

Координационная химия, 2019, T. 45, № 8, стр. 451-460
Синтез и строение трис(пиразолил)боратных оксотиолатных комплексов рения
И. В. Скабицкий 1, *, С. Г. Сахаров 1, А. А. Пасынский 1, Р. С. Эшмаков 1
1 Институт общей и неорганический химии им. Н.С. Курнакова РАН
Москва, Россия
* E-mail: skabitskiy@gmail.com
Поступила в редакцию 21.01.2019
После доработки 12.03.2019
Принята к публикации 14.03.2019
Аннотация
Двумя методами, по аналогии с известными тиофенильными комплексами, получены комплексы рения TpReOCl(StBu) (I), TpReO(StBu)2 (II) и TpReO(SnC3H7)2 (III) с более электронодонорными алкилтиолатными лигандами, охарактеризованные ИК- и ЯМР-спектроскопией. Cтроение I–III определено методом РСА (СIF files CCDC № 1892096 (I), 1892097 (II), 1892098 (III)). Температурная зависимость спектров бис-тиолатных комплексов позволяет определить энергию активации для заторможенного вращения вокруг связи Re–тиолатный лиганд. Выделен и структурно охарактеризован побочный продукт реакции TpReOCl2 с NaS-трет-Bu – биядерный кислородмостиковый комплекс TpReIVCl(S-трет-Bu)O(S-трет-Bu)2ReIVTp (СIF files CCDC № 1892099).
Трис(пиразолил)боратный анион – тридентатный шестиэлектронодонорный лиганд, образующий стабильные комплексы с рением. Будучи прочно связан с металлом, он не вступает в реакции лигандного обмена, не окисляется и не восстанавливается даже в довольно жестких условиях. Трис(пиразолил)боратные комплексы в значительной степени ведут себя, как аналоги пентаметилциклопентадиенильных комплексов: в частности TpReOCl2 (Tp = = HB(C3H3N2)3) [1] по стерическим и электронным характеристикам напоминает C5Me5ReOCl2, полученый in situ реакцией Cp′ReCl4 с водой [2]. TpReOCl2 можно получить непосредственно из перрената аммония при кипячении в спирте в присутствии соляной кислоты (представляет собой голубой кристаллический порошок, ИК-спектр: Re–O и B–H при 978 и 2528 см–1 соответственно [3].
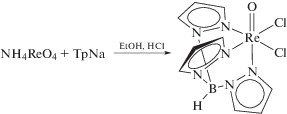
Атомы хлора легко замещаются на тиолатные группировки в реакциях с тиолами в присутствии оснований: например, хелатный комплекс TpReO(SC2H4S) c сильно укороченой связью Re=O (1.69 Å) получали реакцией TpReOCl2 с HSC2H4SH в присутствии триэтиламина [4]. Аналогично получали соединения с одним и двумя фенил- и этилтиолатными лигандами. При этом последовательное замещение хлоридных лигандов на один и два фенилтиолата приводит к смещению полосы колебания мсвязи Re–O в ИК-спектрах комплексов (978, 956 и 945 см–1 соответственно) [5].
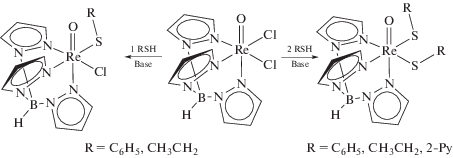
Цель настоящей работы – синтез трис-пиразолилборат-оксо-трет-бутилтиолатных комплексов рения как перспективных металлсодержащих лигандов для получения гетерометаллических комплексов, в которых трет-бутилтиолатные группы обеспечивают лучшую растворимость, а также более легкий термораспад комплексов в процессе получения наноразмерных неорганических составов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали коммерческие перренат аммония, трет-бутилмеркаптан и ди-трет-бутилдисульфид (Acros organics) без дополнительной очистки. Синтезы TpReOCl2 [1] и TpNa [6] проводили согласно литературным методикам. Все операции в ходе синтеза и выделения продуктов вели в абсолютных растворителях в атмосфере чистого аргона. Для тонкослойной хроматографии (ТСХ) применяли пластины фирмы MERCK из силикагеля 60 F254 на алюминиевой основе. ИК-спектры снимали на спектрометре Bruker Alpha с использованием приставки НПВО Platinum ATR. Спектры ЯМР 1H и 13C регистрировали на приборе Bruker AV 400; химические сдвиги приведены относительно тетраметилсилана.
Синтез TpReOCl(S-трет-Bu) (I) и TpReO(S-трет-Bu)2 (II). Методика 1. К суспензии 100 мг (0.21 ммоль) TpReOCl2 в 15 мл толуола добавляли 80 мкл (0.54 ммоль) DBU (диазабициклоундецен) и 50 мкл (0.44 ммоль) трет-бутилмеркаптана. Реакционную смесь перемешивали при 110°C в герметично закрытом сосуде Шленка в течение суток. Летучие компоненты реакционной смеси удаляли в вакууме. Твердый остаток растворяли в 10 мл хлористого метилена, разбавляли 12 мл гексана и наносили на хроматографическую колонку (силикагель 14.5 × 1.5 см), элюируя зеленую и коричневую зоны смесью дихлорметан–гексан = 1 : 1. Зеленый элюат упаривали досуха, остаток растворяли в смеси 3 мл хлористого метилена с 1.5 мл гексана. Смесь выдерживали при –25°C в течение сутoк. Выпавший зеленый порошок отфильтровывали, промывали пентаном (4 раза по 1.5 мл) и высушивали в вакууме. Выход 53 мг (48%).
Коричневую фракцию упаривали досуха, растворяли в 1.5 мл гептана и выдерживали при ‒25°C в течение 3 сут. Выпавшие кристаллические сростки отделяли декантацией, промывали пентаном, охлажденным жидким азотом (4 раза по 4 мл), и высушивали в вакууме. Выход 13 мг (11%).
TpReOCl(S-трет-Bu): ТСХ (CH2Cl2): зеленое пятно, Rf 0.75; TpReO(S-трет-Bu)2: ТСХ (CH2Cl2): желто-коричневое пятно, Rf 0.67.
Синтез I, II и TpReCl(S-трет-Bu)O(S-трет-Bu)2ReTp (IV). Методика 2. К суспензии 0.23 г (10 ммоль) натрия в 20 мл ТГФ добавляли 1.1 мл ди-трет-бутилдисульфида (5.7 ммоль) и 0.5 мл диглима. Реакционную смесь перемешивали в течение сут. К полученной белой суспензии добавляли 2.08 г (4.3 ммоль) TpReOCl2 и нагревали реакционную смесь при 65°C в течение 3 ч. Растворитель удаляли в вакууме, полученное темно-коричневое масло экстрагировали порциями 60, 50 и 4 × 20 мл гептана. Гептановый экстракт упаривали до 20 мл и хроматографировали на колонке с силикагелем (3 × 15 см). Элюировали 3 фракции: зеленую (200 мл смеси CH2Cl2–гексан = 1 : 1), коричневую (250 мл смеси CH2Cl2–гексан = 1 : 1 и 50 мл CH2Cl2) и фиолетовую (300 мл CH2Cl2).
После упаривания зеленой фракции в вакууме получили 65 мг (3%) зеленого порошка I.
Коричневую фракцию упаривали досуха, остаток растворяли в 12 мл гексана. Pаствор выдерживали при –25°С в течение сутoк, выпавшие тонкие игольчатые кристаллы отфильтровывали при ‒70°C, промывали пентаном, охлажденным до начала замерзания (–130°C) в жидком азоте (3 раза по 8 мл), и высушивали в вакууме. Выход II 327.6 мг (13%).
Фиолетовую фракцию упаривали досуха, темное масло экстрагировали горячим гексаном. Образовавшиеся при выдерживании гексанового экстракта в течение суток при комнатной температуре черно-фиолетовые кристаллы IV отделяли декантацией, промывали 3 × 5 мл гексана и высушивали в вакууме. Выход IV 1 мг (0.02%).
Не растворившийся в гептане остаток экстрагировали 40 мл бензола. Полученный экстракт выдерживали при 5°С, образовавшийся коричневый осадок отделяли фильтрованием, промывали 4 × 5 мл гептана и перекристаллизовывали из смеси 10 мл хлористого метилена + 10 мл гексана. Полученый зеленый порошок растворяли в 30 мл CH2Cl2 и фильтровали через слой силикагеля. К зеленому раствору добавляли 5 мл гексана, упаривали досуха и высушивали в вакууме. Выход I 211.5 мг (9%).
I: ТСХ (CH2Cl2): зеленое пятно, Rf 0.75. ИК-спектр (ν, см–1): 3144 о. сл, 3119 о. сл, ш, 2954 о. сл. ш, 2915 о. сл. ш, 2856 о. сл, 2516 о. сл. ш, 1502 сл, 1474 о. сл, 1453 о. сл, 1405 ср, 1388 сл, 1360 о. сл, 1310 ср, 1211 ср, 1186 сл, 1165 сл, 1116 ср, 1073 о. сл, 1046 о. с, 991 сл, 958 о. с, 919 о. сл, 890 о. сл, 863 о. сл, 816 о. сл, 792 сл, 763 о. с, 737 сл, 710 с, 670 о. сл, 651 ср, 613 ср, 578 о. сл, 441 о. сл, 414 о. сл. ЯМР 1H (CDCl3; δ, м.д.): 2.10 (с., 9H), 4.16 (ш. м., 1H), 5.95 (д. д., 3JH–H = 2.3 Гц, 1H), 6.45 (д. д., 3JH–H = 2.3 Гц, 1H), 6.51 (д. д., 3JH–H = 2.35 Гц, 1H), 7.31 (д. д., 3JH–H = = 2.35 Гц, 1 H), 7.58 (д. д., 3JH–H = 2.22 Гц, 1 H), 7.79 (д. д., 4JH–H = 0.60 Гц, δ3JH–H = 2.35 Гц, 1H), 7.87 (д. д., 4JH–H = 0.6 Гц, 3JH–H = 2.35 Гц, 1H), 8.17 (д. д., 3JH–H = 2.2 Гц, 1H), 8.43 (д. д., 3JH–H = = 2.35 Гц, 1H). ЯМР 13С{1H} (CDCl3; δ, м.д.): 63.47; 34.50; 105.77; 107.53; 108.46; 132.68; 163.70; 139.16; 144.03; 145.93; 145.81.
II: ТСХ (CH2Cl2): желто-коричневое пятно, Rf 0.67. ИК-спектр (ν, см–1): 3108 о. сл. ш, 2954 сл, 2915 сл. ш, 2890 сл, 2854 сл, 2488 о. сл. ш, 1501 сл, 1473 о. сл, 1453 сл, 1406 ср, 1390 ср, 1358 сл, 1309 ср, 1217 cр. ш, 1185 сл, 1158 ср, 1118 ср, 1071 сл, 1047 о. с, 983 ср, 936 о. с, 890 cл, 819 сл, 794 сл, 765 с, 756 с. ш, 713 с, 660 сл, 617 ср, 572 сл, 439 сл, 417 сл. ЯМР 1H (CDCl3; δ, м.д.): 1.67 (ш. с., 18H), 4.47 ш. м.; 5.99 (д. д., 3JH–H = 2.2 Гц, 1H), 6.40 (д. д., 3JH–H = = 2.3 Гц, 2H), 7.41 (д. д., 3JH–H = 2.2 Гц, 4JH–H = = 0.7 Гц), 7.70 (д. д., 3JH–H = 2.4 Гц, 4JH–H < 0.4 Гц, 2H) 7.96 (д. д., 3JH–H = 2.2 Гц, 1H), 8.38 (ш. с., 2 H). ЯМР 13С{1H} (CDCl3; δ, м.д.): 105.86; 107.10; 133.89; 136.78; 149.69; 145.67; 60.07; 34.34. ЯМР 1H (d8-толуол; 233 K; δ, м.д.) 1.33 (с., 9H, StBu), 2.41 (с., 9H, StBu), 4.23 (ш. м., 1BH); 5.40 (м., 1H), 5.66 (м., 1H), 5.71 (м., 1H), 7.01 (м., 1H), 8.10 (м., 1H), 8.15 (м., 1H), 8.64 (м., 1H).
IV: ТСХ (CH2Cl2): фиолетовое пятно Rf 0.38. ИК-спектр (ν, см–1): 3144 ср. ш, 3101 ср, 2952 ср, 2910 ср, 2884 ср, 2851 ср. ш, 2482 ср.ш, 1499 ср, 1469 сл, 1451 сл, 1403 с. ш, 1357 ср, 1307 с, 1269 ср, 1207 с. ш, 1153 с, 1117 с, 1098 ср, 1069 ср, 1043 о. с, 983 с, 919 ср, 887 ср, 859 ср, 838 ср, 814 ср, 788 ср, 761 о. с. ш, 733 с, 709 о. с, 666 с, 648 с, 611 с, 574 ср, 485 cр, 436 ср. ш.
Синтез III. К суспензии 0.4 г (17 ммоль) натрия в 20 мл ТГФ добавляли 1.5 мл дипропилдисульфида (9.6 ммоль). Реакционную смесь перемешивали в течение суток. К полученной белой суспензии добавляли 2.1 г (4.3 ммоль) TpReOCl2. Реакционную смесь перемешивали в течение сутoк, растворитель удаляли в вакууме, полученное темно-коричневое масло экстрагировали 60 мл бензола. Бензольный экстракт выливали на 10 см колонку с силикагелем и элюировали 150 мл бензола. Коричневый раствор упаривали до 30 мл, добавляли 30 мл гептана. Полученный раствор выдерживали при 5°С, выпавшие коричневые кристаллы отделяли декантацией, промывали гексаном (3 раза по 5 мл) и высушивали в вакууме. Выход III 1.3 г (53%).
Маточный раствор упаривали досуха, остаток экстрагировали кипящим гептаном (4 раза по 10 мл). Выпадавшие при выдерживании гептанового экстракта при –25°С кристаллы III отделяли декантацией, промывали гексаном (3 раза по 5 мл) и высушивали в вакууме. Выход 0.17 г (7%).
| Найдено, %: | C 32.58; | H 4.56; | N 14.52; | S 11.18. |
| Для C15H24BN6OS2Re (M = 566) | ||||
| вычислено, %: | C 31.86; | H 4.28; | N 14.86; | S 11.34. |
ИК-спектр (ν, см–1): 3146 о. сл, 3129 о. сл, 3109 о. сл, 2959 с, 2927 ср, 2866 ср, 2479 с, 1498 ср, 1460 сл, 1404 с, 1388 ср, 1307 о. с, 1222 ср, 1205 с. ш, 1182 сл, 1114 ср, 1069 сл, 1044 о. с, 986 ср, 938 с, 889 сл, 854 о. сл, 814 о. сл, 789 сл, 766 с, 711 ср, 669 о. сл, 651 сл, 614 сл, 419 о. сл. ЯМР 1H (CDCl3; 298 K; δ, м.д.): 1.06 (т., 3JH–H = 7.26 Гц, 6H, SCH2CH2CH3), 1.91 (м., 4H, SCH2CH2CH3), 2.97–5.04 (ш. м., 5H, SCH2CH2CH3 + BH), 5.94 (т., 3JH–H = 2.2 Гц, 1H), 6.43 (т., 3JH–H = 2.2 Гц, 2H), 7.37 (д. д., 3JH–H = 2.2 Гц, 4JH–H = 0.8 Гц, 1H), 7.67 д., 3JH–H = 1.9 Гц, 2H), 7.79 (д., 3JH–H = 2.0 Гц, 1H), 8.32 (ш.с., 2H). ЯМР 13С{1H} (CDCl3; 298 K; δ, м.д.): 14.1, 26.2, 45.5 о. ш., 106.0, 107.4, 133.9, 136.8, 145.5, 147.9. ЯМР 1H (d8-толуол, 233 K; δ, м.д.): 0.96 (т., 3JH–H = 7.10 Гц 3H, SCH2CH2CH3), 1.27 (т., 3JH–H = = 7.0 Гц 3H, SCH2CH2CH3), 1.79 (м., 1H, SCH2CH2CH3), 1.92 (м., 1H, SCH2CH2CH3), 2.31 (м., 3H, SCH2CH2CH3+ SCH2CH2CH3), 3.45 (м., 1H, SCH2CH2CH3), 4.25 (ш. м., 1H, BH), 4.61 (м., 1H, SCH2CH2CH3), 4.76 (м., 1H, SCH2CH2CH3), 5.38 (т., 3JH-H = 2.1 Гц, 1H), 5.69 (ш.с., 1H), 5.72 (ш. с., 1H), 6.7 (д., 3JH–H = 1.7 Гц 1H), 7.07 (ш. м., 2H), 7.80 (м., 1H), 7.82 (ш. с., 1H), 8.58 (ш. с., 1H). ЯМР 1H (d8-толуол; 353 K; δ, м.д.): 1.06 (т., 3JH–H = = 7.20 Гц 6H, SCH2CH2CH3), 1.96 (м., 4H, SCH2CH2CH3), 3.43 (ш. с., 2H, SCH2CH2CH3), 3.92 (ш. м., 2H, SCH2CH2CH3), 4.3 (ш. м., 1H, BH), 5.51 (т., 3JH–H = 2.2 Гц, 1H), 5.88 (т., 3JH–H = 2.3 Гц, 2H), 6.89 (м., 1H), 7.27 (м., 2H), 7.77 (м., 1H), 8.20 (ш.с., 2H). ЯМР 13С (d8-толуол; 353 K; δ, м.д.): 14.2, 26.9, 45.9 ш., 105.9, 107.2, 133.7, 136.6, 146.1, 148.5.
РСА выполнен на дифрактометре Bruker APEX II CCD. Поглощение учтено методом множественного измерения эквивалентных отражений по программе SADABS [7]. Структуры I–IV определены прямым методом и уточнены методом наименьших квадратов относительно F 2 в анизотропном приближении неводородных атомов с использованием пакетов программ SHELX-2014 [8] и OLEX2 [9]. Положения атомов Н рассчитаны геометрически. Кристаллографические данные и параметры уточнения структуры I–IV приведены в табл. 1. Основные длины связей и валентные углы в I–IV приведены в подписях к рис. 1–4. Координаты атомов и другие параметры структуры I–IV депонированы в Кембриджском банке структурных данных (№ 1892096–1892099; http://www.ccdc.cam.ac.uk/data_request/cif).
Таблица 1.
Кристаллографические данные и параметры уточнения структуры комплексов I–IV
| Параметр | I | II | III | IV |
|---|---|---|---|---|
| Брутто-формула | C20H27BN6OSClRe | C17H28BN6OS2Re | C15H24BN6OS2Re | C31H47B2N12OS3Cl3Re2 |
| M | 631.99 | 593.58 | 565.53 | 1200.35 |
| Излучение (λ , Å) | MoKα (λ = 0.71073) | |||
| Температура съемки, K | 150(2) | 150(2) | 120(2) | 150(2) |
| Сингония | Триклинная | Моноклинная | Триклинная | Тригональная |
| Пр. гр. | P1̅ | Cc | P1̅ | R3̅ |
| а, Å | 9.3684(8) | 15.5182(4) | 7.668(1) | 27.782(1) |
| b, Å | 11.177(1) | 15.3550(3) | 7.882(1) | 27.782(1) |
| c, Å | 12.715(1) | 29.1704(6) | 18.783(2) | 29.663(2) |
| α, град | 111.161(1) | 90 | 82.805(2) | 90 |
| β, град | 93.553(1) | 92.9860(1) | 82.007(2) | 90 |
| γ, град | 93.888(1) | 90 | 65.269(2) | 120 |
| V, Å3 | 1233.4(2) | 6941.3(3) | 1018.3(2) | 19828(2) |
| Z | 2 | 12 | 2 | 18 |
| ρ(выч.), г/cм–3 | 1.702 | 1.704 | 1.844 | 1.809 |
| μ, мм–1 | 5.143 | 5.451 | 6.188 | 5.855 |
| F(000) | 620.0 | 3504.0 | 552.0 | 10512.0 |
| Интервал сканирования, град | 4.17–53.056 | 4.036–55.78 | 4.392–63.996 | 4.36–61.026 |
| Тип сканирования | ω | |||
| Независимых отражений (N1) | 5087 (Rint = 0.0230) | 16 516 (Rint = 0.0419) | 6683 (Rint = 0.0185) | 13 022 (Rint = 0.0458) |
| Отражений с I > 2σ(I) (N2) | 4620 | 15 726 | 6446 | 10 254 |
| Число уточняемых параметров | 284 | 776 | 237 | 496 |
| GOОF (F 2) | 1.064 | 1.037 | 1.057 | 0.933 |
| R1 для N2 | 0.0296 | 0.268 | 0.0171 | 0.0248 |
| wR2 для N1 | 0.0740 | 0.423 | 0.0391 | 0.0543 |
| Δρmax/Δρmin, e Å–3 | 5.21/–0.98 | 0.91/–0.85 | 1.06/–1.12 | 2.14/–1.46 |
Рис. 1.
Молекулярная структура I (сольватная молекула толуола не показана). Некоторые длины связей и валентные углы: Re(1)–Cl(1) 2.355(1), Re(1)–S(1) 2.300(1), Re(1)–O(1) 1.686(4), Re(1)–N(1) 2.101(4), Re(1)–N(3) 2.165(4), Re(1)–N(5) 2.250(4), S(1)–C(10) 1.861(5) Å и S(1)Re(1)Cl(1) 96.89(4)°, O(1)Re(1)Cl(1) 101.5(1)°, O(1)Re(1)S(1) 104.7(1)°, O(1)Re(1)N(1) 93.6(2)°, O(1)Re(1)N(3) 89.6(2)°, O(1)Re(1)N(5) 164.4(2)°, C(10)S(1)Re(1) 119.9(2)°.
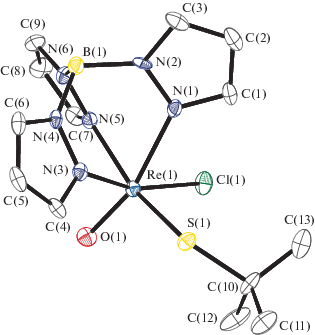
Рис. 2.
Молекулярная структура II (показана одна из трех независимых молекул). Длины связей и валентные углы: Re(1)–S(2) 2.310(2), Re(1)–S(1) 2.311(2), Re(1)–O(1) 1.682(4), Re(1)–N(3) 2.143(5), Re(1)–N(1) 2.261(5), Re(1)–N(5) 2.192(5), S(2)–C(14) 1.857(6), S(3)–C(27) 1.859(6) Å и C(14)S(2)Re(1) 122.8(2)°, C(10)S(1)Re(1) 120.1(2)°, O(1)Re(1)S(1)C(10) ‒95.9(3)°, O(1)Re(1)S(2)C(14) –83.3(3)°.
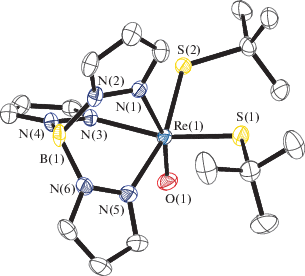
Рис. 3.
Молекулярная структура III. Длины связей и валентные углы: Re(1)–S(1) 2.3045(5), Re(1)–S(2) 2.3018(5), Re(1)–O(1) 1.697(1), Re(1)–N(1) 2.143(2), Re(1)–N(3) 2.268(2), Re(1)–N(5) 2.175(2), S(1)–C(10) 1.832(2) Å и C(10)S(1)Re(1) 115.63(7)°, C(13)S(2)Re(1) 112.36(6)°, O(1)Re(1)S(1)C(10) 87.30(9)°, O(1)Re(1)-S(2)C(13) 98.92(9)°.
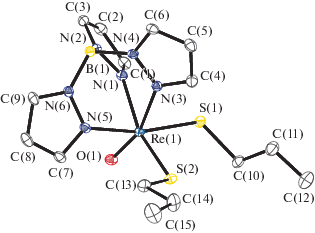
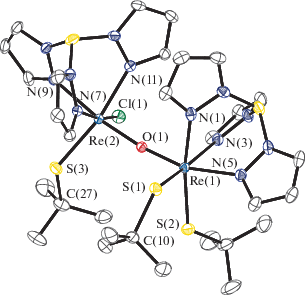
Рис. 4.
Молекулярная структура IV (сольватированная молекула хлористого метилена не показана). Длины связей и валентные углы: Re(1)–S(1) 2.3554(8), Re(1)–S(2) 2.2814(8), Re(1)–O(1) 1.8281(19), Re(1)–N(1) 2.160(3), Re(1)–N(3) 2.154(3), Re(1)–N(5) 2.171(2), Re(2)–Cl(1) 2.4048(8), Re(2)–S(3) 2.2742(8), Re(2)–O(1) 1.8842(19), Re(2)–N(7) 2.110(3), Re(2)–N(9) 2.139(2), Re(2)–N(11) 2.145(2); Å и S(2)Re(1)S(1) 91.39(3)°, O(1)Re(1)S(1) 98.46(7)°, O(1)Re(1)S(2) 104.60(7)°, O(1)Re(1)N(1) 88.09(9)°, O(1)Re(1)N(3) 86.98(9)°, O(1)Re(1)N(5) 163.97(9)°, O(1)Re(2)Cl(1) 96.22(6)°, O(1)Re(2)S(3) 98.90(6)°, O(1)Re(2)N(7) 92.54(9)°, O(1)Re(2)N(9) 170.60(9)°, O(1)Re(2)N(11) 90.07(9)°, C(10)S(1)Re(1) 122.8(1)°, C(14)S(2)Re(1) 122.2(1)°, C(27)S(3)Re(2) 120.3(1)°, Re(1)O(1)Re(2) 170.2(1)°, O(1)Re(1)S(1)C(10) 87.02°, O(1)Re(1)S(2)-C(14) 108.25°, O(1)Re(2)S(3)C(27) 71.33°, N(7)Re(2)-Re(1)N(1) 56.95°.
Квантово-химические расчеты проводили в рамках теории функционала плотности с помощью программного комплекса ORCA 4.01 [10]. Оптимизацию геометрии комплексов и переходных состояний проводили с использованием функционала PBE [11, 12] c дважды расщепленным полноэлектронным базисом def2-SVP [13] и использованием эмпирической поправки к дисперсионным взаимодействиям D3BJ [14, 15]. Энергию активации рассчитывали с использованием гибридного функционала PBE0 [16] с трижды расщепленным полноэлектронным базисом def2-TZVP [13].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Комплексы I и II получали по двум различным методикам. Первый синтез проводили аналогично для TpReOCl(SPh) и TpReO(SPh)2 [5] реакцией TpReOCl2 с трет-бутилмеркаптаном в присутствии DBU в качестве основания в толуоле при 110°C. Ход реакции контролировали при помощи ТСХ на силуфоле в хлористом метилене по исчезновению голубого пятна исходного TpReOCl2 и появлению зеленого (Rf 0.75) и желто-коричневого (Rf 0.67) пятен, наблюдавшихся для вышеуказанных фенилтиолатных комплексов в [5]. Продукты разделяли хроматографически и кристаллизовали из смеси CH2Cl2–гексан (1 : 1) для I и из гексана для II. Пригодные для РСА кристаллы I получали выдерживанием его раствора в толуоле при –70°С.
По методике 2 проводили реакцию TpReOCl2 с трет-бутилтиолатом натрия, полученным в ТГФ из натрия и бис-трет-бутилдисульфида и использованного без выделения.
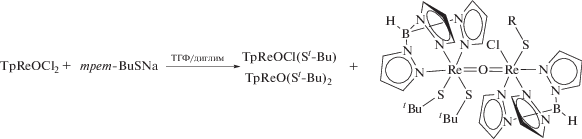
Эта реакция проходит быстрее, чем с меркаптаном, однако, несмотря на использование 20%-ного избытка трет-BuSNa, вместе с дизамещенным также происходит образование монозамещенного продукта. Продукты реакции разделяли хроматографически, при этом кроме моно- и дитиолатного комплексов выделили небольшое количество нового фиолетового биядерного комплекса IV, охарактеризованного РСА, ИК-спектроскопией и ТСХ (фиолетовое пятно, Rf 0.38).
Аналогичный комплексу II пропилтиолатный комплекс III получали вторым способом с использованием двухкратного избытка NaSC3H7, что позволило избежать образования монозамещенного комплекса.
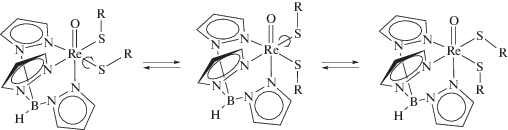
Последовательное замещение хлоридных заместителей на более донорные S-трет-Bu приводит к смещению полосы колебаний Re=O (972, 958, 936 см–1 соответственно), свидетельствующему об ослаблении ее в ряду V, I, II. Cпектры ЯМР 1H комплексов II и III зависят от температуры. При низкой наблюдаются сигналы двух неэквивалентных тиолатных групп и трех неэквивалентных пиразольных колец, тогда как при высокой температуре спектр соответствует эквивалентным тиолатным группам и двум эквивалентным пиразольным группам из трех. По-видимому, это связано с затрудненным вращением вокруг связей Re–S. Наблюдаемые точки коалесценции сигналов пиразольных и тиолатных групп (например, на рис. 5) позволяют оценить барьер активации ($\Delta G_{{259}}^{\ddag }$ = 13.03, $\Delta G_{{281}}^{\ddag }$ = = 12.97, $\Delta G_{{291}}^{\ddag }$ = 13.06 ккал/моль) для II и ($\Delta G_{{278}}^{\ddag }$ = = 12.6 ккал/моль) для III. Для трет-бутильногокомплекса II энергия активации практически не зависит от температуры, что предполагает небольшую энтропию активации.
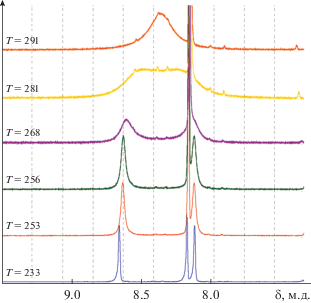
Это свидетельствует о том, что наблюдаемый динамический процесс внутримолекулярный и позволяет сравнить найденные свободные энергии активации с энтальпиями активации, полученными из квантово-химического расчета.
Расчет барьера для данного процесса методом функционала плотности дает энтальпию активации 12.3 и 14.1 ккал/моль для III и II соответственно. При этом расчитанное расстояние Re–S в переходном состоянии для вращающейся тиолатной группы увеличивается до 2.47 Å по сравнению с 2.31 Å для комплекса III, а двугранный угол SReSC 104.5° в переходном состоянии не соответствует максимуму стерического отталкивания каких-либо группировок. Это позволяет предположить, что барьер вращения в данных комплексах имеет в основном электронную природу и связан с существенной кратностью связи Re–S. Для комплекса II переходное состояние для вращения тиолатной группы соответствует конформации, в которой отталкивание трет-бутильной группы и пиразольного фрагмента максимально (SReSC 128.6°) и удлинение связи Re–S менее выраженно.
Структура комплексов I–III определена методом РСА. Расстояния Re–N в I заметно неэквивалентны (2.25, 2.16 и 2.10 Å для связей в транс-положении к O, S-трет-Bu и Cl соответственно), по-видимому, в силу различия транс-влияния трех разных заместителей. Короткое расстояние Re–O 1.686(4) Å, типичное для подобных соединений (табл. 2), свидетельствует о троесвязанности, как и сильно отклоненные от октаэдрических углы OReS 104.7(1)° и OReCl 101.5(1)°. Длина связи Re–S 2.300(1) Å заметно укорочена по сравнению с суммой ковалентных радиусов (СКР) Re и S (2.56 Å) [17]. Это, наряду с углом CSRe 119.9(2)°, значительно большим идеального тетраэдрического, позволяет предположить кратный характер связи Re–S в соответствии с подачей неподеленной пары атома серы на разрыхляющую орбиталь тройной связи Re–O. Близкая к заслоненной конформация трет-бутилтиолатного лиганда по отношению к хлоридному (торсионный угол OReSC 83.56°), по-видимому, также связана с этим взаимодействием.
Таблица 2.
Сравнение структурных и спектроскопических параметров TpReOCl(S-трет-Bu) с известными аналогами* и с TpReOCl2
| Соединение | Торсионный угол OReSC, град | ν(Re–O), см–1 | Re–O, Å | Re–S, Å | Литература |
|---|---|---|---|---|---|
| TpReO(SC2H4S) | 96.79; 81.00 | 951 | 1.694(9) | 2.280(4) | [4, 5] |
| 2.299(4) | |||||
| TpReO(SC2H5)Cl | 961 | [4] | |||
| TpReO(SC2H5)2 | 932 | [4] | |||
| TpReO(SPh)Cl | 78.46 | 956 | 1.691(5) | 2.301(2) | [5] |
| TpReO(SPh)2 | 84.86; 95.12 | 945 | 1.668(5) | 2.312(2); 2.310(2) | [5] |
| TpReO(SC6H4S) | 952 | [5] | |||
| TpReO(SPy)2 | 93.23; 83.14 | 957 | 1.683(6) | 2.317(2); 2.333(2) | [18] |
| TpReOCl2 | 978 | [5] | |||
| TpReOS2Fc | 89.64; 78.93 | 953 | 1.688(3) | 2.316(1); 2.309(1) | [19] |
| TpReOCl(S-трет-Bu) | 83.56 | 958 | 1.686(4) | 2.300(1) | |
| TpReO(S-трет-Bu)2 | 95.9(3) | 936 | 1.682(4) | 2.310(2) | |
| 83.3(3) | 2.311(2) | ||||
| TpReO(S-n-C3H7)2 | 87.30(9); 98.92(9) | 938 | 1.697(1) | 2.3045(5); 2.3018(5) |
В бис-тиолатном комплексе II расстояние Re–N в транс-положении к кислороду (2.261(5) Å) также заметно больше, чем расстояния в транс-положении к тиолатным группам (2.143(2), 2.192(5) Å). Расстояния Re–S 2.310(2) и 2.311(2) Å в II практически не отличаются от расстояния в комплексе I, что, наряду с незначительным отклонением трет-бутильных групп от плоскости ReS2 и увеличенными по сравнению с тетраэдрическими углами ReSC 120.1(2)°, 122.8(2)°, свидетельствует о заметно кратной связи Re–S, такой же как в монотиолатном комплексе.
Замена трет-бутильных заместителей на н-пропильные в III практически не меняет геометрию комплекса. Уменьшение стерических затруднений приводит только к незначительному дополнительному укорочению связей Re–S (до 2.3045(5) и 2.3018(5) Å) и уменьшению углов ReSC (115.63(7)°, 112.36(6)°). При этом конформация тиолатных лигандов практически не изменяется.
Комплекс IV содержит дирениевый фрагмент Re=O=Re, расстояния Re–O в котором (1.828(2) и 1.884(2) Å) хотя и удлинены по сравнению с концевым Re=O в моноядерном комплексе (1.686(4) Å), заметно короче СКР Re и O (2.17 Å) [17]. Некоторое отклонение угла Re(1)O(1)Re(2) 170.2(1)° от строго линейного, по-видимому, связано со стерическими и упаковочными эффектами трет-бутилтиолатных и трис-пиразолилборатных групп, находящихся в син-положении. Связи Re–N с атомами азота в транс-положении к мостиковому кислороду (2.171(2) и 2.139(2) Å) укорочены по сравнению с таковыми в мономерном комплексе (Re–N 2.250(4) Å), по-видимому, из-за уменьшения транс-влияния мостикового кислорода по сравнению с концевым. Об уменьшении порядка связи Re–O в димере также свидетельствует меньшее отклонение углов O(1)Re(2)Cl(1), O(1)Re(2)S(3) (96.22(6)° и 98.90(6)° соответственно) в сравнении с аналогичными углами в TpReOCl(S-трет-Bu), (101.5(1)° и 104.7(1)°). Связи Re–S (2.3554(8), 2.2814(8), 2.2742(8) Å) в среднем практически не меняются по сравнению с мономерным комплексом, оставаясь заметно укороченными. Углы ReSC, близкие к 120°, и торсионные углы OReSC (табл. 3) также свидетельствуют о существенном вкладе дополнительного π-связывания тиолатных лигандов с атомом Re.
Таблица 3.
Сравнительная таблица некоторых параметров структуры Tp2Re2OCl(S-трет-Bu)3 с известными аналогами*
| Соединение | Угол ReORe, град |
Re(1)–O, Å | Re(2)–O, Å | Конфор-мация | Литература |
|---|---|---|---|---|---|
| [${\text{L}}_{2}^{{\text{'}}}\operatorname{Re} _{2}^{{{\text{IV}}}}$Cl2(μ-O)][ZnCl4] L' = 1,4,7-триметил-(1,4,7-триазо)циклонан |
180.0(1) | 1.878(1) | 1.878(1) | Анти- | [21] |
| [$\operatorname{Re} _{2}^{{{\text{III}}}}$O(CH3CN)10]4+ | 180.000 | 1.7993(3) | 1.7993(3) | [22] | |
| [${\text{Re}}_{{\text{2}}}^{{{\text{III,IV}}}}$Cl2O(Pa)2]3+ Тpa = = трис-(2-пиридилметил)амин | 178.0(1) | 1.85(2) | 1.84(2) | Анти- | [20] |
| ${\text{Re}}_{{\text{2}}}^{{\text{V}}}$(μ-O)O2Cl2(BpaO2)2 BpaO2 = дипиколиноиламид |
180 | 1.9122(1) | 1.9122(1) | Анти- | [23] |
| Tp2Re2OCl(S-трет-Bu)3 | 170.17(12) | 1.828(2) | 1.884(2) | Син- |
Аналогичное образование биядерного оксомостикового комплекса рения(III) из моноядерных фрагментов, содержащих Re(IV), наблюдали в реакции ReOCl3(PPh3)2 с тетрадентантным лигандом Tpa (трис-(2-пиридилметил)амин); при этом восстановителем является отщепляющийся трифенилфосфин [20].
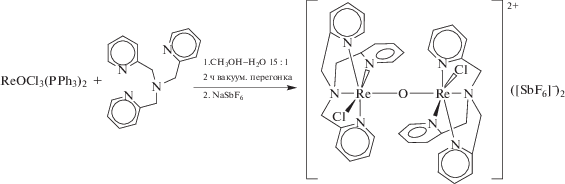
Основные структурные параметры других известных μ-оксо-комплексов рения приведены в табл. 3.
Таким образом, двумя методами, по аналогии с известными тиофенильными комплексами, получены комплексы рения TpReOCl(S-трет-Bu), TpReO(S-трет-Bu)2 и TpReO(SC3H7)2 с более электронодонорными алкилтиолатными лигандами, охарактеризованные ИК- и ЯМР-спектроскопией, а также методом РСА. Исследование температурной зависимости спектров ЯМР в сочетании с квантово-химическими расчетами позволяет предположить, что в дитиолатных комплексах вращение вокруг коротких (~2.3 Å) связей Re–S затруднено с барьером порядка 12.7 и 13.0 ккал/моль для пропилтиолатного и трет-бутилтиолатного комплексов соответственно. Слабая зависимость барьера от размера алкильного заместителя и удлинение расчетного расстояния Re–S в переходном состоянии позволяют предположить, что барьер имеет в основном электронную природу и связан с заметно кратным характером связи Re–S. Также выделен и структурно охарактеризован побочный продукт реакции TpReOCl2 с NaS-трет-Bu – биядерный кислородмостиковый комплекс TpReIVCl(S-трет-Bu)O(S-трет-Bu)2ReIVTp.
Конфликт интересов между авторами статьи отсутствует.
Список литературы
Abrams M.J., Davison A., Jones A.G. // Inorg. Chim. Acta. 1984. V. 82. P. 125.
Cohen E.A., McRae G.A., Goldwhite H. et al. // Inorg. Chem. 1987. V. 26. P. 4000.
Brown S.N., Mayer J.M. // Inorg. Chem. 1992. V. 31. P. 4091.
Tisato F., Bolzati C., Duatti A. et al. // Inorg. Chem. 1993. V. 32. P. 2042.
Degnan I.A., Behm J., Cook M.R., Herrmann W.A. // Inorg. Chem. 1991. V. 30. P. 2165.
Trofimenko S., Long J.R., Nappier T., Shore S.G. // Inorg. Synth. 1970. V. 12. P. 99.
SADABS (version 2008/1). Madison (WI, USA): Bruker AXS Inc., 2008.
Sheldrick G.M. // Acta Crystallogr. A. 2008. V. 64. P. 112.
Dolomanov O.V., Bourhis L.J., Gildea R.J. et al. // J. Ap-pl. Cryst. 2009. V. 42. P. 339.
Neese F. // WIREs Comput. Mol. Sci. 2018. V. 8. P. e1327. https://doi.org/10.1002/wcms.1327
Perdew J. P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1996. V. 77. P. 3865.
Perdew J. P., Burke K., Ernzerhof, M. // Phys. Rev. Lett. 1997. V. 78. P. 1396. Erratum.
Weigend F., Ahlrichs R. // Phys. Chem. Chem. Phys. 2005. V. 7. P. 3297.
Grimme S., Ehrlich S., Goerigk L. // J. Comput. Chem. 2011. V. 32. P. 1456.
Grimme S., Antony J., Ehrlich S., Krieg H. // J. Chem. Phys. 2010. V. 132. P. 154104.
Adamo C., Barone V. // J. Chem. Phys. 1999. V. 110. P. 6158.
Cordero B., Gómez V., Platero-Prats A.E. et al. // Dalton Trans. 2008. V. 21. P. 2832.
Kettler P.B., Chang Y.-D., Chen Q. et al. // Inorg. Chim. Acta. 1995. V. 231. P. 13.
Herberhold M., Jin G.-X., Milius W. // J. Organomet. Chem. 1996. V. 512. P. 111.
Sugimoto H., Takahira T., Yoshimura T. et al. // Inorg. Chim. Acta. 2002. V. 337. P. 203.
Boehm G., Wieghardt K., Nuber B., Weiss J. // Inorg. Chem. 1991. V. 30. P. 3464.
Bera J.K., Schelter E.J., Patra S.K. et al. // Dalton Trans. 2006. V. 33. P. 4011.
Kochel A., Hołyńska M. // Inorg. Chem. Commun. 2010. V. 13. P. 782.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия