрием, затем тщательно дегазировали и конденсировали в вакууме в реакционную ампулу непосредственно перед использованием. Комплекс [ArNC(=CH2)C(Me)=NAr]Y(CH2SiMe3)2(THF) (I) получали в соответствии с ранее опубликованной методикой [19].
Спектры ЯМР 1H и 13С{1H} регистрировали на приборах Bruker DPX 200 MHz и Bruker Avance III 400 MHz. Химические сдвиги приведены в миллионных долях по отношению к известным сдвигам остаточных протонов дейтерированных растворителей. Элементный анализ выполняли на приборе Perkin-Elmer Series II CHNS/O Analyser 2400. Содержание лантанида определяли методом комплексонометрического титрования (Трилон Б) с использованием ксиленолового оранжевого в качестве индикатора.
Синтез {[2,6-изо-Pr2C6H3NCH(Me)CH(Me)NC6H3-изо-Pr2-2,6]Y[2,6-изо-Pr2C6H3-NC(=CH2)CH(Me)- N(H)C6H3-изо-Pr2-2,6]}, {[2,6-изо-Pr2C6H3NCH(Me)- C(=CH2)NC6H3-изо-Pr2-2,6]Y[2,6-изо-Pr2C6H3NC (=CH2)CH(Me)N(H)C6H3-изо-Pr2-2,6]} $({\mathbf{I}}{{{\mathbf{I}}}^{{{{{\mathbf{MeMe}}} \mathord{\left/ {\vphantom {{{\mathbf{MeMe}}} {{\mathbf{MeC}}{{{\mathbf{H}}}_{{\mathbf{2}}}}}}} \right. \kern-0em} {{\mathbf{MeC}}{{{\mathbf{H}}}_{{\mathbf{2}}}}}}}}}).$ В вакуумированную ампулу, снабженную тефлоновым краном и магнитной мешалкой, помещали раствор комплекса I (0.637 г, 0.86 ммоль) в 20 мл гексана и заполняли Н2 до давления 2 атм. Реакционную смесь перемешивали в течение 6 сут при комнатной температуре. Перекристаллизацией продукта реакции из смеси ТHF–гексан (1 : 1) выделяли желто-зеленые кристаллы. Выход 0.318 г (38%).
| Найдено, %: | C 74.39; | H 9.52; | N 5.58; | Y 9.22. |
| Для C60H91N4OY (М = 972.30) | ||||
| вычислено, %: | C 74.04; | H 9.42; | N 5.76; | Y 9.13. |
ЯМР 1H для IIMeMe (400 MГц; пиридин-d5; 293 K; δ, м.д. (J, Гц)): 1.09 (д., 3JHH = 6.8 Гц, 6H, CH3-изо-Pr), 1.11 (д., 3JHH = 6.8 Гц, 6H, CH3-изо-Pr), 1.16 (м., 15H, CH3-изо-Pr и CHCH3), 1.21 (д., 3JHH = 6.8 Гц, 12H, CH3-изо-Pr), 1.23 (д., 3JHH = 6.8 Гц, 12H, CH3-изо-Pr), 1.28 (м., 6H, CHCH3), 1.56 (м., 4H, β‑CH2THF), 2.82 (септ., 3JHH = 6.8 Гц, 2H, CH-изо-Pr), 2.90 (септ., 3JHH = 6.8 Гц, 2H, CH-изо-Pr), 3.60 (м., 10H, NCHCH3, CH-изо-Pr и α-CH2THF), 3.87 (м., 2H, N(H)CHCH3 и C=CH2), 4.37 (м., 1H, C=CH2), 5.08 (д, 3JHH = 8.8 Гц, NH), 7.20 (м., 12H, CHC6H3). ЯМР 13C{1H} для IIMeMe (400 MГц; пиридин-d5; 293 K; δ. м.д.): 15.6 (с., NHCHCH3), 18.8 (c., NCHCH3), 22.6 (с., CH3-изо-Pr), 22.7 (с., CH3-изо-Pr), 23.2 (с., CH3-изо-Pr), 23.3 (с., CH3-изо-Pr), 23.8 (с., CH3-изо-Pr), 23.9 (с., β-CH2THF), 24.1 (с., CH3-изо-Pr), 27.7 (с., CH-изо-Pr), 27.9 (с., CH-изо-Pr), 28.1 (с., CHiPr), 59.7 (с., NHC(H)Me), 60.6 (с., NC(H)Me), 67.6 (с., α-CH2THF), 112.3 (с., NC=CH2 Ar), 116.6 (с., CHAr), 122.7 (с., CHAr), 123.7 (с., CHAr), 123.8 (с., CHAr), 128.4 (с., CHAr), 129.2 (с., CHAr), 136.3 (с., CAr), 141.6 (с., CAr), 142.4 (с., CAr), 143.2 (с., CAr), 145.9 (с., CAr), 158.3 (с., CAr), 173.3 (с., NC=CH2). Содержание ${\text{I}}{{{\text{I}}}^{{{\text{MeC}}{{{\text{H}}}_{{\text{2}}}}}}}$ в кристаллическом порошке, согласно данным спектроскопии ЯМР, менее 5%, что не позволяет корректно описать его спектры ЯМР 1H и 13С{1H}.
РCA II проведено на дифрактометре Agilent XcaliburE (ω-сканирование, МоКα-излучение, λ = = 0.71073 Å, T = 100 K). Измерение и интегрирование экспериментальных наборов интенсивностей, учет поглощения и уточнение структур проведены с использованием программных пакетов CrysAlisPro [29] и SHELX [30]. Структуры решены прямым методом и уточнены полноматричным МНК по $F_{{hkl}}^{2}$ в анизотропном приближении для неводородных атомов. Водородные атомы помещены в геометрически рассчитанные положения и уточнены изотропно с фиксированными тепловыми параметрами U(H)изо = 1.2U(C)экв(U(H)изо = 1.5U(C)экв для метильных групп). Координаты атомов в разупорядоченном диамидном фрагменте найдены из разностного синтеза электронной плотности. C помощью инструкций DFIX, RIGU, ISOR и EADP ограничены некоторые длины связей в нeм и анизотропные параметры атомных смещений разупорядоченных атомов углерода. Кристаллографические данные, параметры рентгеноструктурного эксперимента и уточнения структуры приведены в табл. 1, значения основных длин связей и валентных углов – в табл. 2.
Координаты атомов и другие параметры структуры II депонированы в Кембриджском банке структурных данных (CCDC № 1873206; ccdc.cam.ac.uk/getstructures).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Гидридные комплексы РЗМ, содержащие реакционноспособные связи Ln−H, представляют интерес как с точки зрения фундаментальных исследований, так и использования данных комплексов в реакциях превращений ненасыщенных субстратов [31–37]. Первые примеры гидридных производных РЗМ – металлоценовые комплексы сэндвичевого и полусэндвичевого рядов [38–40]. В дальнейшем исследования в области синтеза гидридных производных РЗМ были сфокусированы на получении гидридов в нециклопентадиенильном лигандном окружении [40–43]. В ряде работ наглядно продемонстрировано, что стабильность гидридных производных РЗМ в значительной степени определяется именно природой вспомогательного лигандного окружения, варьирование которого позволяет модулировать реакционную способность связей Ln−H.
Самый распространенный синтетический метод получения гидридных комплексов лантанидов – реакция метатезиса σ-связи между соответствующим алкильным комплексом и молекулярным водородом или фенилсиланом. С целью получения дигидридного производного иттрия, стабилизированного амидо-иминным лигандом на основе диазабутадиена, мы исследовали реакции бис(алкильного) комплекса I с молекулярным водородом и PhSiH3.
Установлено, что реакция комплекса I с молекулярным водородом легко проходит в растворе гексана при комнатной температуре (P(H2) = 2 атм, 6 сут) (cхема 2) приводит к образованию мелкокристаллического желтого порошка. При перекристаллизации твердых продуктов реакции из смеси ТHF–гексан (1 : 1) медленной диффузией гексана в раствор комплекса в ТHF были выделены зеленовато-желтые кристаллы с выходом 38% в расчете на исходное количество I (схема 2 ). Полученные кристаллы хорошо растворимы в ТHF и пиридине, ограниченно растворимы в толуоле и нерастворимы в гексане, чувствительны к кислороду и влаге воздуха.
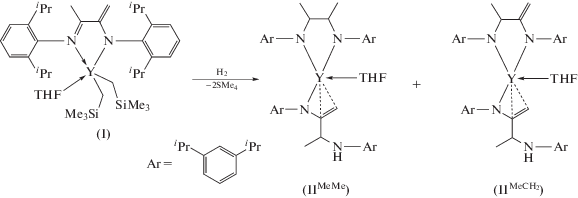
Схема 2 .
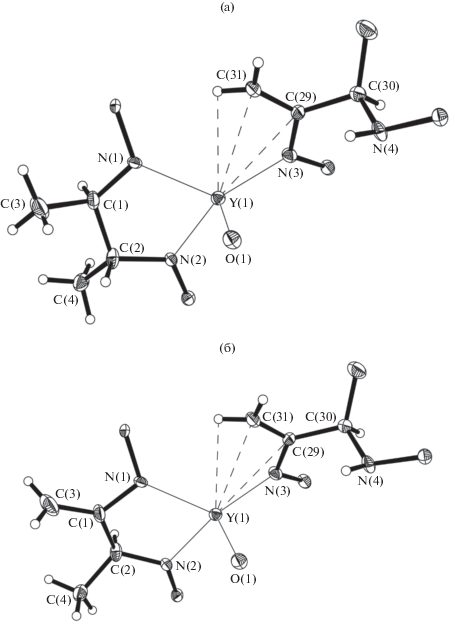
Согласно данным РСА, в монокристаллическом образце продукта реакции II в независимой области кристаллической ячейки располагаются две молекулы двух различных комплексов Y, разупорядоченные в соотношении 50 : 50% и находящиеся в суперпозиции относительно друг друга: {[ArNCH(Me)CH(Me)NAr]Y[ArNC(=CH2)CH(Me)-N(H)Ar](THF)} (IIMeMe; Ar = C6H3-изо-Pr2-2,6; рис. 1а) и {[ArNCH(Me)C(=CH2)NAr]Y[ArNC(=CH2)- CH(Me)N(H)Ar](THF)} (${\text{I}}{{{\text{I}}}^{{{\text{MeC}}{{{\text{H}}}_{{\text{2}}}}}}};$ Ar = C6H3-изо-Pr2-2,6; рис. 1б). Соединения IIMeMe и ${\text{I}}{{{\text{I}}}^{{{\text{MeC}}{{{\text{H}}}_{{\text{2}}}}}}}$ различаются заместителями на атомах углерода в диамидном лиганде: [ArN−CH(Me)−CH(Me)−NAr], либо [ArN−CH(Me)−C(=CH2)−NAr] соответственно. Образование соединений IIMeMe и ${\text{I}}{{{\text{I}}}^{{{\text{MeC}}{{{\text{H}}}_{{\text{2}}}}}}}$ – результат гидрогенолиза связей Y−C, приводящего к промежуточным гидридным комплексам, а также гидрирования кратных связей C=C и C=N амидо-иминного лиганда и перераспределения N-содержащих лигандов. К сожалению, выделить другие иттрий-содержащие продукты в индивидуальном виде из реакционной смеси не удалось.
Рис. 1.
Молекулярная структура комплекса IIMeMe (а) и ${\text{I}}{{{\text{I}}}^{{{\text{MeC}}{{{\text{H}}}_{{\text{2}}}}}}}$ (б). Тепловые эллипсоиды приведены с 30%-ной вероятностью. 2,6-Диизопропилфенильные заместители и атомы углерода молекулы ТHF не приведены.
Несмотря на наличие двух различных соединений, находящихся в монокристаллическом образце продукта реакции (в примерно равных количествах), в спектре ЯМР 1Н мелкокристаллического порошка II (С5D5N, 293 K), присутствуют сигналы, соответствующие комплексу IIMeMe, тогда как содержание комплекса ${\text{I}}{{{\text{I}}}^{{{\text{MeC}}{{{\text{H}}}_{{\text{2}}}}}}}$ незначительно (<5%). По-видимому, основным продуктом реакции является комплекс IIMeMe, содержащий диамидный лиганд с полностью гидрированными связями C=N и C=C, в то время как соединение ${\text{I}}{{{\text{I}}}^{{{\text{MeC}}{{{\text{H}}}_{{\text{2}}}}}}}$ – минорный продукт.
Молекулярное строение соединений IIMeMe и ${\text{I}}{{{\text{I}}}^{{{\text{MeC}}{{{\text{H}}}_{{\text{2}}}}}}}$ представлено на рис. 1. РСА показал, что соединения IIMeMe и ${\text{I}}{{{\text{I}}}^{{{\text{MeC}}{{{\text{H}}}_{{\text{2}}}}}}}$ кристаллизуются в пр. гр. P¯1 (табл. 1). Оба соединения мономерны и содержат моноанионный ен-амидо-аминный лиганд [ArN−C(=CH2)−CH(Me)−N(H)Ar]−, координированный с ионами иттрия через один из анилидных атомов азота и двойной связью С=СH2, приводя в результате к η3-азааллильному типу координации. Образование ен-амидо-аминного лиганда [ArN−C(=CH2)−CH(Me)−N(H)Ar]−, по-видимому, – результат гидрирования в исходном комплексе I связей C=N. Разрыв связи Y−N под действием H2 приводит, в свою очередь, к образованию аминогруппы, которая не принимает участия во взаимодействии металл–лиганд. Кроме того, ион иттрия в обоих соединениях связан с дианионным диамидным лигандом, также являющимся результатом гидрирования скелета исходного амидо-иминного лиганда [ArN−C(=CH2)−C(Me)=NAr]. Однако в соединении IIMeMe связи C=N и С=С в исходном фрагменте гидрированы до [N(Ar)− C(H)Me−C(H)Me−N(Ar)]2−, тогда как в ${\text{I}}{{{\text{I}}}^{{{\text{MeC}}{{{\text{H}}}_{{\text{2}}}}}}}$ во фрагменте [ArN−C(=CH2)−C(H)Me−NAr]2− остается непрореагировавшей связь С=С. Также в обоих соединениях ион иттрия координирован одной молекулой ТHF.
Азааллильный тип координации не характерен для комплексов РЗМ, однако в литературе известны примеры образования азааллильных комплексов в результате присоединения кетиминов либо нитрилов по σ-связям Ln−C [44, 45]. Сравнение расстояний Y−N и расстояний Y−C во фрагменте YN−C=CH2 с ранее известными примерами однозначно указывает на его азааллильный тип координации. Так, длина связи Y−N между атомами иттрия и азота η3-координированного ен-амидо-аминного лиганда в комплексах IIMeMe и ${\text{I}}{{{\text{I}}}^{{{\text{MeC}}{{{\text{H}}}_{{\text{2}}}}}}}$ составляет 2.299(2) Å, что больше величин, характерных для ковалентных связей Y−N в металлациклических ен-диамидных (2.167(3)–2.256(2) Å) [8, 9, 16, 22, 23, 25] и диамидных (2.149(3)–2.219(4) Å) [46, 47] производных, однако сравнимы со значениями, обнаруженными в азааллильных производных иттрия (2.295(6)–2.414(3) Å) [44, 45]. Короткие расстояния между атомами иттрия и углерода фрагмента N−C−CH2 ен-амидо-иминного лиганда в IIMeMe и ${\text{I}}{{{\text{I}}}^{{{\text{MeC}}{{{\text{H}}}_{{\text{2}}}}}}}$ (Y(1)−C(31) 2.749(5), Y(1)−C(29) 2.828(4) Å) также согласуются со значениями, найденными в азааллильных производных (2.613(6)–2.927(8) Å) [44, 45]. Вследствие делокализации отрицательного заряда по азааллильному фрагменту N−C−CH2 происходит укорачивание связи C−N (C(29)–N(3) 1.371(5) Å) и удлинение C=CH2 (С(29)−С(31) 1.355(4) Å) по сравнению со стандартными длинами одинарной C−N (1.469 Å) и двойной С=С (1.322 Å) связей [48].
Второй лиганд, координированный с ионами иттрия в молекулах IIMeMe и ${\text{I}}{{{\text{I}}}^{{{\text{MeC}}{{{\text{H}}}_{{\text{2}}}}}}}$ по типу κ2-N,N является дианионным диамидным. Расстояния Y−N в IIMeMe и ${\text{I}}{{{\text{I}}}^{{{\text{MeC}}{{{\text{H}}}_{{\text{2}}}}}}}$ составляют 2.191(3) и 2.209(3) Å, что соответствует значениям ковалентной связи Y−N [8, 9, 16, 22, 23, 25, 46, 47]. Дианионные диамидные лиганды в комплексах IIMeMe и ${\text{I}}{{{\text{I}}}^{{{\text{MeC}}{{{\text{H}}}_{{\text{2}}}}}}}$ имеют различное строение. Молекула IIMeMe содержит лиганд [ArN−CH(Me)− CH(Me)−NAr]2−, в котором кратные связи C=N и C=CH2 прогидрированы до одинарных (CH−N 1.467(5) и 1.492(12); CH−CH3 1.525(9) и 1.533(8) Å). При этом метильные группы при центральных атомах углерода ориентированы в разные стороны относительно плоскости фрагмента NCCN. Молекула ${\text{I}}{{{\text{I}}}^{{{\text{MeC}}{{{\text{H}}}_{{\text{2}}}}}}}$ содержит лиганд [ArN− C(=CH2)−CH(Me)−NAr]2−, в котором произошло гидрирование только связи C=N (C(1)−N(1) 1.467(5) Å), в то время как кратная связь C=CH2 остается двойной (С(1)−С(3) 1.367(8) Å).
В спектре ЯМР 1H мелкокристаллического порошка продукта реакции, записанного в пиридине-d5, присутствуют сигналы, относящиеся к комплексу IIMeMe. Метильные протоны изопропильных групп, а также протоны метильных заместителей фрагмента NCCN комплекса IIMeMe наблюдаются в спектре ЯМР 1H в виде перекрывающихся дублетов в области от 1.05 до 1.30 м.д. Метиновым протонам изопропильных групп соответствуют четыре септета. Два из них имеют химические сдвиги 2.82 и 2.90 м.д. (3JHH = 6.8 Гц), остальные два перекрываются с сигналом, соответствующим метиленовым протонам α-СH2 молекулы ТHF (3.60 м.д.). К метиновым протонам NCCN-фрагмента лиганда относится комплексный мультиплет в области между 3.51–3.68 м.д., также перекрывающийся с сигналом метиленовых протонов α-СH2 координированной молекулы ТHF. Диастереотопным метиленовым протонам азааллильного фрагмента соответствуют сигналы с химическими сдвигами 3.87 и 4.37 м.д. Ароматические протоны проявляются в виде набора сигналов в области 6.89–7.35 м.д.
Таблица 1.
Кристаллографические данные, параметры рентгеноструктурного эксперимента и уточнения структуры комплекса II
| Параметр | Значение |
|---|---|
| Формула | 1/2(C60H89N4OY), 1/2(C60H91N4OY) |
| М | 972.26 |
| Пр. гр. | P1 |
| a, Å | 12.9100(3) |
| b, Å | 14.7126(3) |
| c, Å | 14.8157(3) |
| α, град | 77.906(2) |
| β, град | 87.430(2) |
| γ, град | 85.626(2) |
| V, Å 3 | 2742.41(10) |
| Z | 2 |
| ρ(выч.), мг/м3 | 1.177 |
| μ, мм–1 | 1.105 |
| Область сканирования θ, град | 2.88–26.02 |
| Количество измеренных рефлексов | 41 266 |
| Количество рефлексов c I > 2σ(I) | 10 778 |
| Rint | 0.1176 |
| Число уточняемых параметров | 633 |
| S(F 2) | 1.002 |
| R1 (I > 2σ(I)) | 0.0712 |
| wR2 (по всем данным) | 0.1413 |
| Остаточная электронная плотность (min/max), е/Å3 | –0.756/0.968 |
Таблица 2.
Основные длины связей (d) и валентные углы (ω) в комплексе II
| Связь | IIMeMe | ${\text{I}}{{{\text{I}}}^{{{\text{MeC}}{{{\text{H}}}_{{\text{2}}}}}}}$ |
|---|---|---|
| d, Å | ||
| Y(1)–N(1) | 2.191(3) | |
| Y(1)–N(2) | 2.209(3) | |
| N(1)–C(1) | 1.467(5) | |
| N(2)–C(2) | 1.43(2) | 1.49(2) |
| C(1)–C(2) | 1.501(8) | 1.519(8) |
| C(1)–C(3) | 1.367(8) | 1.533(8) |
| C(2)–C(4) | 1.525(9) | 1.528(9) |
| Y(1)–N(3) | 2.301(3) | |
| Y(1)–C(29) | 2.828(4) | |
| Y(1)–C(31) | 2.749(5) | |
| Y(1)–O(1) | 2.363(3) | |
| Угол | ω, град | |
| N(1)Y(1)N(2) | 77.8(2) | |
| N(1)Y(1)O(1) | 103.7(2) | |
| N(2)Y(1)O(1) | 103.9(2) | |
| O(1)Y(1)N(3) | 117.4(2) | |
| N(1)C(1)C(2) | 114.0(5) | 114.1(5) |
| N(1)C(1)C(3) | 119.5(8) | 114.0(8) |
| C(2)C(1)C(3) | 126.4(9) | 102.5(8) |
| N(2)C(2)C(1) | 116.1(7) | 111.4(7) |
| N(2)C(2)C(4) | 117(2) | 111(2) |
| C(4)C(2)C(1) | 116(2) | 109(2) |
Реакции алкильных производных РЗМ с PhSiH3 – также одни из часто употребляемых синтетических подходов к соответствующим гидридам [40, 41]. Взаимодействие I с 2 мол. экв. PhSiH3 в гексане при 0°С приводит к образованию вязких маслянистых продуктов. К сожалению, все наши попытки выделить индивидуальные продукты из реакционной смеси обернулись неудачей. Проведение реакции комплекса I с 2 мол. экв. PhSiH3 в растворе бензола-d6 под контролем спектроскопии ЯМР свидетельствовало о ее прохождении. В спектре ЯМР 1H реакционной смеси через 1 ч после приготовления образца наблюдалось полное исчезновение сигналов, относящихся к алкильным группам, связанным с атомом иттрия, и появление соответствующего количества продукта метатезиса σ-связей Y−C – PhSiH2CH2SiMe3. Однако появление сигналов, относящихся к гидридным лигандам, связанным с атомом иттрия, не наблюдалось.
Таким образом, было установлено, что взаимодействие бис(алкильного) комплекса иттрия I, содержащего амидо-иминный лиганд на основе диазабутадиена, с молекулярным водородом сопровождается гидрогенолизом связей Y−C, а также гидрированием связей C=C и C=N амидо-иминного лиганда. Продуктами реакции являются комплексы иттрия, содержащие новый ен-амидо-аминный лиганд ([ArNC(=CH2)CH(Me)- N(H)Ar]−) и дианионный диамидный ([ArNCH(Me)- CH(Me)NAr]2− либо [ArNCH(Me)C(=CH2)NAr]2−) лиганды.
Работа выполнена в рамках госзадания (тема № 44.4, рег. № АААА-А16-116122110054-8) с использованием научного оборудования Центра коллективного пользования “Аналитический центр ИМХ РАН”. Рентгенодифракционные исследования проведены в рамках госзадания (тема № 44.2, рег. № АААА-А16-116122110053-1).
Список литературы
Trifonov A.A. // Eur. J. Inorg. Chem. 2007. P. 3151.
Walter M.D., Berg D.J., Andersen. R.A. // Organometallics. 2007. V. 26. P. 2296.
Trifonov A.A., Borovkov I.A., Fedorova E.A. et al. // Chem. Eur. J. 2007. V. 13. P. 4981.
Booth C.H., Walter M.D., Kazhdan D. et al. // J. Am. Chem. Soc. 2009. V. 131. P. 6480.
Trifonov A.A., Shestakov B.G., Lyssenko K.A. et al. // Organometallics. 2011. V. 30. P. 4882.
Basalov I.V., Lyubov D.M., Fukin G.K. et al. // Organometallics. 2013. V. 32. P. 1507.
Shestakov B.G., Mahrova T.V., Larionova J. et al. // Organometallics. 2015. V. 34. P. 1177.
Li J., Huang H., Wang F., Cui C. // Organometallics. 2015. V. 34. P. 683.
Nagae H., Kundu A., Tsurugi H. et al. // Organometallics. 2017. V. 36. P. 3061.
Van Koten G., Vrieze K. // Adv. Organomet. Chem. 1982. V. 21. P. 151.
Moore J. A., Cowley A. H., Gordon J. C. // Organometallics. 2006. V. 25. P. 5207.
Cardiner M.G., Hanson G.R., Henderson M.J. et al. // Inorg. Chem. 1994. V. 33. P. 2456.
Haaf M., Schmiedl A., Schmedake T.A. et al. // J. Am. Chem. Soc. 1998. V. 120. P. 12714.
Panda T.K., Kaneko H., Pal K. et al. // Organometallics. 2010. V. 29. P. 2610.
Scholz J., Richter B., Goddard R., Krüger C. // Chem. Ber. 1993. V. 126. P. 57.
Chen R., Tatsumi K. // J. Coord. Chem. 2002. V. 55. P. 1219.
Liu Y., Yang P., Yu J. et al. // Organometallics. 2008. V. 27. P. 5830.
Kaneko H., Tsurugi H., Panda T.K., Mashima K. // Organometallics. 2010. V. 29. P. 3463.
Kissel A.A., Lyubov D.M., Mahrova T.V. et al. // Dalton Trans. 2013. V. 42. P. 9211.
Bhadbhade M., Clentsmith G.K.B., Field L.D. // Organometallics. 2010. V. 29. P. 6509.
Makhrova T.V., Fukin G.K., Cherkasov A.V., Trifonov A.A. // Russ. Chem. Bull. Int. Ed. 2008. V. 57. P. 2285.
Mahrova T.V., Fukin G.K., Cherkasov A.V. et al. // Inorg. Chem. 2009. V. 48. P. 4258.
Kaneko H., Nagae H., Tsurugi H., Mashima K. // J. Am. Chem. Soc. 2011. V. 133. P. 19626.
Du G., Wei Y., Ai L. et al. // Organometallics. 2011. V. 30. P. 160.
Kissel A.A., Mahrova T.V., Lyubov D.M. et al. // Dalton Trans. 2015. V. 44. P. 12137.
Li J., Zhao C., Liu J. et al. // Inorg. Chem. 2016. V. 55. P. 9105.
Trifonov A.A., Shestakov B.G., Long J. et al. // Inorg. Chem. 2015. V. 54. P. 7667.
Long J., Shestakov B.G., Liu D. et al. // Chem. Commun. 2017. V. 53. P. 4706.
CrysAlis Pro. Yarnton (Oxfordshire, England): Agilent Technologies Ltd., 2014.
Sheldrick G.M. // Acta Crystallogr. C. 2015. V. 71. P. 3.
Ringelberg S.N., Meetsma A., Hessen B., Teuben H. // J. Am. Chem. Soc. 1999. V. 121. P. 6082.
Werkema E.L., Messines E., Perrin L. et al. // J. Am. Chem. Soc. 2005. V. 127. P. 7781.
Maron L., Werkema E.L., Perrin L. et al. // J. Am. Chem. Soc. 2005. V. 127. P. 279.
Jeske G., Lauke H., Mauermann H. et al. // J. Am. Chem. Soc. 1985. V. 107. P. 8111.
Conticello V.P., Brard L., Giardello M.A. et al. // J. Am. Chem. Soc. 1992. V. 114. P. 2761.
Molander G.A., Romero J.A.C. // Chem. Rev. 2002. V. 102. P. 2161.
Oyamada J., Nishiura M., Hou Z. // Angew. Chem. Int. Ed. 2011. V. 50. P. 10720.
Nishiura M., Hou Z. // Nature Chem. 2010. V. 2. P. 257.
Hou Z. // Bull. Chem. Soc. Jpn. 2003. V. 76. P. 2253.
Trifonov A.A., Lyubov D.M. // Coord. Chem. Rev. 2017. V. 340. P. 10.
Konkol M., Okuda J. // Coord. Chem. Rev. 2008. V. 252. P. 1577.
Fegler W., Venugopal A., Kramer M. et al. // Angew. Chem. Int. Ed. 2015. V. 54. P. 1724.
Okuda J. // Coord. Chem. Rev. 2017. V. 340. P. 2.
Zhang Z., Bu X., Zhang J. et al. // Organometallics. 2010. V. 29. P. 2111.
Hong J., Li Z., Chen Z. et al. // Dalton Trans. 2016. V. 45. P. 6641.
Avent A.G., Cloke F.G.N., Elvidge B.R., Hitchcock P.B. // Dalton Trans. 2004. P. 1083.
Shibata Y., Nagae H., Sumiya S. et al. // Chem. Sci. 2015. V. 6. P. 5394.
Allen F.H., Kennard O., Watson D.G. et al. // Perkin Trans. 2. 1987. P. S1.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия