

Координационная химия, 2019, T. 45, № 4, стр. 235-241
Синтез нового объемного бисамидина с конформационно жестким мета-фениленовым мостиком и его дилитиевого производного [1,3-C6H4{NC(Ph)N(2,6-изо-Pr2C6H3)}2]Li2(TMEDA)2
А. О. Толпыгин 1, А. В. Черкасов 1, Г. К. Фукин 1, А. А. Трифонов 1, 2, *
1 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
2 Институт элементоорганических соединений им. А.Н. Несмеянова РАН
Москва, Россия
* E-mail: trif@iomc.ras.ru
Поступила в редакцию 01.11.2018
После доработки 22.11.2018
Принята к публикации 27.11.2018
Аннотация
По реакции 2,6-диизопропиланилина c м-фенилендибензимидоил хлоридом 1,3-C6H4{N=C(Ph)Cl}2 в присутствии триэтиламина в растворе толуола синтезирован бис(амидин)-1,3-C6H4{NC(Ph)NH(2,6-изо-Pr2C6H3)}2 (H2L), в котором два амидинатных фрагмента связаны между собой конформационно жестким м-фениленовым линкером. Металлирование бис(амидина) I н-бутиллитием в присутствии тетраметилэтилендиамина (TMEDA) легко протекает в растворе тетрагидрофурана при –70°С с образованием соответствующего амидинатного комплекса лития 1,3-C6H4{NC(Ph)N(2,6-изо-Pr2C6H3)}2Li2(TMEDA)2 (I). Методом РСА (CIF file CCDC № 1873201) установлено, что комплекс I мономерный. Каждый амидинатный фрагмент NCN связан с одним атомом лития по хелатному типу κ2-N,N.
На протяжении последних двух десятилетий моноанионные амидинатные лиганды [RC(NR')2]– широко применяются для синтеза комплексов металлов как главных групп, так и ранних и поздних переходных металлов [1–6]. Амидинатные лиганды ведут себя как четырехэлектронные донорные системы, обеспечивая высокую электрофильность металлоцентра в соответствующих координационных соединениях [7, 8]. Благодаря многообразию способов координации с ионами металлов, а также легкости процесса настройки их электронных и стерических свойств путем варьирования заместителей при атомах азота и углерода, введения в боковую цепь дополнительных донорных группировок [9–13] или связывания двух фрагментов NCN между собой подходящей мостиковой группировкой [14–17], амидинатные и бис(амидинатные) лиганды являются перспективной платформой для синтезаширокого круга координационных соединений металлов различной природы. Амидинаты лития – широко используемые прекурсоры для синтеза соответствующих комплексов различных металлов по обменным реакциям.
В настоящей работе сообщается о синтезе нового бис(амидина) 1,3-C6H4{NC(Ph)NH(2,6-изо-Pr2C6H3)}2 (H2L), в котором амидинатные фрагменты связаны конформационно жeсткой мостиковой группой 1,3-C6H4, а также о синтезе и строении его дилитиевого производного 1,3-C6H4{NC(Ph)N(2,6-изо-Pr2C6H3)}2Li2(TMEDA)2 (I) (TMEDA = тетраметилэтилендиамин).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Все операции по синтезу и выделению продуктов проводили в атмосфере аргона или вакуумной аппаратуре с использованием стандартной техники Шленка. Диэтиловый эфир, тeтрагидрофуран (ТГФ) после осушки гидроксидом калия перегоняли над кетилом бензофенон натрия. Гексан и толуол сушили кипячением и перегонкой над металлическим натрием. Бензол-d6 сушили металлическим натрием, дегазировали и конденсировали в вакууме. Пиридин-d5 сушили над гидридом кальция. Мета-фенилендибензимидоил хлорид 1,3-C6H4{N=C(Ph)Cl}2 получали в соответствии с опубликованной методикой [18]. Пиридин-d5, бензол-d6, CDCl3, 1,3-C6H4(NH2)2, изо-PrC6H3NH2 – коммерческие реактивы (Acros).
ИК-спектры регистрировали на приборе Bruker-Vertex 70. Образцы соединений готовили в атмосфере сухого аргона в виде суспензий в вазелиновом масле. Спектры ЯМР 1H, 13C, 7Li регистрировали на приборах Bruker Avance DPX-200 и Bruker Avance III (400 MHz, 25°С, C6D6, пиридин-d5, CDCl3). Химические сдвиги приводили в миллионных долях по отношению к известным сдвигам остаточных протонов дейтерированных растворителей.
Синтез 1,3-фенилено-бис{(2,6-диизопропилфенил)-фенил}-амидина (H2L). К раствору м-фенилендиамина (5.85 г, 54.08 ммоль) в 100 мл дихлорметана приливали 15.0 мл триэтиламинa (10.95 г, 108.16 ммоль), затем 12.5 мл хлористого бензоила (15.20 г, 108.16 ммоль). Реакционную смесь перемешивали в течение 12 ч при 40°С и промывали водой. Органическую фазу отделяли и сушили прокаленным хлоридом кальция. Растворители удаляли на ротационном испарителе, твердый остаток промывали и сушили в вакууме при 50°C до постоянной массы. Выход 15.57 г (91%).
К суспензии N,N'-(1,3-фенилен)дибензамида (15.57 г, 49.22 ммоль) в 50 мл хлорбензола прибавляли пентахлорид фосфора (20.50 г, 98.45 ммоль) и перемешивали при 60°C. По окончании выделения хлороводорода растворитель удаляли в вакууме, остаток растворяли в толуоле. К полученному раствору 17.39 г (49.22 ммоль) 1,3-C6H4{N=C(Ph)Cl}2 в 70 мл толуола добавляли Et3N (9.96 г, 98.46 ммоля) и 2,6-диизопропиланилин (17.45 г, 98.46 ммоль). Реакционную смесь перемешивали при 110°C в течение 72 ч. Растворитель удаляли в вакууме, твердый остаток растворяли в 100 мл диэтилового эфира и промывали 1%-ным водным раствором Na2CO3 (3 × 100 мл). Эфирный слой отделяли и сушили над прокаленным MgSO4. Растворитель удаляли в вакууме, твердый остаток перекристаллизовывали из гексана. Выход бис(амидина) H2L в виде белого аморфного порошка 16.87 г (54%).
Масс-спектр (ЭУ; 70 эВ; m/z (Iотн, %)): 634.3 [M]+ (15). ИК-спектр (ν, см–1): 3390 с, 1632 с, 1596 с, 1538 с, 1351 с, 1263 с, 1094 с, 1031 с, 777 с. Спектр ЯМР 1Н (200 МГц; 25°C; CDCl3; δ, м.д. (J, Гц)): 1.30 (д., 24H, (CH3)2CH, 3JH,H = 6.9 Гц); 2.96 (септ., 4H, (CH3)2CH, 3JH,H = 6.9 Гц); 6.08 (уш. с., 2H, NH); 6.83 (т., 2H, изо-Pr2C6H3, 3JH,H = 7.7 Гц); 7.07 (д., 4H, изо-Pr2C6H3, 3JH,H = 7.7 Гц); 7.14 (уш. с., 2H, C6H4, изо-Pr2C6H3); 7.21 (д., 4H, C6H4, изо-Pr2C6H3, 3JH,H = 6.1 Гц); 7.34 (с., 5H, C6H4, изо-Pr2C6H3); 7.49 (с., 3H, C6H4, изо-Pr2C6H3). Спектр ЯМР 13C (100 МГц; 25°C; CDCl3; δ, м.д.): 22.5 ((CH3)2CH); 27.9 ((CH3)2CH); 118.6, 122.8, 123.6, 128.3, 128.8, 129.8, 132.5 (C6H4, изо-Pr2C6H3); 140.3 (NCN).
Синтез 1,3-фенилено-бис{(2,6-диизопропилфенил)-фенил}-амидинат дилития бис(тетраметилэтилендиамината) (I). К раствору 4.05 г (6.33 ммоль) I в 5 мл тетрагидрофурана при –70°С добавляли 1.14 M раствор н-BuLi в гексане (11.2 мл, 12.75 ммоль). Реакционную смесь медленно доводили до комнатной температуры и перемешивали в течение 2 ч. Добавление к реакционной смеси 5 мл ТМЭДА приводило к выпадению желтого порошка I. Выход 82% (3.37 г). Перекристаллизацией полученного порошка из смеси ТГФ–гексан (1 : 5) получили желтые кристаллы комплекса I.
ИК-спектр (ν, см–1): 1640 с, 1617 с, 1575 с, 1315 с, 1254 с, 1221 с, 1103 с, 1037 с, 946 с, 913 с, 865 с, 783 с, 741 с. Спектр ЯМР 1Н (400 МГц; 25°C, пиридин-d5; δ, м.д. (J, Гц)): 0.85 (д., 12H, (CH3)2CH, 3JH,H = = 6.2 Гц); 0.99 (д., 12H, (CH3)2CH, 3JH,H = 6.2 Гц); 2.11 (с., 24H, CH3, ТМЭДА); 2.33 (с., 8H, CH2, ТМЭДА); 3.62 (септ., 4H, (CH3)2CH, 3JH,H = 6.2 Гц); 6.22–7.28 (м., 20H, C6H4, изо-Pr2C6H3). Спектр ЯМР 1Н (400 МГц; 25°C, бензол-d6; δ, м.д. (J, Гц)): 1.13, 1.22 (д., 12H, (CH3)2CH, 3JH,H = 6.8 Гц); 1.97 (уш. с., 32H, ТМЭДА); 3.60 (септ., 4H, (CH3)2CH, 3JH,H = 6.8 Гц); 6.78–7.11 (м., 16H, C6H4, изо-Pr2C6H3); 7.34–7.36 (м., 4H, C6H4, изо-Pr2C6H3). Спектр 13C ЯМР (50 МГц; 2°C, пиридин-d5; δ, м.д.): 22.8, 24.7 ((CH3)2CH); 27.9 ((CH3)2CH); 45.7 (CH3, ТМЭДА); 57.9 (CH2, ТМЭДА); 114.2, 120.6, 122.5, 123.6, 126.9, 130.6, 141.2, 154.9 (C6H4, изо-Pr2C6H3); 167.6 (NCN). Спектр ЯМР 7Li (155.5 МГц; 25°C, пиридин-d5; δ, м.д.): 3.3.
РСА I проведен на дифрактометре Bruker Smart Apex (ω-сканирование, МоКα-излучение, λ = = 0.71073 Å, T = 150 K). Интегрирование экспериментальных наборов интенсивностей, учет поглощения и уточнение структуры проведены по программным пакетам SMART [19], TWINABS [20] и SHELX [21]. Структура решена прямым методом и уточнена полноматричным МНК по $F_{{hkl}}^{2}$ в анизотропном приближении для неводородных атомов. Водородные атомы помещены в геометрически рассчитанные положения и уточнены изотропно с фиксированными тепловыми параметрами U(H)iso = 1.2U(C)eq (U(H)iso = 1.5U(C)eq для метильных фрагментов). Кристаллографические данные и параметры уточнения структуры II приведены в табл. 1, основные длины связей и валентные углы – в табл. 2.
Таблица 1.
Кристаллографические данные и параметры уточнения структуры I
| Параметр | Значение |
|---|---|
| М | 879.16 |
| Пр. гр. | C2/c |
| a, Å | 20.8453(18) |
| b, Å | 15.7829(15) |
| c, Å | 17.4531(15) |
| β, град | 99.539(3) |
| V, Å 3 | 5662.7(9) |
| Z | 4 |
| ρ(выч.), мг/м3 | 1.031 |
| μ, мм–1 | 0.060 |
| Область сканирования θ, град | 1.98–26.02 |
| Количество измеренных рефлексов | 12 351 |
| Количество рефлексовc I > 2σ(I) | 4226 |
| Rint | 0.0515 |
| Число уточняемых параметров | 309 |
| GOOF (F 2) | 1.050 |
| R1 (I > 2σ(I)) | 0.0805 |
| wR2 (по всем данным) | 0.1992 |
| Остаточная электронная плотность (min/max), е/Å3 | –0.35/0.55 |
Таблица 2.
Основные длины связей (d) и валентные углы (ω) в комплексе I
| Связь | d, Å | Угол | ω, град |
|---|---|---|---|
| Li(1)–N(2) | 1.994(7) | N(2)Li(1)N(3) | 123.9(4) |
| Li(1)–N(3) | 2.035(9) | N(2)Li(1)N(1) | 68.0(2) |
| Li(1)–N(1) | 2.036(8) | N(3)Li(1)N(1) | 131.4(4) |
| Li(1)–N(4) | 2.073(8) | N(2)Li(1)N(4) | 114.8(4) |
| N(1)–C(1) | 1.331(4) | N(3)Li(1)N(4) | 91.3(3) |
| N(2)–C(1) | 1.330(4) | N(1)Li(1)N(4) | 129.0(4) |
| N(1)–C(8) | 1.423(4) | N(2)Li(1)C(1) | 34.0(2) |
| N(2)–C(20) | 1.393(4) | N(3)Li(1)C(1) | 137.9(4) |
Координаты атомов и другие параметры структуры I депонированы в Кембриджском банке структурных данных (CCDC № 1873201; ccdc.cam.ac.uk/getstructures).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Из литературных данных известно несколько типов связывания амидинатных лигандов с ионами металлов (cхема 1) [6]. Бис(амидинатные) лиганды демонстрируют большее разнообразие координационных возможностей. В зависимости от природы и протяженности группировки, связывающей два амидинатных фрагмента, они могут выступать в качестве три- и тетрадентатных дианионных лигандов, позволяющих получать различные мономерные, димерные и тримерные структуры [14–17, 22, 23].
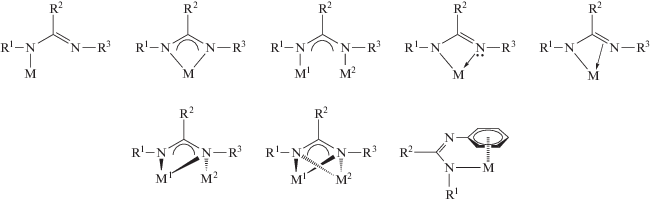
Схема 1 .
Бис(амидин) H2L получен по методике, разработанной Арнольдом [24] для синтеза других представителей данного класса соединений. Так, в результате реакции м-фенилендиамина с хлористым бензоилом получен N,N'-(1,3-фенилен)дибензамид. Далее реакция соответствующего амида бензойной кислоты с пентахлоридом фосфора приводит к N,N'-(1,3-фенилен)дибензимидоил хлориду (1,3-C6H4{N=C(Ph)Cl}2) [18]. Заключительная стадия синтеза протекает при взаимодействии м-фенилендибензимидоил хлорида с 2 мол. экв. 2,6-диизопропиланилина в толуоле в присутствии триэтиламина (схема 2 ). Бис(амидин) H2L был выделен в виде белого аморфного порошка с выходом 54% и охарактеризован методами элементного анализа, масс-спектроскопии, а также спектроскопии ИК и ЯМР.
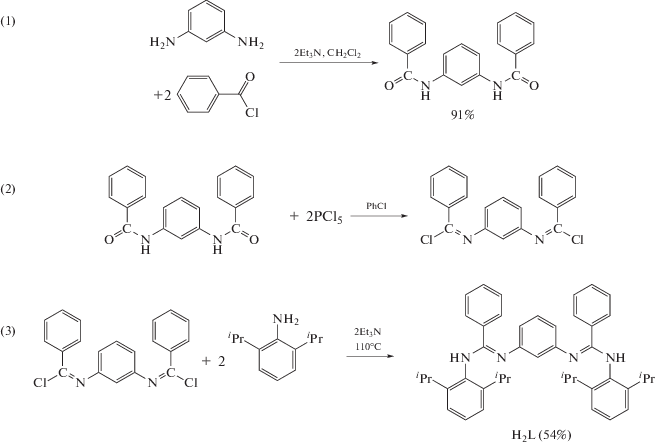
Схема 2 .
В спектре ЯМР 1H соединения H2L метильные протоны изопропильных групп проявляются в виде дублета при 1.30 м.д. с константой спин-спинового взаимодействия, равной 6.9 Гц. Метиновые протоны изопропильных групп дают септет с химическим сдвигом 2.96 м.д. и константой спин-спинового взаимодействия 3JH,H = 6.9 Гц. Уширенный синглет при 6.08 м.д. соответствует двум протонам NH амидиновых фрагментов. Ароматические протоны проявляются в характерной области слабого поля (6.83–7.49 м.д.).
В ИК-спектре соединения H2L присутствует интенсивная полоса поглощения при 1632 см−1, соответствующая ассиметричным колебаниям кратных связей C–N в лиганде, широкая полоса поглощения при 3390 см-1 отвечает колебаниям связей N–H.
Бис(амидин) H2L легко депротонируется н-бутиллитием в растворе ТГФ при –70°C (cхема 3). После обработки реакционной смеси ТМЭДА и перекристаллизации продуктов реакции из смеси ТГФ–гексан (в объемном соотношении 1 : 5) был выделен бис(амидинат)литий I в виде желтых кристаллов с выходом 82%.
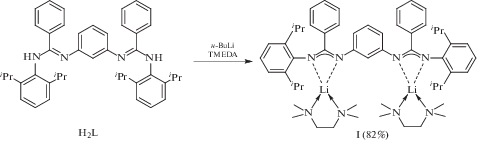
Схема 3 .
В спектре ЯМР 1H (бензол-d6) соединения I метильные протоны изопропильных групп проявляются в виде двух дублетов (3JH,H = 6.8 Гц) при 1.13 и 1.22 м.д. Метиновые протоны изопропильных групп дают септет с химическим сдвигом 3.60 м.д. и константой спин–спинового взаимодействия, равной 6.8 Гц. Ароматическим протонам соответствует набор сигналов в характерной области слабого поля 6.78−7.36 м.д. Метильные и метиленовые протоны координированных молекул ТМЭДА проявляются в виде уширенного синглета при 1.97 м.д. Согласно данным ЯМР 1H спектроскопии, I диссоциирует в растворе пиридина-d5, о чем свидетельствует присутствие в протонном спектре синглетов при 2.11 и 2.33 м.д. с соотношением интегральных интенсивностей сигналов 12 : 4, относящихся к метильным и метиленовым протонам свободного ТМЭДА. В спектре ЯМР 7Li комплекса I, записанном в пиридине-d5, ядра лития проявляются в виде единственного сигнала при 3.3 м.д. В ИК-спектре I присутствует интенсивная полоса поглощения при 1640 см−1, соответствующая ассиметричным колебаниям кратных связей CN амидинатных фрагментов.
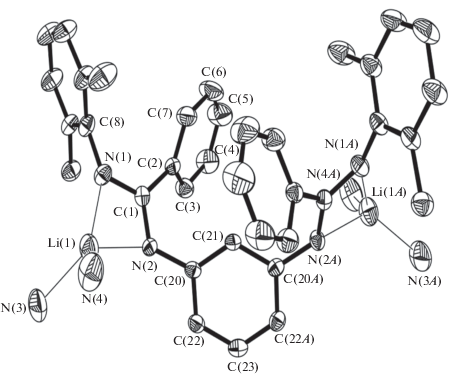
Кристаллы комплекса I, пригодные для РСА, получены в результате медленного конденсирования гексана к его насыщенному раствору в ТГФ при комнатной температуре. Молекулярное строение бис(амидинат)литиевого производного I представлено на рис. 1. В независимой области кристаллической ячейки I находится одна молекула комплекса: в частном положении на оси симметрии второго порядка, проходящей через атомы С(21) и С(23). Атом лития в I координирован двумя атомами азота анса-бис(амидинатного) лиганда [1,3-C6H4{NC(Ph)N(2,6-${\text{Pr}}_{{\text{2}}}^{i}{{{\text{C}}}_{{\text{6}}}}{{{\text{H}}}_{{\text{3}}}}$)}2]2– и двумя атомами азота молекулы ТМЭДА. Формальное КЧ(Li) в I равно четырем. Амидинатные фрагменты развернуты относительно друг друга; атомы лития лежат по разные стороны плоскости, в которой располагается м-фениленовая группа. Длины связей Li−N в комплексе I (1.994(7), 2.036(8) Å) сопоставимы с аналогичными расстояниями в известных амидинатных комплексaх лития [12, 25–32]. Длины связей C−N в амидинатных фрагментах (C(1)−N(1) и C(1)−N(2) (1.330(4), 1.331(2) Å)) близки между собой и свидетельствуют о делокализации отрицательных зарядов внутри NCN-групп. Двугранный угол между NCN-плоскостями амидинатных фрагментов составляет 55.7(3)°.
Рис. 1.
Молекулярная структура соединения I. Тепловые эллипсоиды приведены с 30%-ной вероятностью. Атомы водорода и углероды молекул ТМЭДА не приведены для ясности. Молекула лежит на оси симметрии второго порядка, проходящей через атомы C(21) и C(23) (–x, y, 1/2 – z).
Таким образом, синтезирован новый бис(амидин) 1,3-C6H4{NC(Ph)NH(2,6-изо-Pr2C6H3)}2, содержащий конформационно жесткий 1,3-C6H4 линкер, способный координироваться на ионы лития по хелатному типу κ2-N,N. Было установлено, что использование бидентатного хелатирующего основания Льюиса (ТМЭДА) позволило выделить бис(амидинат)литиевый комплекс 1,3-C6H4{NC(Ph)N(2,6-изо-Pr2C6H3)}2Li2(TMEDA)2 (I) в виде мономера.
Работа выполнена в рамках госзадания (тема № 44.4, рег. № АААА-А16-116122110054-8) с использованием научного оборудования Центра коллективного пользования “Аналитический центр ИМХ РАН”. Рентгенодифракционные исследования проведены в рамках госзадания (тема № 44.2, рег. № АААА-А16-116122110053-1)
Список литературы
Edelmann F.T. // Coord. Chem. Rev. 1994. V. 137. P. 403.
Edelmann F.T. // Adv. Organomet. Chem. 2008. V. 57. P. 183.
Patai S., Rappoport Z. The Chemistry of Amidinesand Imidates.V. 2. N.Y.: Wiley Intersci., 1991.
Barker J., Kilner M. // Coord. Chem. Rev. 1994. V. 133. P. 219.
Coles M.P. // Dalton Trans. 2006. № 8. P. 985.
Junk P.C., Cole M.L. // Chem. Commun. 2007. P. 1579. doi 10.1039/B613984A
Edelmann F.T. // Chem. Soc. Rev. 2012. V. 41. P. 7657.
Hagadorn J.R., Arnold J. // Dalton Trans. 1997. № 18. P. 3087.
Bambirra S., Otten E., Leusen D. van et al. // Z. Anorg. Allg. Chem. 2006. V. 632. P. 1950.
Rad’kov V.Y., Skvortsov G.G., Lyubov D.M. et al. // Eur. J. Inorg. Chem. 2012. V. 2012. P. 2289.
Bambirra S., Brandsma Menno J.R., Brussee Edward A.C. // Organometallics. 2000. V. 19. P. 3197.
Скворцов Г.Г., Толпыгин А.О., Любов Д.M. // Изв. АН. Сер. хим. 2016. № 12. С. 2832 (Skvortsov G.G., Tolpygin A.O., Lyubov D.M. // Russ. Chem. Bull. Int. Ed. 2016. V. 65. № 12. P. 2832).
Rad’kova N.Yu., Tolpygin A.O., Rad’kov V.Yu. et al. // Dalton Trans. 2016. № 45. P. 18572.
Tolpygin A.O., Skvortsov G.G., Cherkasov A.V. // Eur. J. Inorg. Chem. 2013. № 35. P. 6009.
Bambirra S., Meetsma A., Hessen B., Teuben J.H. // Organometallics. 2001. V. 20. P. 782.
Yakovenko M.V., Trifonov A.A., Kirillov E. et al. // Inorg. Chim. Acta. 2012. V. 383. P. 137.
Pan C.-L., Chen W., Song S. // Inorg. Chem. 2009. V. 48. P. 6344.
Матвеева Е.В., Лысенко K.A., Петровский П.В. // Изв. АН. Сер. хим. 2009. № 4. P. 800 (Matveeva E.V., Lyssenko K.A., Petrovskii P.V. // Russ. Chem. Bull., Int. Ed. 2009. V. 58. P. 817).
SMART. Madison (WI, USA): Bruker AXS Inc., 2001.
TWINABS. Madison (WI, USA): Bruker AXS Inc., 2001.
Sheldrick G.M. // Acta Crystallogr. C. 2015. V. 71. P. 3.
Skvortsov G.G., Cherkasov A.V., Long J. // Mendeleev Commun. 2018. V. 28. P. 521.
Tolpygin A.O., Luconi L., Shavyrin A.S. // Eur. J. Inorg. Chem. 2017. V. 2017. P. 2736.
Whitener G.D., Hagadorn J.R., Arnold J. // Dalton Trans. 1999. № 8. P. 1249.
Chivers T., Downard A., Parvez M. // Inorg. Chem. 1999.V. 38. P. 4347.
Cole M.L., Davies A.J., Jones C., Junk P.C. // J. Organomet. Chem. 2004. V. 689. P. 3093.
Yao S., Chan H.-S., Lam C.-K., Lee H.K. // Inorg. Chem. 2009. V. 48. P. 9936.
Tacke R., Kobelt C., Baus J.A. et al. // Dalton Trans. 2015. № 44. P. 14959.
Skvortsov G.G., Fukin G.K., Ketkov S.Yu. et al. // Eur. J. Inorg. Chem. 2013. № 24. P. 4173.
Hill M.S., Hitchcock P.B., Mansell S.M. // Dalton Trans. 2006. № 15. P. 1544.
Bai S.-D., Guo J.-P., Liu D.-S. // Dalton Trans. 2006. № 18. P. 2244.
Novotný M., Svec P., Ruzickova Z., Ruzicka A. // J. Organometallic. Chem. 2017. V. 849–850. P. 88.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия