

Координационная химия, 2019, T. 45, № 6, стр. 323-340
На пути к тонкому неорганическому синтезу: манипуляции с мостиковыми лигандами в халькогенидных кластерах
М. Н. Соколов 1, 2, А. Л. Гущин 1, 2, *
1 Институт неорганической химии им. А.В. Николаева СО РАН
Новосибирск, Россия
2 Новосибирский государственный университет
Новосибирск, Россия
* E-mail: gushchin@niic.nsc.ru
Поступила в редакцию 20.11.2018
После доработки 06.12.2018
Принята к публикации 13.12.2018
Аннотация
В данном обзоре рассмотрены подходы к направленному, многостадийному синтезу координационных соединений на примере халькогенидных и оксидных кластерных и полиядерных соединений металлов V и VI групп. Рассмотрены методы введения двух разных атомов халькогена, а также способы синтеза смешанных оксидно-халькогенидных комплексов.
Синтез новых соединений является “сердцевиной” химии. Это не только эффективное средство познания химических законов, но и необходимый этап на пути к непрерывно декларируемой цели – созданию веществ и материалов с заранее заданными свойствами. Для демонстрации успехов синтеза обычно приводят яркие примеры из органической химии, где уже несколько десятилетий назад стало возможным планировать и осуществлять многостадийный (десятки стадий!) синтез сложных соединений, преимущественно природных. Стало возможным говорить о тонком органическом синтезе. Возможно ли что-либо подобное в координационной химии – области, которую традиционно относят к неорганической химии, но которая по своей идеологии (показательно, что две знаковые для становления координационной химии фигуры – Вернер и Чугаев – начинали как органики) близка к органической химии? Действительно, оба исследователя во главе своих школ выполнили большой объем работ по координационной химии Cr(III), Co(III), Pt(II), Pt(IV), руководствуясь классической методологией органического синтеза – “замещение”, “присоединение”, “отщепление”, “изомеризация” [1]. Формулировка принципа транс-влияния и виртуозное его применение позволило, например, целенаправленно синтезировать комплексы Pt(IV) с шестью разными лигандами [2]. Однако достаточно скоро обозначились границы применимости классического подхода, ограниченного только кинетически инертными к замещению комплексами с электронными конфигурациями d3 и низкоспиновые d6 и d8. Именно инертность таких комплексов и делала для них применимой методологию, разработанную для органического синтеза. В русле классического подхода до сих пор развиваются исследования по химии нитрозильных комплексов Ru(II) и Os(II) (низкоспиновая d6-конфигурация обеспечивает необходимую инертность) [3, 4]. В остальных случаях лабильность координационной сферы делает невозможным такой подход. Исключением являются низкоспиновые карбонильные комплексы переходных металлов и их производные π-комплексы, которые роднит с органическими соединениями, помимо “препаративной” инертности, предсказуемость электронного состояния (правило 18 электронов) и постоянство координационного числа. Эту аналогию ярко иллюстрирует концепция изолобальности, предложенная и развитая Р. Хоффманом [5]. Поэтому металлоорганическая химия переходных металлов в настоящее время обладает высокой степенью предсказуемости и возможностью реализаций достаточно многоступенчатых цепочек превращений [6, 7]. Недавно наметилось многообещающее развитие в области химии полиядерных карбоксилатов, где показана возможность достаточно прогнозируемой поэтапной сборки полиядерных систем из устойчивых гомо- и гетерометаллических фрагментов меньшей нуклеарности и связывания таких фрагментов в координационные полимеры. При этом возможно использование комбинаций 3d- и 4f-элементов и даже d и s-элементов [8, 9]. Несколько особняком стоит супрамолекулярная координационная химия, современное состояние которой делает возможным синтез топологически сложных объектов, молекулярных машин и устройств; однако она в значительной степени опирается на тонкий многостадийный органический синтез [10–12].
Новые возможности для сложного, многостадийного неорганического синтеза открылись в результате бурного развития химии кластерных соединений в 1970–1980-е гг. [13]. Возможности, связанные с модификацией количества и природы атомов металла в металлоостове были широко использованы прежде всего в химии карбонильных и близких по природе металлорганических кластеров, включая синтез хиральных кластерных каркасов [14–18].
Несколько позднее была разработана химия халькогенидных кластеров переходных металлов, которая в настоящее время позволяет проводить достаточно сложные многостадийные манипуляции по синтезу кластеров с заданным строением и составом металлоостова, включая синтез моде-лей нитрогеназы на основе железо-сульфидных и железо-молибден-сульфидных кластеров [19–23].
Особенностью химии некарбонильных, так называемых “высоковалентных” кластеров является наличие прочно связанных с металлоостовом мостиковых (или шапочных) лигандов, чаще всего галогенидных, оксидных или халькогенидных. В отличие от манипуляций с числом и качественным составом атомов металла разработка селективных методов синтеза кластеров переходных металлов с разными типами мостиков является нетривиальной задачей и своеобразным вызовом для химика-синтетика. Трудности на этом пути хорошо иллюстрирует недавно опубликованный обзор [24]. В случае металлокластеров, в которых присутствуют мостиковые атомы халькогена, различающиеся по геометрическому расположению, селективность может быть достигнута либо путем кластерной самосборки, когда два разных атома халькогена входят в специфические позиции, либо с помощью селективного обмена благодаря различной реакционной способности структурно неэквивалентных атомов халькогена.
Для селективного получения смешанно-халькогенидных кластеров можно использовать два принципиально разных подхода. Если мостиковые атомы халькогена в кластере неэквивалентны, селективность может быть достигнута либо путем кластерной самосборки, при которой два разных атома халькогена входят в их специфические позиции, отвечающие минимальной энергии (термодинамический подход), либо с помощью селективного обмена атомов халькогена благодаря различной реакционной способности структурно неэкивалентных атомов халькогена (кинетический подход). Следует отдельно выделять случаи, когда в кластере присутствуют мостиковые полихалькогенидные лиганды (как правило, дихалькогенидные), при этом возможно возникновение связей между двумя разными атомами халькогена (лиганды SeS2–, TeS2–, TeSe2–). Такая ситуация реализуется в ряде биядерных комплексов железа, ванадия, хрома и ниобия. Например, взаимодействие смеси Na2TeO3 и Na2SeO3 с [Fe(CO)5] в присутствии KOH в метаноле приводит к смеси трехъядерных комплексов [Fe3(CO)9(μ3-Se)(μ3-Te)], [Fe3(CO)9(μ3-Se)2] и [Fe3(CO)9(μ3-Te)2], которую не удается разделить хроматографически. Если же эту смесь последовательно обработать метилатом натрия, а затем кислотой, получается смесь биядерных комплексов [Fe2(CO)6(μ-SeTe)], [Fe2(CO)6(μ-Se2)] [Fe2(CO)6(μ-Te2)], которую удается разделить и получить в чистом виде [Fe2(CO)6(μ-SeTe)] – первый пример комплекса с лигандом SeTe2– [25]. Облучение раствора [Fe2(CO)6(μ-SeTe)] в присутствии [Fe(CO)5] количественно приводит к [Fe3(CO)9(μ3-Se)(μ3-Te)]. Аналогично получены [Fe2(CO)6(μ-SSe)] [26] и [Fe2(CO)6(μ-STe)] [27]. Все комплексы типа [Fe2(CO)6(μ-QQ')] (Q = Q', Q ≠ Q') очень реакционноспособны по отношению к внедрению атомов других переходных металлов по связи Q–Q, что было использовано для получения гетерометаллических кластеров [25, 26, 28].
В реакции селенохлорида ниобия NbSe2Cl2, которому отвечает кристаллохимическая формула ∞[Nb2(Se2)2Сl8/2], с расплавом роданида калия, происходит замещение селена на серу, а в качестве промежуточных продуктов образуются комплексы, содержащие лиганды SeS2– [29]. Однако процесс протекает не селективно и не может служить для получения индивидуальных представителей семейства {Nb2SxSe4 –x}4+. Возможно и обратное замещение серы на селен: дитиокарбаматный комплекс [Nb2(S2)2(Et4NCS2)4] взаимодействует с SePEt3 в присутствии PEt3 с образованием [Nb2(Sе2)2(Et4NCS2)4] [30]. Реакция требует присутствия некоторого количества свободного фосфина. Предполагаемая последовательность стадий представлена на схеме 1 и включает образование промежуточных продуктов с лигандами SeS2–.
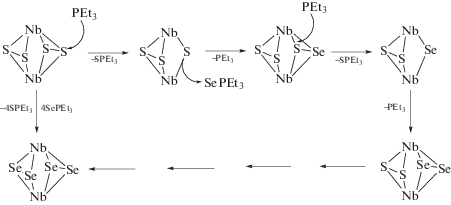
Схема 1 .
Выделить индивидуальные промежуточные комплексы с лигандом SeS2– не удается. Однако при проведении аналогичной реакции между [Nb2(S2)2(Et4NCS2)4] и TePEt3 удалось выделить комплекс [Nb2(S)(Te2)(Et4NCS2)4], содержащий одновременно лиганды μ-S и μ-Te2 [30].
Промежуточные стадии этой реакции включают атаку источника селена (PEt3Se) на моносульфидный лиганд, образующийся в результате отрыва атома серы свободным фосфином из дисульфидного лиганда. Аналогично, видимо, протекает взаимодействие [${\text{Cp}}_{{\text{2}}}^{ * }$V2(μ2-S)3] с Na2Se5, которое приводит к комплексу [${\text{Cp}}_{{\text{2}}}^{ * }$V2(μ2-S)2(μ2-SSe)] (схема 2 ), выделенному в чистом виде [31].
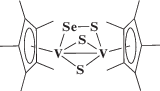
Схема 2 .
Спектроскопическими методами было показано, что реакция моноселенидного комплекса хрома [Cp(CO)2Cr≡Se≡Cr(CO)2Cp] c S8, конечным продуктом которой является [Cp(CO)2Cr≡S≡Cr(CO)2Cp], протекает через образование [Cp2Cr2(CO)5(SSe)] и [Cp2Cr2(CO)4(SSe)] c лигандом SеS2–, которые не выделялись [32].
Трехъядерные кластеры {M3(μ3-Q)(μ2-Q2)3}4+ (M = Mo, W; Q = S, Se, Te) – наиболее интересные объекты для изучения селективного введения двух разных атомов халькогена в кластерное ядро. Они содержат три сорта атомов халькогена: апикальный атом (Qaп или μ3-Q), три экваториальных атома, лежащих практически в плоскости M3 (μ2-Qэкв), три аксиальных атома, расположенных над плоскостью треугольника, противоположной апикальному атому (μ2-Qакс) (рис. 1).
Рис. 1.
Строение кластера {M3(μ3-Q)(μ2-Q2)3}4+ (M = Mo, W; Q = S, Se, Te): вид сверху (а) и сбоку (б).
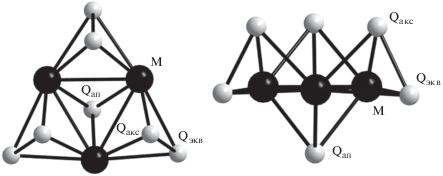
Таким образом, каждый мостиковый лиганд μ2-Q2 можно представить как μ2-QэквQакс. Поэтому в случае атомов халькогена двух сортов a priori возможны различные варианты селективного встраивания одного из них в любую из трех позиций, включая изомерию ${{{{\mu }_{{\text{2}}}}{\text{ - Q}}_{{{\text{э к в }}}}^{'}{\text{Q}}_{{{\text{а к с }}}}^{{''}}} \mathord{\left/ {\vphantom {{{{\mu }_{{\text{2}}}}{\text{ - Q}}_{{{\text{э к в }}}}^{'}{\text{Q}}_{{{\text{а к с }}}}^{{''}}} {{{\mu }_{{\text{2}}}}{\text{ - Q}}_{{{\text{э к в }}}}^{{''}}{\text{Q}}_{{{\text{а к с }}}}^{'}}}} \right. \kern-0em} {{{\mu }_{{\text{2}}}}{\text{ - Q}}_{{{\text{э к в }}}}^{{''}}{\text{Q}}_{{{\text{а к с }}}}^{'}}}.$ Интерес к поиску селективных способов получения смешанно-халькогенидных комплексов {M3(μ3-Q)(μ2-Q2)3}4+ заключается в том, что при этом открывается возможность тонкого регулирования физико-химических свойств комплексов путем замены одного атома халькогена на другой. В частности, это важно при дизайне молекулярных проводников на основе халькогенидных кластеров, в которых реализуются многочисленные специфические межмолекулярные невалентные взаимодействия Q…Q, играющие важную роль в транспорте электронов [33–35]. В качестве примера можно привести хорошо известный полупроводник [Mo3S7(Dmit)3] с величиной Ea = 12–22 мэВ, в структуре которого молекулы [Mo3S7(Dmit)3] связаны контактами μ3-S…μ2-S2, образуя проводящие стопки [33]. С другой стороны, соли с переносом заряда на основе кластерных анионов [Mo3(μ3-S)(μ2-S2)3Cl6]2– и [Mo3(μ3-S)(μ2-SаксSeэкв)3Br6]2– и бис(этилендитио)тетратиафульвалена могут быть как полупроводниками, так и изоляторами, что определяется различными типами взаимодействий халькоген–халькоген и халькоген–галоген [36, 37]. Очевидно, что синтез смешанно-халькогенидных трехъядерных кластеров, в которых атомы халькогена разного сорта будут занимать строго определенные позиции, поможет лучше понять индивидуальные вклады каждого типа невалентных взаимодействий в механизм проводимости и оптимизировать проводящие свойства.
Для получения смешанно-халькогенидных трехъядерных кластеров можно использовать несколько подходов. Один из них основан на том эмпирическом факте, что в реакциях замещения халькогена в первую очередь замещаются экваториальные атомы как наиболее реакционноспособные. Большая лабильность атомов халькогена в экваториальных позициях может быть связана с большей длиной соответствующей связи металл–халькоген: расстояния до экваториальных атомов систематически на ~0.1 Å длиннее, чем до аксиальных [38]. Такой подход был использован для получения кластеров с ядром {M3(μ3-S)(μ2-SаксSeэкв)3}4+ (схема 3 ). Впервые их идентифицировали в галогенидных комплексах, образующихся с высокими выходами при нагревании [Mo3S7X6]2– (Х = Cl, Br) с SePPh3. Атомы селена занимают исключительно экваториальные положения. Замещение аксиальных и тем более центрального атома серы не происходит даже в условиях избытка SePPh3. Соль (Ph3EtP)2[M3(μ3-S)- (μ2-SаксSeэкв)3Cl6] – первый пример структурно охарактеризованного соединения, содержащего лиганд SеS2– [39]. Позднее определены кристаллические структуры для (PPN)2[M3(μ3-S)(μ2-SаксSeэкв)3Cl6] [40] и (TTF)(TBA)[M3(μ3-S)(μ2-SаксSeэкв)3Br6] (TTF = бис(этилендитио)тетратиафульфален, TBA = тетрабутиламмоний) [36]. Последняя соль получена в результате электрохимического окисления TTF в присутствии анионов [Mo3(μ3-S)(μ2-SаксSeэкв)3Br6]2–.
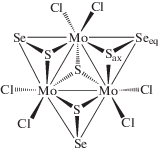
Схема 3 .
Акватация (Et4N)2[Mо3(μ3-S)(μ2-SаксSeэкв)3Br6] в 4 М HРts (пара-толуолсульфуновая кислота) дает аквакомплекс [Mо3(μ3-S)(μ2-SаксSeэкв)3(H2O)6]4+, реагирующий c водорастворимым фосфином ${\text{PR}}_{{\text{3}}}^{{{\text{3}}--}}$ (R = C6H4SO3) c количественным образованием [Mo3S4(H2O)9]4+. Константа скорости этой реакции (2 M HРts) составляет 1.48 × 105 М–1 с–1 [41, 42]:
Аналогично при взаимодействии комплекса вольфрама (Ph4P)2[W3S7Br6] с SePPh3 образуется (Ph4P)2[W3(μ3-S)(μ2-SаксSeэкв)3Br6]. За исчезновением исходного тиокомплекса [M3S7X6]2– удобно следить с помощью спектроскопии КР по отсутствию интенсивной полосы ν(S–S) в районе 550 см–1 и появлению новой интенсивной полосы ν(S–Sе) при 450 см–1.
Другой реагент для введения селена в состав треугольного кластера – селеноцианат калия. Если проводить реакции [M3S7X6]2– с KNCSe в присутствии избытка KNCS, образуются [M3(μ3-S)- (μ2-SаксSeэкв)3(NCS)6]2– (M = Mo, W; X = Cl, Br). Эти реакции необратимы, и даже избыток KNCS или PPh3S не превращает {M3(μ3-S)(μ2-SаксSeэкв)3}4+ обратно в {M3S7}4+. В случае вольфрама наблюдается побочная реакция отщепления селена от [W3(μ3-S)(μ2-SаксSeэкв)3(NCS)6]2– с образованием кластера {W3S4}4+. В отличие от галогенидных комплексов дитиокарбаматный комплекс [Mo3S7(Dtc)3]Dtc (Dtc = диэтилдитиокарбамат) не вступает в реакцию с PPh3Se, но может быть превращeн в [M3(μ3-S)(μ2-SаксSeэкв)3(Dtc)3]SeCN обработкой KNCSe в CH3CN при комнатной температуре. В структуре этой соли присутствуют почти линейные цепочки (S–Se…Se…S–Se…Se…)n, образованные за счeт коротких (2.9–3.3 Å) катион-анионных контактов. Расстояния S–Se в лиганде SSe составляют 2.16–2.23 Å [43].
Схожий дитиокарбаматный комплекс [M3(μ3-S)-(μ2-SаксSeэкв)3(Dtc)3]Br получается при взаимодействии [Mo3S4(CH3CN)9]4+ с Se(Dtc)2 [42]. В этом случае лиганды S2– превращаются в SeS2–.
Высокая степень региоселективности этих реакций была продемонстрирована экспериментами с изотопомерными соединениями. Реакции изотопного обмена между (Et4N)2[M334S7Br6] (М = Mo, W) и K32SCN в мольном соотношении 1 : 100 приводят только к комплексам (Et4N)2-[M3(μ3-34S)(μ2-34Sакс32Sэкв)3(NCS)6], содержащим лиганд 32Sэкв–34Sакс, на основании данных изотопных сдвигов в спектрах КР. Аксиальные атомы серы и атом μ3-S в этом обмене не затрагиваются [44]:
Низкоплавкий KNCSe (Tпл = 100°С с разложением) использовали в реакциях лигандного обмена с кластерными полимерами $[{{{\text{M}}}_{{\text{3}}}}{\text{T}}{{{\text{e}}}_{{\text{7}}}}{{{\text{I}}}_{{\text{2}}}}{{{\text{I}}}_{{{4 \mathord{\left/ {\vphantom {4 2}} \right. \kern-0em} 2}}}}]_{\infty }^{{\text{2}}}$ и их сульфидными и селенидными аналогами из того же семейства треугольных кластеров путем нагревания твердых халькогалогенидов в расплаве KNCSe при 200°C с последующей экстракцией водой [45]. В случае Mo3Te7I4 происходит полное замещение теллура в кластерном ядре {Mo3Te7}4+:
К этому же продукту приводит и реакция Mo3Se7Br4 с расплавом KCNSe – наиболее подходящий способ получения [Mo3Se7(CN)6]2–. Данные масс-спектрометрии свидетельствуют о том, что реакция Mo3Te7I4 с KCNSe протекает ступенчато с образованием [Mo3Te4Se3(CN)6]2– на промежуточной стадии. Замещение халькогена в Mo3S7Br4 в расплаве KCNSe также идет через [Mo3S4Se3(CN)6]2– и [Mo3SSe6(CN)6]2– с образованием [Mo3Se7(CN)6]2– в качестве конечного продукта.
Если предположить, что обмен Te на Se такой же региоселективный, как S на Se в рассмотренных выше примерах, то атомы Se должны сначала входить в экваториальные позиции в ядре {M3(μ3-Q)(μ-Qакс–Qэкв)3}4+. В реакции W3Te7Br4 с KNCSe методом масс-спектрометрии доказано последовательное образование [W3Te4Se3(CN)6]2–, [W3Te3Se4(CN)6]2–, [W3Te2Se5(CN)6]2–, [W3TeSe6(CN)6]2– и [W3Se7(CN)6]2– (схема 4 ). В отличие от молибденового аналога [W3Se7(CN)6]2– легко теряет селен из экваториальных позиций в условиях реакции, и конечным продуктом оказывается [W3Se4(CN)9]5– [45].
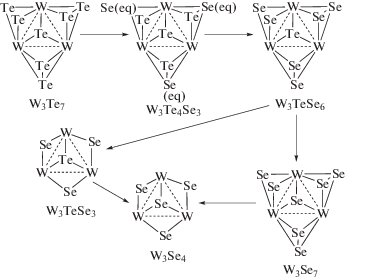
Схема 4 .
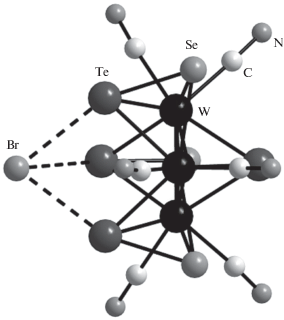
Тщательный контроль за протеканием реакции позволяет выделить смешанный Se/Te комплекс K3{[W3(μ3-Te)(μ2-TeаксSeэкв)3(CN)6]Br} ⋅ 6H2O (рис. 2), в котором атомы Se селективно входят в экваториальные позиции дихалькогенидных лигандов. На сегодняшний день этот комплекс – единственное структурно охарактеризованное соединение с лигандом TeSe2–.
Рис. 2.
Строение ассоциата {[W3Te4Se3(CN)6]Br}3– в структуре K3{[W3(μ3-Te)(μ2-TeаксSeэкв)3(CN)6]Br} ⋅ · 6H2O.
Полимеры W3Se7Br4 и W3S7Br4 в реакции с расплавом KCNSe превращаются в [W3Se4(CN)9]5– и [W3S4(CN)9]5– соответственно, в согласии с меньшей стабильностью ядер {W3Q7}4+, легко теряющих три атома халькогена лигандов μ2-Q2, образуя более устойчивые кластеры {W3Q4}4+.
При добавлении KCN к водному экстракту продуктов реакции W3Te7Br4 с KNCSe был выделен комплекс K5[W3(μ3-Te)(μ2-Se)3(CN)9] [45], содержащий кластерное ядро {W3(μ3-Te)(μ2-Se)3}4+ (рис. 3). Можно предположить, что ион CN– атакует либо атом Te в [W3(μ3-Te)(μ2-TeаксSeэкв)3-CN)6]2– (ион TeCN–, образующийся на начальной стадии, неустойчив в водном растворе и разлагается на CN– и Te), либо атом Se в [W3(μ3-Te)- (μ2-SeаксSeэкв)3(CN)6]2–. Нельзя исключать и нуклеофильную атаку SeCN– по Te с последовательным замещением теллура на селен. Известно, что кластерные ядра {M3Q4}4+ (в крайне редких случаях) могут быть чувствительны к обмену халькогена, о чем свидетельствует неожиданное получение комплексов [Mo3(μ3-S)Te3(изо-PrO)2PS2)3-(PAB)(PBu3)], PAB = пара-аминобензоат, из [Mo3Te7(Dtp)3]I (источником атома серы служит дитиофосфатный лиганд) [46].
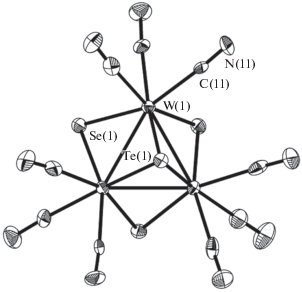
Вероятно, такая лабильность объясняется тем, что связь M–Te – наиболее слабая из всех связей металл–халькоген, что делает возможным замещение теллура на атомы более легких халькогенов в сравнительно мягких условиях.
Следует иметь в виду, что реакционная способность дисульфидных и деселенидных лигандов в кластерах {M3Q7}4+ резко отличается от дителлуридных лигандов. Мостиковые группировки S2 и Se2 в {M3Q7}4+ взаимодействуют с такими нуклеофилами, как CN–, триарилфосфины, тиолы с образованием кластеров {M3Q4}4+, а кластеры {М3Те7}4+ в этих условиях либо не затронуты, либо требуют более основных триалкилфосфинов для отрыва теллура [46, 47]. Напротив, сульфидные и селенидные кластеры весьма устойчивы по отношению к электрофильным реагентам (H+, дигалогены) [48]. Дителлуридные лиганды восприимчивы к электрофилам. Так, реакция [Мо3Te7((EtO)2PS2)3]I с S2Cl2 в мольном соотношении 1 : 5 приводит к частичному региоселективному замещению теллура на серу, давая продукт состава [Мо3Te4.74S2.26((EtO)2PS2)3]I0.72Cl0.28 [49]. По данным РСА, положения Теакс и Теэкв заняты атомами теллура на ~90% (Teакс) и ~50% (Teэкв). В масс-спектре [Мо3Te4.74S2.26((EtO)2PS2)3]I0.72Cl0.28 распределение кластерных форм {Mo3Te7–xSx}4+ варьируется в пределах x от 0 до 5. Реакция [Мо3Te7((EtO)2PS2)3]I с большим избытком S2Cl2 (мольное соотношение 1 : 15) привела к продукту состава [Mo3Te4S3((EtO)2PS2)3]I. Это не индивидуальное соединение, а смесь комплексов [Mo3Te2S5((EtO)2PS2)3]+, [Mo3Te3S4((EtO)2PS2)3]+, [Mo3Te4S3((EtO)2PS2)3]+ и [Mo3Te5S2((EtO)2PS2)3]+ с лигандами S22–, TeS2– и Te22–. Таким образом, степень замещения атомов теллура в аксиальных позициях возрастает при увеличении количества S2Cl2, но при этом “шапочная” позиция μ3-Q остается неизменной и отвечает 100% содержанию Te. Наблюдаемая картина находится в соответствии с рядом уменьшения реакционной способности: Teэкв > Teакс > μ3-Te. Из анализа расстояний Q–Q в кластерах {M3Q7} ожидаемая длина связи S–Te 2.35–2.40 Å. Действительно, найденные расстояния S–Te в обоих продуктах (2.35–2.39 Å) попадают в этот диапазон.
При взаимодействии [W3(μ3-Te)(μ2-Te2)3-((EtO)2PS2)3]Br с S2Cl2 выделен продукт состава [W3Te4.30S2.70((EtO)2PS2)3]Br0.62Cl0.38 [49]. Его образование можно объяснить следующим образом:
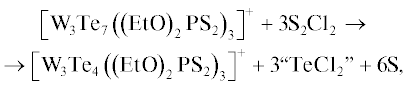
Замещение теллура на серу происходит преимущественно в экваториальных положениях: позиции Qакс занимают преимущественно атомы Te, в то время как позиции Qeq заняты и S, и Te. В масс-спектрах продукта доминируют пики от [W3Te4S3((EtO)2PS2)3]+. Наблюдаются сигналы средней интенсивности от [W3Te3S4((EtO)2PS2)3]+ и [W3Te7((EtO)2PS2)3]+ и минорные сигналы от [W3Te5S2((EtO)2PS2)3]+ и [W3Te6S((EtO)2PS2)3]+.
Таким образом, региоселективность в реакциях замещения и элиминирования является характерной особенностью всех кластеров типа {M3Q7} (Q = S, Se, Те), для которых реакции элиминирования или обмена халькогена затрагивают предпочтительно экваториальные позиции, вероятно, из-за более слабого связывания халькогена в них [50].
Комплекс [W3(μ3-Te)(μ2-Te2)3((EtO)2PS2)3]Br не вступает в реакции с мягкими электрофилами Ph3PX2 (X = Cl, Br), но реагирует с Br2 с образованием продукта состава [W3Te4.25S2.75((EtO)2PS2)3]Br с лигандом TeS2– [49]. Образование смешанно-халькогенидного комплекса [W3Te4S3((EtO)2PS2)3]+ можно объяснить следующей последовательностью превращений:
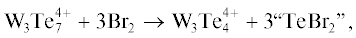
Кластеры {M3(μ3-Sе)(μ2-SеаксSэкв)3}4+, содержащие лиганды ${{\mu }_{{\text{2}}}}{\text{ - S}}{{{\text{е }}}_{{{\text{а к с }}}}}{\text{S}}_{{{\text{э к в }}}}^{{{\text{2}}--}},$ изомерны рассмотренным выше, поскольку атомы селена в этих лигандах занимают аксиальные положения (схема 5 ). Такой тип координации лиганда SeS2– впервые обнаружен в комплексе [Mo3(μ3-S0.65Se0.35)(μ-SеаксSэкв)3((EtO)PS2)3]I, полученном в реакции [Mo3(μ-S0.65Se0.35)Sе3((EtO)PS2)4(Рy)] с Н2S в присутствии I2 [51]. Селенсодержащие кластеры {M3(μ3-Sе)(μ2-SеаксSэкв)3}4+ не вступают в реакции обмена халькогена с источниками серы (Ph3PS, KNCS) в мягких условиях. Поэтому для получения кластеров {M3(μ3-Sе)(μ2-SеаксSэкв)3}4+ подходящей оказалась реакция селенидных кластеров {M3(μ3-Q)(μ2-Sе)3}4+ с источниками “активной” серы (H2S + I2, полисульфиды). В реакции [M3Sе4(H2O)9]4+ с P4S10 в ROH селективно образуются [M3(μ3-Sе)(μ2-SеаксSэкв)3((RO)2PS2)3]Cl, (M = Mo, W; R = Et, изо-Pr) [52]. Здесь сульфид фосфора выступает как источник лиганда (RO)2PS2 (согласно уравнению P4S10 + 8EtOH → → 4(EtO)2P(S)SH + 2H2S), так и в качестве источника “активной” серы, превращающей лиганд μ2-Se в μ2-SеаксSэкв. Наиболее вероятно, что источником этой “активной” серы является смесь ди-, три- и тетрасульфидов, (RO)2P(S)SnP(S)(OR)2, присутствующей в продуктах реакции P4S10 и ROH [53].
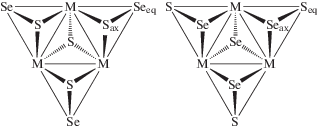
Схема 5 .
Селективное введение одного из атомов халькогена в “шапочную” μ3-позицию. Поскольку атом халькогена в “шапочной” позиции удерживается наиболее прочно, реакции обмена халькогена мало пригодны для селективного получения кластеров типа {M3(μ3-Q')(μ2-Q'')3}. Альтернативой являются реакции самосборки кластеров в присутствии источников двух разных атомов халькогена, где можно надеяться на то, что разные атомы халькогена будут предпочитать позиции с разной связностью. Например, это имеет место в высокотемпературных синтезах кубановых кластеров рения {Re4(μ3-Q)4} (Q = S, Se) из простых веществ в присутствии двух разных халькогенов (S + Te или Se + Te): более легкий халькоген неизменно занимает “внутреннюю” позицию (μ3-Q) в кластере. В соединениях Re4S4Te4 и Re4S4Cl8(TeCl2)4 только атомы серы участвуют в образовании кубанового остова {Re4(μ3-S)4}, а атомы теллура формируют внешнее координационное окружение кластера [54, 55]. В структуре Cs4[Re6S9.45Se3.55], полученного при нагревании смеси Re, Cs2CO3, H2S и Se при 850°C, μ3-позиции в кластере {Re6(μ3-Q)8} заняты только атомами серы, в то время как атомы селена присутствуют только в мостиках μ-Q2 между октаэдрическими кластерами [56]. Тиотеллурохлорид состава Mo2S4Te4Cl22, который образуется при взаимодействии MoTe2 с S2Cl2 при 100°С, как предполагается, содержит кластерное ядро {Mo2(μ2-S2)2}6+, содержащее только атомы серы [57]. Во всех перечисленных примерах кластерное ядро состоит из атомов халькогена только одного типа (серы или селена).
В случае трехъядерных кластеров с помощью реакций самосборки становится возможным, что два разных халькогена занимают определенные положения в кластерном ядре. При этом более легкий атом всегда занимает μ3-позицию. Так, в системах Re2O7–S–Se2Cl2, Re2O7–Se–S2Cl2 и OsO4–Se–S2Cl2 (при 200°С) получены халькогалогениды состава Re3S5Se2Cl7, Re3S6.5Se0.5Cl7 и Os3S7SeCl8 соответственно, содержащие трехъядерные кластеры {M3(μ3-S)(μ-Q2)3} (M = Re, Os; Q = S, Se), в которых μ3-позиция занята атомом серы, а группы μ-Q2 состоят из атомов серы и селена [58]. Реакция между MoCl3 · 3H2O, ZnSe и Me4N(S2P(OEt)2) протекает менее селективно и приводит к смеси трехъядерных дитиофосфатных комплексов [Mo3(μ3-S)(μ2-Se2)3((EtO)PS2)3] и [Mo3(μ3-Se)(μ2-Se2)3((EtO)PS2)3], из которой выделен продукт состава [Mo3(μ3-S0.65Se0.35)(Se2)3-((EtO)PS2)3] [51].
С целью получения кластеров {Mо3(μ3-S)(μ2-Se2)3}4+ и {Mо3(μ3-S)(μ2-Se)3}4+ мы проводили реакции Mo, S, Se и Br2 в мольном соотношении 3 : 1 : 6 : 2 при 350°С, в соответствии со стехиометрией (по аналогии с синтезом Mo3S7Br4 и Mo3Se7Br4), которая должна была обеспечить селективное вхождение серы в “шапочную” позицию:
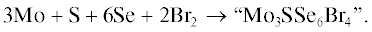
Взаимодействие “Mo3SSe6Br4” с Bu4NBr в условиях механохимической активации в вибрационной мельнице приводит к образованию бромидного комплекса [Mо3(μ3-S)(μ2-Se2)3Br6]2–, выделенного в виде соли (Bu4N)3{[Mo3(μ3-S)(μ2-Se2)3Br6]Br} [59]. Спектр КР (Bu4N)3{[Mo3(μ3-S)- (μ2-Se2)3Br6]Br} показывает полосу при 448 cм–1, относящуюся к колебаниям фрагмента Mo3–μ3-S [60]. В спектре ЯМР 77Se этого комплекса наблюдаются только два сигнала с δ = 256.3 и –132.0 м.д., относящиеся к аксиальным и экваториальным атомам селена в лиганде μ2-Se2.
Акватация [Mo3(μ3-S)(μ2-Se2)3Br6]2– в 4 M HРts с последующим добавлением стехиометрического количества PPh3 приводит к образованию аквакомплекса [Mo3(μ3-S)Se3(H2O)9]4+ [59]:
что отвечает превращению кластерного ядра {Mo3(μ3-S)(μ2-Se2)3}4+ в {Mo3(μ3-S)(μ2-Se)3}4+. Для подтверждения индивидуальности полученного продукта аквакомплекс вводили в реакцию с ацетилацетоном (HАcac) и пиридином с образованием [Mo3(μ3-S)Se3(Аcac)3(Рy)3](PF6). Электроспрей-масс-спектр раствора комплекса в ацетонитриле показал исключительно наличие пика от [Mo3(μ3-S)Se3(Аcac)3(Рy)3]+ и продуктов потери пиридина вплоть до [Mo3(μ3-S)Se3(Аcac)3]+. Кластерное ядро {Mo3(μ3-S)Se3}4+ не фрагментирует в этих экспериментальных условиях, демонстрируя высокую прочность. Пиков, отвечающих другому соотношению селена и серы, не наблюдалось.
Известно, что аквакомплексы кластеров {M3Q4}4+ (M = Mo, W) образуют супрамолекулярные аддукты с макроциклическими кавитандами, в частности с кукурбит[6]урилом (CB[6]). Эти аддукты хорошо кристаллизуются даже из милимолярных водных растворов [61]. Применяя этот подход, был получен и структурно охарактеризован аддукт состава {[Mo3(μ3-S)(μ2-Se)3(H2O)6Cl3]2CB[6]}Cl2 · 11H2O (рис. 4а) из раствора [Mo3(μ3-S)Se3(H2O)9]4+ в 2 M HCl.
Рис. 4.
Строение [Mo3(μ3-S)(μ2-Se)3(H2O)6Cl3]+ (а) и [W3(μ3-S)(μ2-Se)3(H2O)6Cl3]+ (б) (эллипсоиды 50% вероятности).
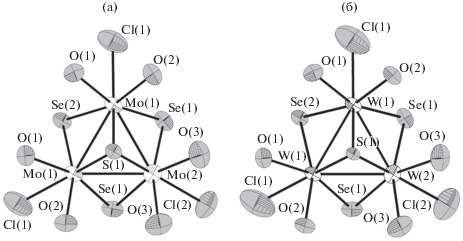
Поведение вольфрамого аналога более сложное. Оптимальный метод синтеза аква-комплексов вольфрама [W3Q4(H2O)9]4+ (Q = S, Se) состоит в реакциях полимеров W3S7Br4 или W3Se7Br4 с H3-PO2 в концентрированной соляной кислоте [62]. Аналогично продукт (или смесь продуктов) брутто-состава “W3SSe6Br4” из реакции вольфрама, смеси серы и селена и брома вводился в реакцию с H3PO2. Ожидалась следующая реакция:
Однако продуктами оказались все формы в ряду [W3SxSe4 –x(H2O)9]4+, что говорит о низкой селективности реакции самосборки кластера в данном случае. Наличие в растворе сразу пяти форм, демонстрирующих схожее поведение на катионообменной смоле, затруднило их полное разделение [59]. Использование стандартных условий разделения на смоле Dowex (комнатная температура, 2 М HCl в качестве элюента) приводит к образованию пурпурной (преимущественно [W3S4(H2O)9]4+ на основании ЭСП), синей и зеленой (основной) фракций. При добавлении CB[6] к синему раствору получены кристаллы [W3S3Se(H2O)7Cl2]2Cl2(CB[6]) ⋅ 15H2O [63]. Из зеленого раствора кристаллизуется [W3S1..5Se2..5Cl1..5(H2O)7..5]2Cl5(CB[6]) ⋅ 18.5H2O [63]. В обоих случаях μ2-мостиковые позиции заняты как серой, так и селеном. Это говорит о том, что исходные растворы содержат смесь аквакомплексов, для разделения которых требуются специальные условия. Очень медленное элюирование с помощью 0.5 M H2SO4 при 4°C позволило выделить зеленую фракцию, обогащенную [W3(μ3-S)Se3(H2O)9]4+ (74%), но содержащую также [W3Se4(H2O)9]4+ (20%) и [W3S2Se2(H2O)9]4+ (6%) [59]. Полное разделение этих продуктов не было достигнуто из-за их малой устойчивости в условиях длительного времени элюирования (дни и даже недели). Так, элюирование с помощью 0.2 M H2SO4 при 4°C в течение нескольких недель приводит к тому, что форма с меньшем содержанием селена ([W3S2Se2(H2O)9]4+) становится доминирующей, а доля [W3(μ3-S)Se3(H2O)9]4+ падает.
Зеленые кристаллы {[W3(μ3-S)(μ2-Se)3-(H2O)7Cl2]2CB[6]}Cl4 · 12H2O (рис. 4б) получены при добавлении СB[6] к зеленому раствору в 2 M HCl с преимущественным содержанием [W3(μ3-S)Se3(H2O)9]4+ [59]. В этом соединении μ2-мостиковые позиции заняты атомами селена, т.е. соединение содержит исключительно кластерное ядро {W3(μ3-S)(μ2-Se)3}4+.
Интересно отметить, что если на стадии высокотемпературной реакции вводить количество серы, соответствующее стехиометрии “W3S4Se3Br4”, то конечным продуктом реакции с H3PO2 оказывается [W3S4(H2O)9]4+ [59]. Образование [W3S4(H2O)9]4+ можно представить следующим уравнением:
Рассмотренный выше подход для селективного введения серы в “шапочную” позицию можно использовать и для получения смешанно-халькогенидных кластеров тантала. Известно, что тантал образует квадратные кластеры {Ta4(μ4-Q)(μ2-Q2)4}8+ (Q = S, Se) – своеобразные высшие “гомологи” рассматриваемых в обзоре треугольных кластеров [64, 65]. Для получения смешанного по халькогену (S/Se) кластера нагревали смесь Ta, S, Se и I2 в мольном соотношении 4 : 1 : 8 : 4, в результате чего был получен халькоиодид состава Ta4S1.5Se7.5I8, в котором μ4-позицию занимает исключительно атом серы [66].
Кластеры {Mo3(μ3-Q)(μ2-Te2)3} (Q = O, S, Se). В системах Mo–Se–Te–Br2 и Mo–Se–Te–I2 при 350°C образуются халькогалогениды [Mo3Se7(TeBr3)Br2]2-[Te2Br10] и [Mo3Se7(TeI3)I2]I (рис. 5), содержащие кластерное ядро {Mo3Se7} [67]. Атомы теллура не входят в состав кластерного остова, а присутствуют в виде лигандов ${\text{TeX}}_{{\text{3}}}^{--},$ координирующихся к одному из атомов молибдена.
Рис. 5.
Строение [Mo3Se7(TeI3)I2]I. Все атомы представлены в термических эллипсоидах с 50%-ной вероятностью. Пунктирными линиями показаны контакты Te(TeI2)…I и 3Seакс…I.
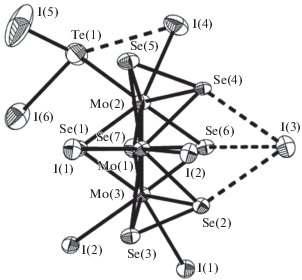
Для введения серы или селена в μ3-позицию мы проводили реакции Mo, Te + Q (Q = S, Se) и Br2 в мольном соотношении 3 : 6 : 1 : 2. Полученные продукты подвергали твердофазным реакциям с KDtp (Dtp = диэтилдитиофосфат) в условиях механической активации, что является эффективным способом “вырезания” теллуридных кластеров из полимерной цепочки [68]. В результате были выделены дитиофосфатные комплексы [Mo3(μ3-Q)(μ2-Te2)3(Dtp)3]+ (схема 6 ), содержащие в μ3-позиции атом серы или селена [69]:
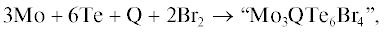
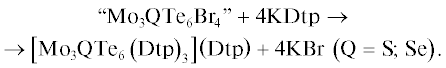
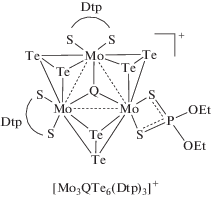
Схема 6 .
Побочным продуктом этих реакций оказалось оксопроизводное [Mo3(μ3-O)(μ2-Te2)3(Dtp)3]+. Однако данную смесь не удалось разделить хроматографически.
Изомерия в [Mo3(µ3-S)(µ2-SSe)3(Dtp)3]Cl. Получение двух изомеров с разным типом координации лиганда µ2-SSe. Описанные выше закономерности позволили нам превратить кластер {Mo3(μ3-S)-(μ2-Se)3}4+ в {Mo3(μ3-S)(μ2-SeаксSэкв)3}4+. Последний можно рассматривать как связевый изомер по отношению к {Mo3(μ3-S)(μ2-SаксSeэкв)3}4+ и целенаправленно синтезировать уникальную пару связевых изомеров состава [Mo3(µ3-S)(µ2-SSe)3(Dtp)3]Cl, отличающуюся лишь способом координации лиганда µ2-SSe [70].
Для синтеза изомера [Mo3(μ3-S)(μ2-SeаксSэкв)3(Dtp)3]Cl (схема 7 ) использовали реакцию аквакомплекса [Mo3(µ3-S)(µ2-Se)3(H2O)9]4+ со свежеприготовленным раствором P4S10 в этаноле, аналогично получению [Mo3(µ3-Se)(µ2-SeаксSэкв)3-(Dtp)3]Cl из [Mo3(µ3-Se)(µ2-Se)3(H2O)9]4+ [52]. Для получения второго изомера использовали реакцию хлоридного комплекса [Mo3S7Cl6]2– с SePPh3 с образованием [Mo3(µ3-S)(µ2-SаксSeэкв)3Cl6]2–. При его взаимодействии с P4S10 в этаноле образуется изомерный дитиофосфатный комплекс [Mo3(µ3-S)(µ2-SаксSеэкв)3(Dtp)3]Cl (схема 7 ), в котором атомы серы находятся исключительно в аксиальных позициях [70].
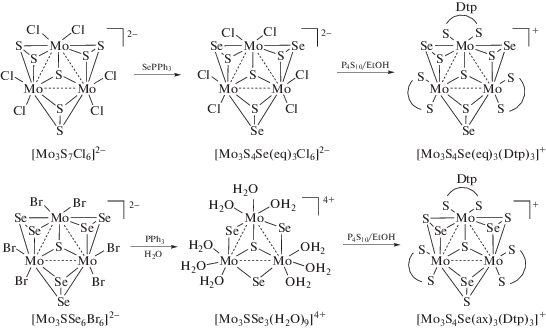
Схема 7 .
Масс-спектры обоих изомеров идентичны и показывают основной пик, относящийся к катиону [Mo3S4Se3(Dtp)3]+. Тем не менее, изомеры можно различить по характеристичным продуктам фрагментации в условиях диссоциации, индуцированной столкновениями (CID). Известно, что для кластеров типа {Mo3Q7}4+ газофазное селективное элиминирование молекулы “Q2” из экваториальных позиций позволяет надежно установить природу халькогена в этих положениях [71, 72]. В спектре CID катиона [Mo3(μ3-S)(μ2-SаксSeэкв)3(Dtp)3]+ наблюдается отщепление фрагмента Se2. Это подтверждает то, что атомы селена занимают экваториальные позиции. Напротив, масс-спектр CID катиона [Mo3(μ3-S)(μ2-SeаксSэкв)3(Dtp)3]+ показывает элиминирование фрагмента S2 в этих же условиях [70].
Весьма чувствительными к расположению атомов халькогена в μ2-SSе2– оказались химические сдвиги в спектрах ЯМР 77Se. Для изомера с μ2-SeаксSэкв наблюдается сигнал при 131 м.д., в то время как для изомера с μ2-SаксSeэкв сигнал сдвинут в отрицательную область (–107 м.д.), что говорит о большем экранировании позиции Seэкв. Для оценки относительной устойчивости двух изомеров были проведены расчеты методом DFT. Оптимизировали структуры комплексов с атомами серы (изомеры I и II) и селена (изомеры III и IV) в “шапочной” позиции: [Mo3(μ3-X)(QэквQакс)3(H2O)6]+ (I: X = S, Qэкв = S, Qакс = Se; II: X = S, Qэкв = Se, Qакс = S; III: X = Se, Qэкв = Se, Qакс = S; IV: X = Se, Qэкв = S, Qакс = Se). Рассчитанные энергии для комплексов I, II, III и IV составили: –2.480517, –2.480518, –3.005715 и –3.005713 × 10–7 кДж/моль соответственно. Исходя из этих данных можно сделать вывод, что для каждой пары изомер с атомами селена в экваториальных позициях стабильнее [70].
Селективное введение атома кислорода в треугольные халькогенидные кластеры. Почти одновременно с открытием очень устойчивого к гидролизу в сильнокислых средах кластера [Mo3(μ3-S)- (μ-S)3(H2O)9]4+ было доказано, что Mo(IV) существует в кислых водных растворах исключительно в виде аналогично построенного оксидного кластера [Mo3(μ3-О)(μ-О)3(H2O)9]4+ [73]. Были получены также вольфрамовые аналоги обоих кластеров [74, 75], а также селенидные кластеры как для молибдена, так и для вольфрама [62]. Очевидный интерес представляло получение промежуточных, оксидно-сульфидных и оксидно-селенидных кластеров. Действительно восстановление биядерного оксо-мостикового комплекса Mo(V) [Mo2O2S2(H2O)6]2+ различными реагентами привело к желаемым продуктам [Mo3OxS4 –x(H2O)9]4+ (x = 0–3) [76, 77] в виде смеси, выделение из которой индивидуальных членов этого семейства – достижимая, но очень трудоемкая задача.
Возможен ли селективный синтез таких кластеров? Здесь можно использовать ретросинтетический подход, хорошо развитый в органическом синтезе. Если взять, к примеру, кластер [Mo3(μ3-S)- (μ-S)(μ-O)2(H2O)9]4+, то его можно рассматривать как продукт координации фрагмента {Mo(H2O)3}2+ к двум атомам серы и одному атому кислорода аквакомплекса [Mo2O2S2(H2O)6]2+. Известно, что реакции конпропорционирования между [Mo2O2(μ-S)2(Сys)2]2– и [MoCl6]3–, или между [Mo2O2(μ-O)- (μ-S)(Сys)2]2– и [MoCl6]3– в 2 М HCl или HРts приводят к образованию [Mo3O2S2(H2O)9]4+ и красного [Mo3O3S(H2O)9]4+ соответственно [77]. Предшественником {Mo(H2O)3}2+ мог бы выступить Mo(CO)6. В этом случае весь молибден встроится в кластер, в то время как в случае гексахлоромолибдата часть реагента должна служить лишь источником электронов. Действительно в жестких условиях (140°С, 4 M HCl) реакция [Mo2O2S2(H2O)6]2+ с Мо(СО)6 приводит практически исключительно к [Mo3(μ3-S)(μ-S)(μ-O)2(H2O)9]4+ (схема 8 ). Любопытно, что в этих условиях даже порошок металлического молибдена реагирует с образованием этого же продукта с выходом 90%. Аналогично был получен [W3(μ3-S)(μ-S)(μ-O)2(H2O)9]4+ (схема 8 ):
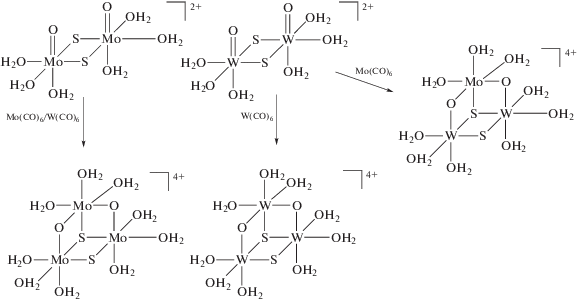
Схема 8 .
Схему 3 ([M2O2S2(H2O)6]2+ + источник M' (M' = Fe, Co, Ni, Pd, Pt, Cu и др.) в низкой степени окисления в принципе можно использовать для синтеза гетерометаллических кластеров. Действительно из [W2O2S2(H2O)6]2+ и Mo(CO)6 был получен кластер [W2Mo(μ3-S)(μ-S)(μ-O)2(H2O)9]4+, выделенный и структурно охарактеризованный в виде [W2Mo(μ3-S)(μ-S)(μ-O)2(Асас)3(Рy)3]PF6. Интересно, что другие комбинации реагентов ([M2O2S2(H2O)6]2+ + Cr(CO)6, [Mo2O2S2(H2O)6]2+ + + W(CO)6) приводят к образованию исключительно гомометаллических треугольных кластеров [M3(μ3-S)(μ-S)(μ-O)2(H2O)9]4+, причем весь металл берется из биядерного комплекса [78]. Неожиданно оказалось, что комбинация [Mo2O2S2(H2O)6]2+/Re(CO)5Cl дает моносульфидный кластер [Mo3(μ3-S)(μ-O)3(H2O)9]4+ [79]. Маршрут этих превращений неясен, но примечательно, что во всех случаях образуется только один трехъядерный кластер, а не смесь. Можно ожидать, что аналогичные реакции биядерных моносульфидных кластеров [M2O2(μ-O)(μ-S)(H2O)6]2+ c соответствующими карбонилами должны давать треугольные кластеры [M3(μ3-S)(μ-O)3(H2O)9]4+. Косвенное подтверждение возможности схемы 8 – получение селенидных кластеров [M3(μ3-Sе)(μ-O)3(H2O)9]4+ в реакциях самосборки из моноядерных [MoOCl5]2– или WO42– и H2Se (схема 9 ). Селеноводород является сильным восстановителем (E0 Se/H2Se –0.4 В). Можно предположить, что на определенном этапе образуется биядерный комплекс [M2O2(μ-O)(μ-Sе)(H2O)6]2+, координация атома металла к которому с последующим восстановлением приводит к [M3(μ3-Sе)(μ-O)3(H2O)9]4+, ранее практически недоступному (известный метод синтеза Мо кластера давал выход 3%; кластер вольфрама не был известен). Примечательна высокая селективность этих реакций, в которых не образуются треугольные кластеры с иным соотношением кислород : халькоген, и выделение целевого продукта не вызывает затруднений. Это позволило провести ряд реакций замены лигандов и изучить электрохимическое поведение оксосульфидных и оксоселенидных кластеров молибдена и вольфрама [80, 81].
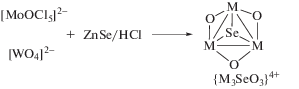
Схема 9 .
Этот же подход можно распространить и на кластеры металлов V группы. В частности, восстановление NbCl5 в присутствии ZnSe/HCl приводит к образованию единственного продукта с кластерным ядром {Nb4(μ4-Se)(μ2-O)5}4+ [82]. Таким образом, анион Se2–, по-видимому, играет ключевую роль в реакциях самосборки кластеров, выполняя функцию лиганда с максимальной связностью, µ3 – в случае трехъядерных кластеров Mo и W и µ4 – в случае четырехъядерных кластеров Nb.
Смешанные по металлу кластеры {MoxW3 –xQ4} и {MoxW4 –xQ4} (Q = S, Se). Единственный способ синтеза смешанных кластеров такого типа мето-дом самосборки – восстановление эквимолярной смеси (NH4)2WS4 и [Mo2O2(μ-S)2(Сys)2]2– избытком NaBH4. При этом образуется смесь продуктов, из которой после хроматографического разделения выделены зеленый [Mo2WS4(H2O)9]4+ (17%) и серый [MoW2S4(H2O)9]4+ (3%) [83]. Очевидно, что эту методику нельзя рассматривать в качестве препаративной. Проблема селективного получения кластеров {MoxW3 –xQ4} (Q = S, Se) оказалась тесно связанной с получением кубановых кластеров {MoxW4 –xQ4} и их селективной деградацией.
Мы показали, что кубановые кластеры можно получить c высоким выходом по реакции “достройки” треугольных кластеров (схема 10 ):
Эта реакция основана на том, что треугольные и кубановые кластеры тесно связаны друг с другом как структурно, так и химически. Три мостиковых атома халькогена треугольного кластера можно рассматривать как лиганды, способные координировать атом металла и тем самым “достроить” треугольный кластер до кубанового. При нагревании на воздухе растворов кубановых аквакомплексов происходит обратная реакция – окислительная деградация кластеров с образованием трехъядерных комплексов, причем в случае гетерометаллических кластеров всегда селективно “теряется” атом вольфрама, а не молибдена. Таким образом, становятся доступны кластеры [W2MoQ4(H2O)9]4+ (Q = S, Se) (схема 10 ):
Примечательно то, что это цикл можно повторить:
и даже перейти к следующему этапу:
Интересно, что если исходить из вольфрамовых треугольных кластеров, то, осуществив последовательно все три цикла достройки–деградации, на выходе получается треугольный молибденовый кластер, который как бы последовательно “собирается” на матрице из атомов серы. Это можно рассматривать как уникальный пример передачи структурной информации при полной замене одного атома металла в кластере на другой [84, 85].
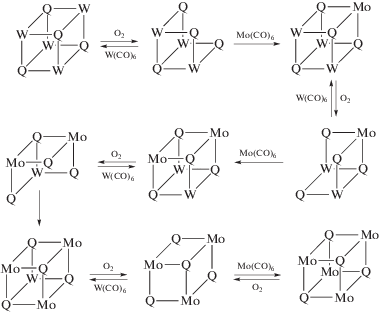
Схема 10.
Смешанный по халькогену треугольный кластер [Mo3(μ3-S)Se3(H2O)9]4+ реагирует с Mo(CO)6 при 130°C в растворе HCl в запаянной ампуле с образованием красно-коричневого комплекса [Mo4(μ3-S)(μ3-Se)3(H2O)12]4+, который на воздухе окисляется в зеленый [Mo4(μ3-S)(μ3-Se)3(H2O)12]5+ (схема 11 ). Нагревание раствора [Mo4(μ3-S)(μ3-Se)3(H2O)12]5+ на воздухе приводит к деградации комплекса и образованию исходного комплекса [Mo3(μ3-S)Se3-(H2O)9]4+, идентифицированного с помощью ЭСП [59].
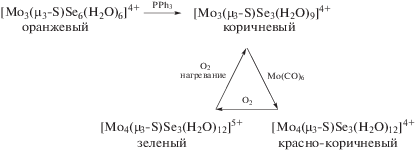
Возможны два направления деградации кубанового кластера. Во-первых, куб Mo4SSe3 может потерять одну вершину в результате разрыва как связи Mo–S, так и Mo–Se. Это должно приводить к кластеру Mo3SSe3, в котором один атом серы занимает μ2-позицию (такой кластер неизвестен). Во-вторых, возможен разрыв трех связей Mo–Se. Это приведет к исходному кластеру Mo3SSe3, что и наблюдается в нашем случае. Второй путь фрагментации предпочтителен, вероятно, из-за относительной легкости разрыва связей Mo–Se [59].
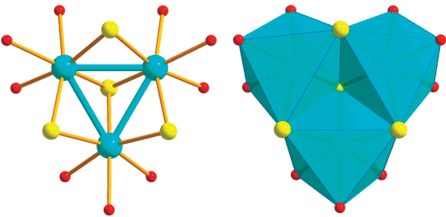
Халькогенсодержащие полиоксометаллаты (ПОМ) представляют собой полиядерные комплексы, структурная основа которых – атомы переходных металлов (Mo, W, V, Nb, Ta) в высшей степени окисления, связанные через мостиковые атомы кислорода. Как правило, замещение атомов кислорода на атомы халькогена вызывает полное разрушение структуры ПОМ. Редким исключением являются реакции ниобий-замещенных поливольфраматов ([W5O18{NbO}]3–, [PW11O39{NbO}]3–) с источниками серы или селена ((Me3Si)2S, (Me3Si)2Se, R2P2S4), приводящие к селективному замещению кислорода при атоме ниобия на халькоген с сохранением структуры ПОМ [86]. Альтернативный подход к синтезу гибридных халькогенсодержащих ПОМ был предложен в [87]. Авторы получили серусодержащий аналог γ-[SiW12O40]4– реакцией γ-[SiW10O36]8– с биядерным аквакатионом [Mo2S2O2(H2O)4]2+. В этой простой и изящной реакции силиковольфрамат выступает как тетрадентатный лиганд, замещающий четыре молекул воды в биядерном тиокатионе. Мы обратили внимание на другой потенциальный строительный блок – аквакомплексы [Mo3S4(H2O)9]4+ и их аналоги [88]. Очевидна их структурная аналогия с фрагментом [W3O13]8– – основным строительным блоком многих полиоксометаллатов со структурой Кеггина и Доусона (рис. 6).
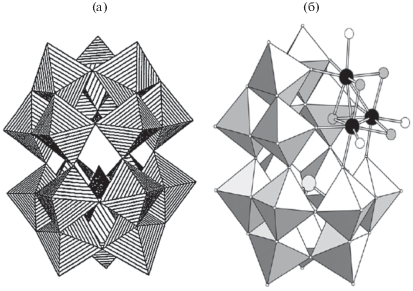
На практике внедрение [Mo3S4(H2O)9]4+ удалось реализовать следующим образом. Известно, что трилакунарные анионы ${\text{E}}{{{\text{W}}}_{{\text{9}}}}{\text{O}}_{{{\text{33}}}}^{{{\text{9}}--}}$ (E = As, Sb) и ${\text{Te}}{{{\text{W}}}_{{\text{9}}}}{\text{O}}_{{{\text{33}}}}^{{{\text{8}}--}}$ способны достраиваться в присутствии избытка вольфрамата до более крупных ${\text{E}}{{{\text{W}}}_{{{\text{15}}}}}{\text{O}}_{{{\text{6}}0}}^{{{\text{9}}--}}$ (E = As, Sb) и ${\text{Te}}{{{\text{W}}}_{{{\text{15}}}}}{\text{O}}_{{{\text{6}}0}}^{{{\text{8}}--}}$ (рис. 7).
Рис. 7.
а – строение ${\text{E}}{{{\text{W}}}_{{{\text{15}}}}}{\text{O}}_{{{\text{60}}}}^{{{\text{9}} - }}$ (E = As, Sb) и ${\text{Te}}{{{\text{W}}}_{{{\text{15}}}}}{\text{O}}_{{{\text{6}}0}}^{{{\text{8}}--}},$ где нижняя половина отвечает структуре исходных анионов, “достроенных” тремя фрагментами {W3O13} до 15-ядерного аниона; б – та же структура с заменой одного из фрагментов {W3O13} на структурно эквивалентный {Mo3S4(H2O)3O6}.
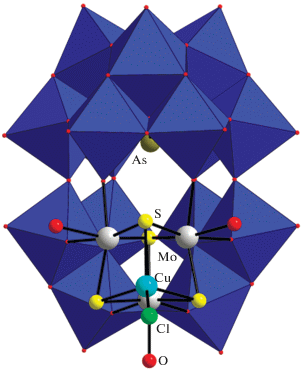
Нам удалось осуществить синтез[AsW15Mo3S4-(H2O)3O53]9–, [SbW15Mo3S4(H2O)3O53]9–, [TeW15Mo3S4-(H2O)3O53]8– добавлением необходимого количества вольфрамата и [Mo3S4(H2O)9]4+ к соответствующим нонавольфраматным производным As(III), Sb(III), Te(IV). Полученные гибридые халько-полиокосметаллаты (халько-ПОМ) соответствуют максимально достигнутой в настоящее время степени замещения атомов кислорода на атомы халькогена в структуре ПОМ. Введение атомов серы резко меняет реакционную способность ПОМ, делая возможным координацию “мягких” ионов металла, таких как Cu(I) или Au(I). Действительно [AsW15Mo3S4(H2O)3O53]9– реагирует с CuCl с образованием гетерометаллического кластера [AsW15Mo3(CuCl)S4(H2O)3O53]9–, выделенного и структурно охарактеризованного в виде цезиевой соли (рис. 8) [89, 90].
Таким образом продемонстрирована возможность и необходимость разработки рациональных подходов к синтезу сложных кластерных и полиядерных соединений. В качестве иллюстративного материала использованы главным образом кластерные комплексы молибдена и вольфрама, содержащие различные по своей природе атомы халькогена в ядре. Для них удалось также разработать синтетические пути для целенаправленного получения смешанных халькогенидных и оксидно-халькогенидных кластеров и полиядерных комплексов. Синтез таких соединений не только стимулирует методологию синтеза сложных (разнолигандных, полиядерных и кластерных) координационных соединений, но и дает возможность “пошаговых” модификаций физических и химических свойств, что, безусловно, важно для создания функциональных материалов с прогнозируемыми свойствами.
Список литературы
Кукушкин Ю.Н. Химия координационных соединений. М.: Высшая школа, 1985. 457 с.
Эссен Л.Н. Геометрические изомеры платины и транс-влияние. М.: Наука, 1969. 117 с.
Синицын Н.М. Проблемы координационной химии. Транс-влияние в химии координационных соединений. М.: Наука, 1979. 341 с.
Kabin E.V., Emel’yanov V.A., Vorob’yev V.A. et al. // Russ. J. Inorg. Chem. 2012. V. 57. P. 1146. https://doi.org/10.1134/ S0036023612050105
Hoffmann R. // Angew. Chem. Int. Ed. 1982. V. 21. P. 711.
Губин С.П., Шульпин Г.Б. Химия комплексов со связями металл–углерод. Новосибирск: Наука. Сиб. отд-ние, 1984. 282 с.
Эльшенбройх К. Металлорганическая химия. М.: БИНОМ. Лаборатория знаний, 2014. 748 с.
Eremenko I.L., Sidorov A.A., Kiskin M.A. // Magnetic Nanoparticles. Weinheim: Wiley-VCH, 2009. P. 349.
Sapianik A.A., Zorina-Tikhonova E.N., Kiskin M.A. et al. // Inorg. Chem. 2017. V. 56. P. 1599.
Balzani V., Credi A., Venturi M. Molecular Devices and Machines: Concepts and Perspectives for the Nanoworld. Weinheim: Wiley-vch, 2008. 588 p.
Beer P.D., Gale P.A., Smith D.K. // Supramolecular Chemistry. Oxford: Oxford Univ. Press, 1999. 96 p.
Meshkov I.N., Bulach V., Gorbunova Y.G. et al. // Inorg. Chem. 2016. V. 55. P. 10774.
Губин С.П. Химия кластеров. Основы классификации и строение. M.: Наука, 1987. 262 с.
Пасынский A.A., Eременко ИЛ., Гасанов Г.Ш. и др. // Коорд. химия. 1984. V. 10. № 5. P. 634.
Pasynskii A.A., Skabitski I.V., Torubaev Y.V. et al. // J. Organomet. Chem. 2003. V. 671. P. 91.
Konchenko S.N., Virovets A.V., Tkachev S.V., Podberezskaya N.V. // Polyhedron. 1997. V. 16. P. 707.
Vahrenkamp H. // Comments Inorg. Chem. 1985. V. 4. P. 253.
Zhang Y.H., Yuan J.C., Yin Y.Q. et al. // New J. Chem. 2001. V. 25. P. 939.
Holm R.H. Comprehensive Coordination Chemistry II. Amsterdam: Elsevier, 2004. V. 8. P. 61.
Malinak S.M., Coucouvanis D. // Progress in Inorg. Chem. 2001. V. 49. P. 599.
Ohki Y., Tatsumi K. // Z. Anorg. Allg. Chem. 2013. V. 639. P. 1340.
Sokolov M.N., Hernandez-Molina R., Abramov P.A. // Molybdenum: Its Biological and Coordination Chemistry and Industrial Applications. New York: Nova Science Publishers, 2013. P. 105.
Gushchin A.L., Laricheva Y.A., Sokolov M.N., Llusar R. // Russ. Chem. Rev. 2018. V. 87. P. 670.
Naumov N.G., Brylev K.A., Mironov Y.V. et al. // J. Struct. Chem. 2014. V. 55. P. 1371.
Mathur P., Chakrabarty D., Hossain M.M. // J. Organomet. Chem. 1991. V. 401. P. 167.
Mathur P., Sekar P., Satyanarayana C.V.V., Mahon M.F. // Organometallics. 1995. V. 14. P. 2115.
Mathur P., Chakrabarty D., Hossain M.M., Rashid R.S. // J. Organomet. Chem. 1991. V. 420. P. 79.
Chakrabarty D., Hossain M.M., Kumar R.K., Mathur P. // J. Organomet. Chem. 1991. V. 410. P. 143.
Мищенко А.В., Федоров В.Е., Колесов Б.А. и др. // Коорд. химия. 1989. V. 15. № 2. P. 200.
Sokolov M., Imoto H., Saito T., Fedorov V. // Dalton Trans. 1999. № 1. P. 85.
Herberhold M., Kuhnlein M., Schrepfermann M. et al. // J. Organomet. Chem. 1990. V. 398. P. 259.
Goh L.Y. // Dalton Trans. 1989. № 3. P. 431.
Llusar R., Uriel S., Vicent C. et al. // J. Am. Chem. Soc. 2004. V. 126. P. 12076.
Llusar R., Vicent C. // Coord. Chem. Rev. 2010. V. 254. P. 1534.
Gushchin A.L., Llusar R., Vicent C. et al. // Eur. J. Inorg. Chem. 2013. P. 2615.
Alberola A., Llusar R., Triguero S. et al. // J. Mater. Chem. 2007. V. 17. P. 3440.
Alberola A., Fourmigué M., Gómez-García C.J. et al. // New J. Chem. 2008. V. 32. P. 1103.
Virovets A.V., Podberezskaya N.V. // J. Struct. Chem. 1993. V. 34. P. 306.
Fedin V.P., Mironov Y.V., Sokolov M.N. et al. // Inorg. Chim. Acta. 1990. V. 174. P. 275.
Fedin V.P., Sokolov M.N., Fedorov V.Y. et al. // Inorg. Chim. Acta. 1991. V. 179. P. 35.
Saysell D.M., Fedin V.P., Lamprecht G.J. et al. // Inorg. Chem. 1997. V. 36. P. 2982.
Fedin V.P., Mironov Y.V., Sokolov M.N. et al. // Zh. Neorg. Khim. 1992. V. 37. P. 2205.
Fedin V.P., Sokolov M.N., Virovets A.V. et al. // Polyhedron 1992. V. 11. P. 2395.
Fedin V.P., Sokolov M.N., Kibirev O.S. et al. // Zh. Neorg. Khim. 1991. V. 36. P. 3089.
Sokolov M.N., Abramov P.A., Gushchin A.L. et al. // Inorg. Chem. 2005. V. 44. P. 8116.
Lin X., Chen H.Y., Chi L.S. et al. // Polyhedron. 2000. V. 19. P. 925.
Chen H.Y., Lin X., Chi L.S. et al. // Inorg. Chem. Commun. 2000. V. 3. P. 331.
Sokolov M.N., Fedin V.P., Sykes A.G. // Comprehensive Coordination Chemistry II. Amsterdam: Elsevier, 2004. V. 4. P. 768.
Gushchin A.L., Sokolov M.N., Vicent C. et al. // Polyhedron. 2009. V. 28. P. 3479.
Mayor-López M.J., Weber J., Hegetschweiler K. et al. // Inorg. Chem. 1998. V. 37. P. 2633.
Hu J., Zhuang H.H., Liu S.X., Huang J.L. // Trans. Met. Chem. 1998. V. 23. P. 547.
Hernandez-Molina R., Sokolov M., Nunez P., Mederos A. // Dalton Trans. 2002. № 6. P. 1072.
Lippman A.E. // J. Org. Chem. 1966. V. 31. P. 471.
Mironov Y.V., AlbrechtSchmitt T.E., Ibers J.A. // Inorg. Chem. 1997. V. 36. P. 944.
Fedorov V.E., Mironov Y.V., Fedin V.P. et al. // Acta Crystallogr. C. 1996. V. 52. P. 1065.
Bronger W., Miessen H.J., Neugroschel R. et al. // Z. Anorg. Allg. Chem. 1985. V. 525. P. 41.
Герасько О.А. Автореф. дисс. … канд. хим. наук. Новосибирск: ИНХ, 1989. 19 с.
Volkov S.V., Fokina Z.A., Yanko O.G. et al. // Russ. J. Inorg. Chem. 2005. V. 50. P. 1150.
Gushchin A.L., Ooi B.L., Harris P. et al. // Inorg. Chem. 2009. V. 48. P. 3832.
Fedin V.P., Sokolov M.N., Gerasko O.A. et al. // Inorg. Chim. Acta. 1990. V. 175. P. 217.
Gerasko O.A., Sokolov M.N., Fedin V.P. // Pure Appl. Chem. 2004. V. 76. P. 1633.
Fedin V.P., Sokolov M.N., Virovets A.V. et al. // Inorg. Chim. Acta. 1998. V. 269. P. 292.
Abramov P.A., Gushchin A.L., Sokolov M.N., Fedin V.P. // J. Struct. Chem. 2010. V. 51. P. 378.
Sokolov M.N., Gushchin A.L., Virovets A.V. et al. // Inorg. Chem. 2004. V. 43. P. 7966.
Sokolov M.N., Gushchin A.L., Abramov P.A. et al. // Inorg. Chem. 2005. V. 44. P. 8756.
Gushchin A.L., Sokolov M.N., Abramov P.A. et al. // J. Cluster Sci. 2009. V. 20. P. 241.
Sokolov M.N., Gushchin A.L., Abramov P.A. et al. // Inorg. Chem. 2007. V. 46. P. 4677.
Virovets A.V., Gushchin A.L., Abramov P.A. et al. // J. Struct. Chem. 2006. V. 47. P. 326.
Gushchin A.L., Ryzhikov M.R., Virovets A.V., Sokolov M.N. // Russ. J. Coord. Chem. 2013. V. 39. P. 181. https://doi.org/10.1134/ S1070328413020036
Hernandez-Molina R., Gushchin A., Vicent C., Gili P. // J. Cluster Sci. 2015. V. 26. P. 83.
Hegetschweiler K., Caravatti P., Fedin V.P., Sokolov M.N. // Helv. Chim. Acta. 1992. V. 75. P. 1659.
Llusar R., Polo V., Velez E., Vicent C. // Inorg. Chem. 2010. V. 49. P. 8045.
Benory E., Bino A., Gibson D., Cotton F.A. // Inorg. Chim. Acta. 1985. V. 99. P. 137.
Segawa M., Sasaki Y. // J. Am. Chem. Soc. 1985. V. 107. P. 5565.
Shibahara T., Kohda K., Ohtsuji A. et al. // J. Am. Chem. Soc. 1986. V. 108. P. 2757.
Shibahara T., Yamamoto T., Kanadani H., Kuroya H. // J. Am. Chem. Soc. 1987. V. 109. P. 3495.
Martinez M., Ooi B.L., Sykes A.G. // J. Am. Chem. Soc. 1987. V. 109. P. 4615.
Abramov P.A., Sokolov M.N., Hernandez-Molina R. et al. // Inorg. Chim. Acta. 2010. V. 363. P. 3330.
Abramov P.A., Laricheva Y.A., Peresypkina E.V. et al. // Inorg. Chim. Acta. 2012. V. 383. P. 7.
Abramov P.A., Sokolov M.N., Peresypkina E.V. et al. // Inorg. Chim. Acta. 2011. V. 375. P. 314.
Sokolov M.N., Gonzalez-Platas J., Hernandez-Molina R. et al. // Polyhedron. 2013. V. 60. P. 116.
Gushchin A.L., Ooi B.L., Harris P. et al. // Z. Anorg. Allg. Chem. 2019. V. 645. P. 398.
Shibahara T., Yamasaki M., Watase T., Ichimura A. // Inorg. Chem. 1994. V. 33. P. 292.
Sokolov M., Esparza P., Hernandez-Molina R. et al. // Inorg. Chem. 2005. V. 44. P. 1132.
McLean I.J., Hernandez-Molina R., Sokolov M.N. et al. // Dalton Trans. 1998. P. 2557.
Klemperer W.G., Schwartz C. // Inorg. Chem. 1985. V. 24. P. 4459.
Cadet E., Bereau V., Marg B. et al. // Inorg. Chem. 1996. V. 35. P. 3099.
Cadot E., Sokolov M.N., Fedin V.P. et al. // Chem. Soc. Rev. 2012. V. 41. P. 7335.
Sokolov M.N., Kalinina I.V., Peresypkina E.V. et al. // Angew. Chem. Int. Ed. 2008. V. 47. P. 1465.
Sokolov M.N., Peresypkina E.V., Kalinina I.V. et al. // Eur. J. Inorg. Chem. 2010. P. 5446.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия