

Координационная химия, 2020, T. 46, № 11, стр. 672-681
Особенности спектроскопии ЭПР оловоорганических соединений с парамагнитными лигандами о-иминобензосемихинонового типа
М. Г. Чегерев 1, К. В. Арсеньева 2, А. В. Черкасов 2, А. В. Пискунов 2, *
1 Институт физической и органической химии Южного федерального университета
Ростов-на-Дону, Россия
2 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
* E-mail: pial@iomc.ras.ru
Поступила в редакцию 05.05.2020
После доработки 05.06.2020
Принята к публикации 10.06.2020
Аннотация
Синтезированы и исследованы методом спектроскопии ЭПР новые парамагнитные производные олова(IV), содержащие анион-радикал редокс-активного лиганда 2,4,6,8-тетра-трет-бутил-феноксазин-1-она (phenox-imQ). Комплексы I–V парамагнитны в кристаллическом состоянии и в растворе. Геометрия полученных парамагнитных частиц и распределение спиновой плотности в них исследованы методом теории функционала плотности. Синтезирован диамагнитный комплекс VI, на основе дианиона phenox-imQ и исследовано его одноэлектронное окисление. Молекулярное строение комплекса VI · ТHF установлено методом РСА (CIF file CCDC № 2000962 (VI · ТHF).
Химия редокс-активных лигандов развивается на протяжении последних 50 лет. На заре своего появления подобные лиганды и металлокомплексы с ними являлись исключительно объектами ЭПР-спектроскопических исследований [1]. Однако истинный потенциал данных лигандов оказался значительно шире, и в настоящее время они представляют собой универсальные инструменты для расширения реакционной способности как комплексов переходных, так и непереходных металлов. Использование производных редокс-активных лигандов в координационной и элементорганической химии металлов позволило реализовать превращения, ранее считавшиеся неосуществимыми: облегчение окислительно-восстановительных реакций производных s-, p- и d-металлов, реакции кросс-сочетания без изменения степени окисления металлоцентра, феномен редокс-изомерии и его практическое применение [2–8].
На данный момент опубликовано большое количество работ, посвященных синтезу, исследованию строения, каталитических и магнитных свойств комплексов переходных металлов на основе редокс-активных лигандов разной природы – о-хинонов (Q), о-иминохинонов (imQ), α-ди-иминов (DAD) и их замещенных аналогов [9–11]. Введение анион-радикального (о-семихинонового, SQ) лиганда в комплексы металлов открывает возможности изучения динамики координационной сферы комплексообразователя in situ посредством спектроскопии ЭПР. Неспаренный электрон, локализованный на парамагнитном лиганде, не оказывает заметного влияния на металл и его окружение. Эта особенность означает, что о-семихиноны могут быть использованы в качестве спиновых меток. Параметры спектров ЭПР семихиноновых комплексных соединений (g-фактор, сверхтонкая структура (СТС), константы сверхтонкого взаимодействия (СТВ)) весьма чувствительны к природе атома металла и лигандного окружения, геометрической конфигурации комплекса [12–17].
Несмотря на то что о-иминохиноны [18, 19] являются ближайшими структурными аналогами о‑хинонов, их химия заметно отличается. В частности, данный факт отчетливо прослеживается на примере химии элементов 14 группы (Si, Ge, Sn, Pb) с перечисленными выше редокс-активными лигандами. о-Семихиноновые соединения элементов подгруппы кремния малоустойчивы, и в растворе быстро происходит распад парамагнитных частиц по двум наиболее вероятным механизмам: диспропорционирование о-семихинонового комплекса или элиминирование углеводородного радикала (R) при атоме комплексообразователя. При этом соотношение этих двух процессов зависит от природы металла, хинонового лиганда и углеводородного заместителя [20–22]. В то же время использование о-иминохинонов позволило выделить и структурно охарактеризовать большое количество устойчивых парамагнитных производных общего вида imSQMX3, где imSQ – анион-радикал о-иминобензохинона; X – алкил, арил, галоген [23, 24].
Настоящая работа посвящена синтезу новых соединений олова(IV) на основе редокс-активного 2,4,6,8-тетра-трет-бутил-феноксазин-1-она (phenox-imQ) и исследованию физико-химических свойств полученных соединений. Поскольку рассматриваемый замещенный феноксазинон является бидентатным о-иминохиноном, был выполнен сравнительный анализ синтезированных нами комплексов олова(IV) с известными ранее соединениями олова на основе N-арилзамещенных о-иминохинонов. Изучение подобного класса соединений позволит расширить представления о строении, характере связывания и распределении спиновой плотности в о-иминохиноновых производных олова в зависимости от строения редокс-активного лиганда.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Все операции по синтезу феноксазиноновых комплексов олова, а также их химические превращения проводили в условиях отсутствия кислорода и влаги воздуха. Использованные в работе растворители очищали и обезвоживали по стандартным методикам [25].
ИК-спектры регистрировали на ИК-Фурье-спектрометре ФСМ-1201 (диапазон 4000–450 см−1) в вазелиновом масле в кюветах из KBr. Элементный анализ выполняли на анализаторе Elementar Vario El cube. Спектры ЭПР регистрировали на спектрометре Bruker-EMX (рабочая частота ~9.7 ГГц). В качестве стандарта при определении g-фактора использовали дифенилпикрилгидразил (gi = 2.0037). Симуляцию спектров ЭПР проводили при помощи программного пакета Ea-syspin для Matlab [26].
Синтез редокс-активного phenox-imQ выполняли по оригинальной методике, являющейся модификацией, представленной в [27]. В колбу с мешалкой помещали 3.50 г (15.8 ммоль) 4,6-ди-трет-бутил-о-аминофенола [28] и 3.47 г (15.8 ммоль) 3,5-ди-трет-бутил-о-бензохинона и растворяли в метаноле (50 мл), при этом реакционная смесь приобретала интенсивно фиолетовую окраску. К полученному раствору добавляли 5 мл пиридина и кипятили реакционную смесь 2 ч с воздушным холодильником. В результате реакции образовалась смесь окисленной и восстановленной форм phenox-imQ. Добавление в реакционную смесь воды по каплям приводила к выпадению темно-синего осадка. Сухой остаток растворяли в Et2O и переносили в делительную воронку. К полученной смеси добавляли насыщенный водный раствор 20 г K3Fe(CN)6 и 1 г КОН. Полученную реакционную смесь интенсивно встряхивали. Процесс окисления отслеживали методом тонкослойной хроматографии (ТСХ, элюент гексан–этилацетат = = 1 : 30). Окончанием реакции служило исчезновение одного из двух синих пятен на хроматограмме. Полученный продукт кристаллизовали из гексана при –18°С. Выделяющиеся кристаллы phenox-imQ темно-синего цвета отделяли от маточного раствора декантацией. Выход 5.26 г (78%). Спектроскопические данные синтезированного соединения соответствуют представленным в [27, 29].
Динатриевую соль феноксазин-1-она (phenox-APNa2) получали непосредственным восстановлением phenox-imQ избытком металлического натрия в среде тетрагидрофурана (THF). При этом происходит изменение цвета раствора с темно-синего на светло-желтый. Полученный раствор использовали in situ. Мононатриевую соль (phenox-imSQNa) получали взаимодействием phenox-APNa2 с феноксазин-1-оном в мольном соотношении 1 : 1 в среде ТHF. Реакция сопровождается изменением цвета раствора с желтого на коричневый.
Синтез комплексов олова(IV) (I–V) (общая методика). Раствор мононатриевой соли phenoxim-SQNa (1.0 ммоль) в THF (20 мл) медленно приливали к раствору оловоорганического галогенида (1.0 ммоль) в том же растворителе (10 мл), при этом окраска реакционной смеси менялась с коричневой на бордово-красную. Полученный раствор перемешивали при комнатной температуре в течение 30 мин, после чего растворитель удаляли при пониженном давлении. Остаток обрабатывали гексаном (20 мл) и фильтровали на стеклянном фильтре Шотта № 4. Гексановый раствор упаривали вдвое и медленно охлаждали до –18°С. Выделяющиеся мелкокристаллические осадки полученных комплексов олова отделяли от маточного раствора декантацией и сушили при пониженном давлении.
Комплекс phenox-imSQSnMe2Cl (I): выход 75%.
ИК-спектр (ν, см–1) 1597 ср, 1534 с, 1479 с, 1412 с, 1390 сл, 1360 с, 1344 сл, 1300 сл, 1285 сл, 1275 сл, 1255 с, 1246 с, 1206 ср, 1187 сл, 1124 сл, 1076 с, 1050 с, 1015 ср, 992 ср, 956 сл, 930 сл, 916 сл, 889 ср, 858 ср, 838 ср, 813 сл, 770 ср, 745 ср, 684 ср, 645 ср, 620 с, 587 сл, 547ср, 495сл.
Комплекс phenox-imSQSnEt2Cl (II): выход 81%.
ИК-спектр (ν, см–1): 1598 ср, 1533 с, 1478 с, 1412 с, 1390 сл, 1360 с, 1344 сл, 1299 сл, 1285 сл, 1274 сл, 1255 с, 1245 с, 1206 ср, 1186 сл, 1124 сл, 1076 с, 1050 с, 1015 ср, 992 ср, 956 сл, 930 сл, 916 сл, 889 ср, 858 ср, 838 ср, 813 сл, 767 ср, 742 ср, 684 ср, 645 ср, 620 с, 587 сл, 527 сл, 525 ср, 498 сл.
Комплекс phenox-imSQSn(t-Bu)2Cl (III): выход 76%.
ИК-спектр (ν, см–1): 1597 ср, 1534 с, 1479 с, 1412 с, 1390 сл, 1360 с, 1344 сл, 1318 сл, 1285 сл, 1275 сл, 1255 с, 1246 с, 1206 ср, 1187 сл, 1124 сл, 1076 с, 1050 с, 1015 ср, 992 ср, 956 сл, 930 сл, 916 сл, 889 ср, 858 ср, 838 ср, 813 сл, 770 с, 745 ср, 684 ср, 645 ср, 620 с, 587 сл, 547ср, 489 сл.
Комплекс phenox-imSQSnPh2Cl (IV): выход 80%.
ИК-спектр (ν, см–1): 1597 ср, 1534 с, 1479 с, 1412 с, 1390 сл, 1360 с, 1344 сл, 1285 сл, 1275 сл, 1246 с, 1206 ср, 1187 сл, 1124 сл, 1076 с, 1050 с, 1018 сл, 985 ср, 956 сл, 930 сл, 916 сл, 889 ср, 858 ср, 838 ср, 813 сл, 770 с, 733 с, 695 с, 645 ср, 650 сл, 640 ср, 587 сл, 547ср, 492 сл.
Комплекс phenox-imSQSnPh3 (V): выход 82%.
ИК-спектр (ν, см–1): 1597 ср, 1534 с, 1479 с, 1412 с, 1390 сл, 1360 с, 1344 сл, 1285 сл, 1275 сл, 1247 с, 1204 с, 1187 сл, 1124 сл, 1076 с, 1050 с, 1018 сл, 992 с, 956 сл, 931 сл, 916 сл, 889 ср, 858 ср, 838 ср, 813 сл, 770 с, 733 с, 695 с, 645 ср, 651 сл, 642 ср, 587 сл, 547 ср, 492 сл.
Синтез комплекса phenox-APSnEt2 · THF (VI). Раствор phenox-APNa2 (1.0 ммоль) в THF (20 мл) медленно приливали к раствору Et2SnCl2 (1.0 ммоль) в том же растворителе (10 мл), при этом окраска реакционной смеси менялась с желтой на интенсивно оранжевую. Полученный раствор перемешивали при комнатной температуре в течение 10 мин, после чего растворитель удаляли при пониженном давлении. Остаток обрабатывали гексаном (20 мл) и фильтровали на стеклянном фильтре Шотта № 4. Гексановый раствор упаривали вдвое, после чего из него выделили кристаллы комплекса VI. Выход 62%.
Спектр ЯМР 1Н (C6D6; 20°С; δ, м.д.; J, Гц): 7.24 (д., 1H, 2.2, HAr,); 6.89 (c., 1H, HAP); 6.69 (д., 1H, 2.2, HAr,); 3.43 (м., 4H, α-CH2(THF)); 1.67 (с., 9H, (t-Bu)); 1.64 (кв., 4H, 7.5, –CH2–); 1.58 (с., 9H, (t‑Bu)); 1.35 (м., 4H, β-CH2(THF)); 1.32, 1.30 (с., 9H, (t-Bu)); 1.24 (т., 6H, 7.5, –CH3).
ИК-спектр (ν, см–1): 1594 ср, 1541 ср, 1441 с, 1362 с, 1340 ср, 1316 ср, 1302 сл, 1273 сл, 1254 ср, 1236 ср, 1200 ср, 1169 ср, 1119 ср, 1049 с, 1006 ср, 986 сл, 957 сл, 914 сл, 883 ср, 866 сл, 852 ср, 837 ср, 808 сл, 766 сл, 756 сл, 740 ср, 673 ср, 652 ср, 600 сл, 540 сл, 524 сл.
Квантово-химические расчеты выполняли с использованием программного пакета Gaussian09 [30] методом теории функционала плотности (DFT) с применением функционала B3LYP [31] и стандартного базисного набора dgdzvp для всех атомов.
РСА монокристаллического образца VI · THF (0.40 × 0.40 × 0.10 мм) проведен на дифрактометре Rigaku OD Xcalibur (ω-сканирование, МоКα-излучение, λ = 0.71073 Å, T = 100(2) K, 2θ = 60.06°). Измерение и интегрирование экспериментальных наборов интенсивностей, учет поглощения и уточнение структур проведены с использованием программных пакетов CrysAlisPro [32] и SHELX [33]. Соединение VI · THF (C36H57NO3Sn · C4H8O) кристаллизуется в пространственной группе P21/n (a = 14.9485(3), b = 16.3242(2), с = 17.8566(9) Å, β = 112.754(2)°, V = 4018.29(13) Å3, Z = 4, ρрасч = = 1.108 г/см3, μ = 0.665 мм–1). Измерено 83 764 отражения, 11 720 независимых отражений (Rint = 0.0506) были использованы для решения структуры и последующего уточнения 383 параметров полноматричным МНК по $F_{{hkl}}^{2}$ в анизотропном приближении для неводородных атомов. Водородные атомы в VI · THF помещены в геометрически рассчитанные положения и уточнены изотропно с фиксированными тепловыми параметрами U(H)изо = 1.2U(C)экв (U(H)изо = 1.5U(C)экв для метильных групп). Вклад сольватной молекулы THF в структурную модель VI ⋅ THF учтен с помощью процедуры SQUEEZE (Platon) [34]. После финального уточнения wR2 = 0.0915 и S(F2) = 1.038 для всех отражений (R1 = 0.0359 для всех отражений, удовлетворяющих условию I > 2σ(I)). Остаточные максимум и минимум электронной плотности составили 2.59/–0.83 e/Å3.
Структура VI ⋅ THF зарегистрирована в Кембриджском банке структурных данных (CCDC № 2000962; ccdc.cam.ac.uk/structures).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В литературе описано несколько подходов к получению редокс-активного phenox-imQ, однако все они характеризуются достаточно низкими выходами конечного продукта [27, 29]. Мы провели модификацию методики с целью увеличения выхода исследуемого лиганда, добавив дополнительную стадию окисления реакционной смеси ферроцианидом калия. Кипячение эквимольных количеств 4,6-ди-трет-бутил-о-аминофенола и 3,5-ди-трет-бутил-о-бензохинона в среде метанола приводит к образованию смеси продуктов, состоящей из искомого phenox-imQ и его восстановленной формы – phenox-imSQH в соотношении ~1 : 1. Последующее окисление реакционной смеси без выделения гексацианоферратом(III) калия в щелочной среде позволило увеличить выход конечного продукта phenox-imQ вдвое (78%) (схема 1 ).
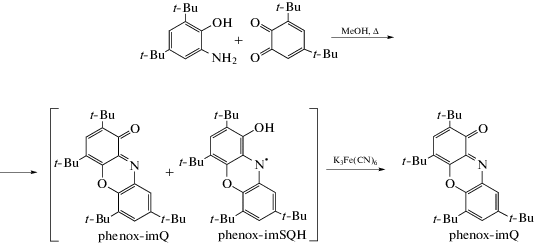
Схема 1 .
Полученный phenox-imQ лиганд постадийно восстанавливается избытком металлического натрия в среде ТHF с образованием моно- и динатриевой соли, при этом окраска реакционной смеси меняется с синей на коричневую (phenox-imSQNa) и далее на желтую (phenox-APNa2) соответственно. Для получения парамагнитных комплексов олова(IV) мы использовали реакцию обмена между мононатриевой солью лиганда и органогалогенидами олова. Взаимодействие phenox-imSQNa с R2SnCl2 (R = Me, Et, t-Bu, Ph) и Ph3SnCl в растворе THF заканчивается в течение нескольких минут при комнатной температуре. При этом реакционная смесь окрашивается в бордово-красный цвет, характерный для анион-радикальной формы феноксазинонового лиганда в комплексах олова(IV). После кристаллизации из реакционных смесей были выделены комплексы I–V с высокими выходами, превышающими 75%. Полученные соединения парамагнитны в растворе и в кристаллическом состоянии и устойчивы к действию кислорода и влаги воздуха (схема 2 ).
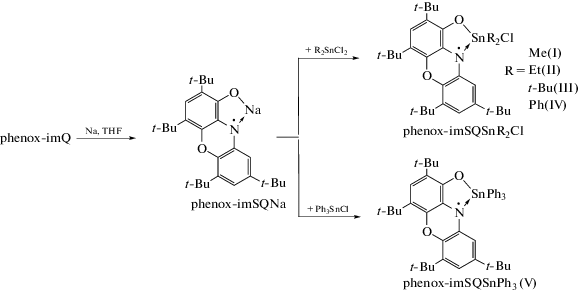
Схема 2 .
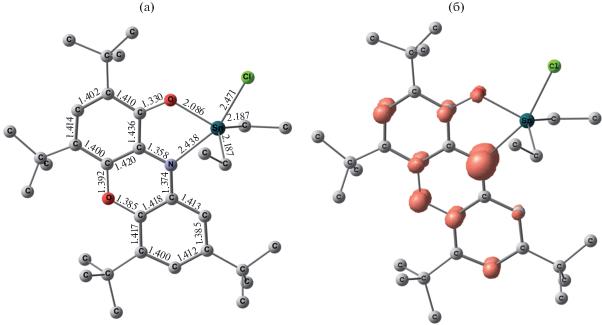
К сожалению, все попытки получить монокристаллы комплексов I–V, пригодных для РСА, не увенчались успехом. С целью установления молекулярного строения и распределения спиновой плотности в исследуемых соединениях мы выполнили квантово-химические расчеты методом DFT (B3LYP/dgdzvp). Согласно расчетам, координационное окружение иона олова во всех случаях I–V представляет собой искаженную тригональную бипирамиду. Рассмотрим подробнее строение серии комплексов на примере соединения II (рис. 1). Атомы азота N и галогена Cl находятся в вершинах бипирамиды, в то время как атом кислорода O и атомы углерода алкильных заместителей образуют ее основание. Атом хлора лежит в плоскости феноксазинонового лиганда. Длины связей в редокс-активном лиганде находятся в интервале, характерном для анион-радикального состояния phenox-imSQ [35, 36]. Необходимо отметить значительную разницу в длинах связей Sn–O (2.086 Å) и Sn–N (2.437 Å), что отличает данный случай от изученных ранее о-иминосемихинолятов олова(IV), в которых расстояния металл–азот и металл–кислород сопоставимы по своей величине [37, 38]. Сравнивая полученные данные, можно заключить, что характер связывания анион-радикала с металлоцентром можно описать, скорее, как “фенолят олова”, дополнительно координированный азот-центрированным радикалом, чем “о-иминосемихинолят” олова. Схожий тип координации демонстрируют о‑иминосемихиноновые производные ртути(II) [39]. Анализ распределения спиновой плотности показывает, что она преимущественно (38%) сосредоточена на атоме азота феноксазинонового лиганда, в то время как на атомах олова и хлора спиновая плотность составляет менее 1%.
Рис. 1.
Оптимизированная геометрия комплекса II (DFT B3LYP/dgdzvp) (а) и распределение спиновой плотности в нем (б). Атомы водорода опущены, длины связей приведены в ангстремах.
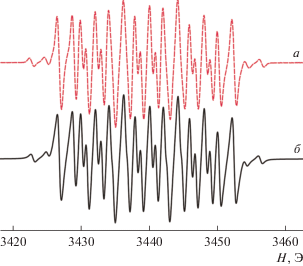
Все соединения имеют хорошо разрешенные спектры ЭПР в растворе. Значения параметров спектров исследованных комплексов I–V приведены в табл. 1. Поскольку спектры близки по своим параметрам, в качестве примера на рис. 2 приведен спектр ЭПР комплекса II. Он представляет собой триплет (1 : 1 : 1) дублетов (1 : 1) дублетов (1 : 1) дублетов (1 : 1). Сверхтонкая структура спектра обусловлена взаимодействием неспаренного электрона с тремя неэквивалентными магнитными ядрами атома водорода 1Н (99.98%, I = 1/2, μN = 2.7928) и одним атомом азота 14N (99.63%, I = 1, μN = 0.4037), кроме того, наблюдается сателлитное расщепление на магнитных изотопах олова 117Sn (7.68%, I = 1/2, μN = 1.000) и 119Sn (8.58%, I = 1/2, μN = 1.046) (рис. 2). Отнесение констант СТВ к протонам в положениях 3, 7 и 9 сделано на основании распределения спиновой плотности, полученного по результатам DFT расчетов. Отсутствие СТВ неспаренного электрона с магнитными изотопами атома 35Cl и 37Cl хорошо коррелирует с расчетными данными, согласно которым атом галогена лежит в плоскости лиганда и на нем отсутствует спиновая плотность.
Таблица 1.
Параметры изотропных спектров ЭПР (Э) для комплексов I–VIII
| Комплекс | ai(H(7)) | ai(H(9)) | ai(H(3)) | ai(14N) | ai(117Sn, 119Sn) | gi |
|---|---|---|---|---|---|---|
| I | 2.16 | 3.28 | 4.11 | 7.93 | 6.50; 6.80 | 2.0030 |
| II | 2.18 | 3.35 | 4.15 | 7.98 | 7.68; 8.04 | 2.0031 |
| III | 2.15 | 3.31 | 4.13 | 7.89 | 5.26; 5.51 | 2.0031 |
| IV | 2.11 | 3.30 | 4.13 | 7.95 | 4.83; 5.05 | 2.0030 |
| V | 2.31 | 3.40 | 4.19 | 7.98 | 1.73; 1.81 | 2.0029 |
| VII | 2.18 | 3.35 | 4.20 | 7.97 | 8.41; 8.79 | 2.0032 |
| VIII | 2.20 | 3.34 | 4.25 | 7.97 | 6.12; 6.40 | 2.0033 |
Рис. 2.
Изотропный спектр ЭПР комплекса phenox-imSQSnEt2Cl (II) в ТHF при Т = 298 К: теоретический (а), экспериментальный (б).
Расположение галогена в плоскости парамагнитного лиганда наблюдалось ранее и в родственных пятикоординационных N-арилзамещенных о-иминосемихинолятах диорганогалогенидов олова(IV) [23, 24]. Однако последние имеют существенное отличие от соединений I–IV в формировании координационного полиэдра. Апикальные позиции тригональной бипирамиды в комплексах типа imSQSnR2Cl, где imSQ – анион-радикал 4,6-ди-трет-бутил-N-арил-о-иминохинона, R – Alk, Ar, занимают атомы галогена и кислорода парамагнитного лиганда, а азот располагается в экваториальной плоскости [23, 24]. Напротив, согласно квантово-химическим расчетам геометрия соединения II характеризуется апикальным расположением азота в тригональной бипирамиде. Конфигурация с азотом в экваториальной плоскости тригональной бипирамиды также имеет минимум на поверхности потенциальной энергии для соединения II, однако она дестабилизирована на 5 ккал/моль относительно приведенной на рис. 1. Наличие атома азота о‑иминосемихинолятного анион-радикала в апикальной позиции в I–IV сопровождается уменьшением значений констант СТВ с магнитными изотопами 117Sn и 119Sn в спектрах ЭПР феноксазиноновых производных в 5–10 раз в сравнении с imSQSnR2Cl. Так, в случае N-арилзамещенных о‑иминосемихинолятов олова(IV) характерное значение констант СТВ варьируется в интервале 40–50 Э [23, 24], в то время как в полученных phenox-imSQSnR2Cl (I–IV) – в интервале 5–8 Э (табл. 1). Учитывая, что спиновая плотность в феноксазиноновом анион-радикале сосредоточена преимущественно на атоме азота, то спиновая σ–π-поляризация связи N–Sn будет вносить значимый вклад в величины констант СТВ на магнитных изотопах металла. Известно [40], что увеличение констант СТВ в спектрах ЭПР наблюдается при взаимодействии орбитали, занятой неспаренным электроном, с имеющей ненулевую плотность на ядре s-орбиталью элемента. Можно ожидать, что при sp3d-гибридизации в тригональной бипирамиде вклад s-орбитали металла в формирование гибридной АО, обеспечивающей связывание с экваториальными заместителями в координационной сфере олова(IV), должен быть гораздо выше, чем для апикальных направлений, носящих скорее pz,d$_{{{{z}^{2}}}}$-характер [41]. Таким образом, перемещение азота анион-радикального лиганда из экваториальной позиции в аксиальную является основной причиной резкого снижения констант СТВ с магнитными изотопами 117Sn и 119Sn в парамагнитных комплексах I–IV по сравнению с N-арилзамещенными о-иминосемихинолятами олова(IV). Другой характерной особенностью, наблюдаемой в спектрах ЭПР таких пятикоординационных комплексов диорганоолова(IV), является зависимость константы СТВ с магнитными изотопами 117, 119Sn от природы углеводородных заместителей на металле. Так, для пятикоординационных комплексов диорганоолова(IV), содержащих атом азота в экваториальной плоскости, наблюдается нарастание константы СТВ с увеличением разветвленности алкильных заместителей [23, 42]. В настоящей работе для производных I–III прослеживается обратная зависимость. Данное наблюдение также, видимо, обусловлено изменением характера АО металла, отвечающей за связывание с азотным центром анион-радикала.
Необходимо также отметить тот факт, что в случае комплекса V, содержащего три фенильных заместителя при атоме олова, значение константы СТВ на магнитных изотопах олова уменьшается в сравнении с комплексами I–IV и составляет 1.75 Э. Данный факт может быть обусловлен стерическими затруднениями, создаваемыми тремя фенильными группами при атоме олова, что приводит к увеличению расстояния между металлоцентром и редокс-активным лигандом. Подобная тенденция наблюдалась и в изученных ранее о-иминосемихинолятах олова, при переходе от imSQSnR2Cl к imSQSnR3 [14]. Трифенильный комплекс V был генерирован в растворе ранее [43], но не был выделен в индивидуальном состоянии, а лишь исследован спектроскопией ЭПР в растворе.
Как было описано выше, phenox-imQ может быть восстановлен двумя эквивалентами металлического натрия с образованием динатриевой соли phenox-APNa2. Взаимодействием полученной соли с Et2SnCl2 в соотношении 1 : 1 в растворе ТHF был получен диамагнитный комплекс олова(IV) – phenox-APSnEt2(THF) (VI) (схема 3 ).
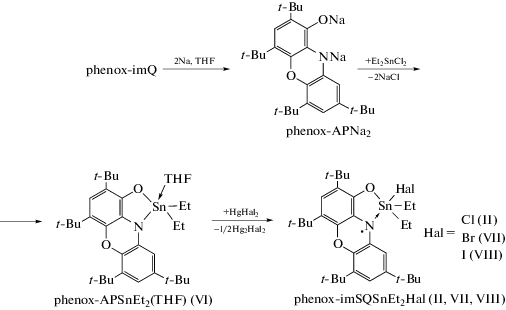
Схема 3 .
В ходе реакции окраска реакционной смеси менилась с желтой на интенсивно-оранжевую. Комплекс VI неустойчив к действию кислорода и влаги воздуха. Кристаллы, пригодные для РСА, были получены из раствора ТHF при медленном упаривании растворителя. На рис. 3 приведена кристаллическая структура соединения VI, значения избранных длин связей и углов – в табл. 2. Согласно данным РСА, VI кристаллизуется в моноклинной пространственной группе P21/n. В независимой области кристаллической ячейки располагается одна молекула комплекса, находящаяся в общем положении, а также одна сольватная молекула ТHF. Таким образом, VI кристаллизуется в виде сольвата VI · THF. Координационное окружение атома олова в VI · THF представляет собой искаженную тетрагональную пирамиду, в вершине которой находится атом углерода C(29), квадратное основание формируют атомы углерода C(31), кислорода O(1), O(THF) и азота N(1). Для анализа координационной сферы пятикоординационных комплексов используется геометрический параметр τ, значение которого в идеальной квадратной пирамиде равно нулю, а в идеальной тригональной бипирамиде – единице [44]. В VI · THF τ = 0.32 и соответствует сильно искаженной тетрагонально-пирамидальной геометрии координационного окружения атома олова. Примечательно, что ближайший аналог соединения VI среди пятикоординационных о-иминохиноновых производных – о-амидофенолятный комплекс dippAPSnEt2THF [45] имеет строение гораздо ближе к тригональной бипирамиде с τ = 0.75. Таким образом, переход от производных 4,6-ди-трет-бутил-N-арил-о-иминобензохинона к близким по строению соединениям на основе phenox-imQ сопровождается существенным изменением типа координационной геометрии.
Рис. 3.
Молекулярная структура комплекса VI. Для ключевых атомов тепловые эллипсоиды приведены с 50%-ной вероятностью. Атомы водорода не изображены для ясности.
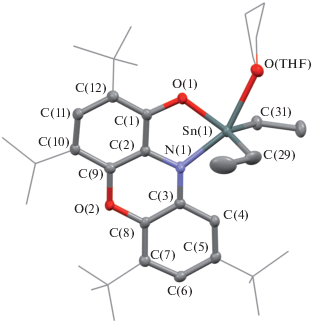
Таблица 2.
Избранные длины связей (Å) и углы (град) в комплексе VI · THF
| Связь | d, Å | Связь | d, Å |
|---|---|---|---|
| Sn(1)–O(1) | 2.018(2) | O(2)–C(9) | 1.408(2) |
| Sn(1)–N(1) | 2.103(2) | C(2)–C(9) | 1.393(2) |
| Sn(1)–C(31) | 2.136(2) | C(3)–C(4) | 1.402(2) |
| Sn(1)–C(29) | 2.137(2) | C(3)–C(8) | 1.405(2) |
| Sn(1)–O(THF) | 2.438(2) | C(4)–C(5) | 1.392(2) |
| O(1)–C(1) | 1.368(2) | C(5)–C(6) | 1.394(2) |
| N(1)–C(3) | 1.388(2) | C(6)–C(7) | 1.404(2) |
| N(1)–C(2) | 1.398(2) | C(7)–C(8) | 1.409(2) |
| C(1)–C(12) | 1.399(2) | C(9)–C(10) | 1.406(2) |
| C(1)–C(2) | 1.406(2) | C(10)–C(11) | 1.406(2) |
| O(2)–C(8) | 1.403(2) | C(11)–C(12) | 1.396(2) |
| Угол | ω, град | Угол | ω, град |
| O(1)Sn(1)N(1) | 80.05(5) | N(1)Sn(1)C(31) | 103.77(7) |
| C(31)Sn(1)C(29) | 123.02(9) | O(1)Sn(1)C(29) | 108.22(8) |
| O(1)Sn(1)O(3) | 76.19(5) | N(1)Sn(1)C(29) | 105.02(8) |
| N(1)Sn(1)O(3) | 155.25(5) | O(1)Sn(1)O(3) | 76.19(5) |
| O(1)Sn(1)C(31) | 124.54(7) |
Длины связей в шестичленных углеродных кольцах (см. табл. 2) близки к ароматическим (~1.40 Å), что наряду с характеристичными расстояниями С(1)–O(1) (1.368(2) Å) и С–N (1.388(2), 1.398(2) Å) [46, 47], однозначно указываeт на дианионное состояние редокс-актиного лиганда в комплексе VI · THF. Длины связей Sn–O(1) (2.018(2) Å) и Sn–N(1) (2.103(2) Å) короче суммы ван-дер-ваальсовых радиусов соответствующих элементов [48] и указывают на ковалентный характер взаимодействия дианиона феноксазинонового лиганда с металлоцентром. Необходимо отметить, что полученное соединение является первым структурно охарактеризованным соединением, содержащим лиганд phenox-imQ в дианионной форме. В литературе имеются сведения о строении комплексов металлов, содержащих данный лиганд в нейтральной [49, 50] и анион-радикальной форме [35, 36, 51–54].
С целью установления возможности окисления дианионного лиганда в исследуемом комплексе, мы выполнили в условиях ЭПР эксперимента взаимодействие VI с галогенидами ртути (HgCl2, HgBr2, HgI2). Как было показано ранее, выбранные реагенты – удобные одноэлектронные окислители, позволяющие получать радикальные производные в реакциях с катехолатными, о-амидофенолятными и диамидными комплексами металлов [55–57]. Продукты этой реакции были исследованы методом спектроскопии ЭПР в растворе in situ и не выделялись в индивидуальном состоянии. При взаимодействии растворов комплекса VI c HgHal2 наблюдается изменение цвета раствора с оранжевого на бордово-красный, а в спектрах ЭПР появляются сигналы, аналогичные рассмотренным выше для комплексов I–V. Параметры спектров ЭПР представлены в табл. 1. Спектр продукта реакции с хлоридом ртути(II) идентичен таковому для комплекса II. Анализ параметров наблюдаемых спектров ЭПР позволяет заключить, что в ходе окисления образуются парамагнитные производные phenox-imSQSnEt2Hal (II, VII, VIII) (схема 3 ), строение которых аналогично обсуждаемому выше. Необходимо отметить, что изменение констант СТВ неспаренного электрона с магнитными изотопами 117, 119Sn, вызванное сменой галогена, не укладывается в какую-либо однозначную зависимость. Наибольшие константы (см. табл. 1) наблюдаются для бромпроизводного. Вероятно, отсутствие корреляции величин констант СТВ с 117, 119Sn в ряду Cl–Br–I может быть вызвано действием сразу нескольких факторов (стерические, электронные эффекты, изменение вкладов атомных орбиталей олова в гибридизацию), различным образом сказывающихся на измеряемый параметр.
В результате проделанной работы показано, что о-иминосемихиноновые анион-радикальные лиганды могут выступать достаточно информативными спиновыми метками, позволяющими исследовать молекулярное и электронное строение элементоорганических соединений в растворе. При этом они обладают существенным преимуществом перед традиционно используемыми для этой цели о-семихинонами, поскольку не вызывают элиминирования карбанионных заместителей у металла. На примере оловоорганических соединений продемонстрировано, что параметры спектров ЭПР, наблюдаемые для их о-иминосемихиноновых производных, оказываются весьма чувствительны к строению координационного полиэдра, к природе алкильных заместителей у атома металла и к их количеству, что сказывается на значениях констант СТВ на магнитных изотопах металла.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Kabachnik M.I. // Pure Appl. Chem. 1980. V. 52. P. 859.
Broere D.L.J., Plessius R., van der Vlugt J.I. // Chem. Soc. Rev. 2015. V. 44. P. 6886.
Tezgerevska T., Alley K.G., Boskovic C. // Coord. Chem. Rev. 2014. V. 268. P. 23.
Jacquet J., Cheaib K., Ren Y. et al. // Chem. Eur. J. 2017. V. 23. P. 1530.
Kaim W., Paretzki A. // Coord. Chem. Rev. 2017. V. 344. P. 345.
van der Vlugt J.I. // Chem. Eur. J. 2019. V. 25. P. 2651.
Khan F.F., Chowdhury A.D., Lahiri G.K. // Eur. J. Inorg. Chem. 2020. P. 1138.
Абакумов Г.А., Пискунов А.В., Черкасов В.К. и др. // Успехи химии. 2018. Т. 87. С. 393 (Abakumov G.A., Piskunov A.V., Cherkasov V.K. et al. // Russ. Chem. Rev. 2018. V. 87. P. 393).
Poddel’sky A.I., Cherkasov V.K., Abakumov G.A. // Coord. Chem. Rev. 2009. V. 253. P. 291.
Chirik P.J., Wieghardt K. // Science. 2010. V. 327. P. 794.
Kaim W., Schwederski B. // Coord. Chem. Rev. 2010. V. 254. P. 1580.
Kozhanov K.A., Bubnov M.P., Cherkasov V.K. et al. // Dalton Trans. 2004. P. 2957.
Kozhanov K.A., Bubnov M.P., Cherkasov V.K. et al. // Dalton Trans. 2008. P. 2849.
Bubnov M.P., Teplova I.A., Kozhanov K.A. et al. // J. Magn. Reson. 2011. V. 209. P. 149.
Kozhanov K.A., Bubnov M.P., Abakumov G.A., Cherkasov V.K. // J. Magn. Reson. 2012. V. 225. P. 62.
Kozhanov K.A., Bubnov M.P., Teplova I.A. et al. // J. Mol. Struct. 2017. V. 1147. P. 541.
Bubnov M.P., Kozhanov K.A., Skorodumova N.A. et al. // J. Mol. Struct. 2019. V. 1180. P. 878.
Чегерев М.Г., Пискунов А.В. // Коорд. химия. 2018. Т. 44. С. 109 (Chegerev M.G., Piskunov A.V. // Russ. J. Coord. Chem. 2018. V. 44. P. 258). https://doi.org/10.1134/S1070328418040036
Ершова И.В., Пискунов А.В. // Коорд. химия. 2020. Т. 46. С. 132 (Ershova I.V., Piskunov A.V. // Russ. J. Coord. Chem. 2020. V. 46. P. 154). https://doi.org/10.1134/S1070328420030021
Razuvaev G.A., Tsarjapkin V.A., Gorbunova L.V. et al. // J. Organomet. Chem. 1979. V. 174. P. 475.
Прокофьев А.И., Помбрик С.И., Касымбекова З.К. и др. // Изв. АН СССР. Сер. хим. 1982. С. 540 (Prokof’ev A.I., Pombrik S.I., Kasymbekova Z.K. et al. // Russ. Chem. Bull. 1982. V. 31. P. 482).
Разуваев Г.А., Абакумов Г.А., Баюшкин П.Я. и др. // Изв. АН СССР. Сер. хим. 1984. С. 2098 (Razuvaev G.A., Abakumov G.A., Bayushkin P.Y. et al. // Russ. Chem. Bull. 1984. V. 33. P. 1915).
Пискунов А.В., Мещерякова И.Н., Баранов Е.В. и др. // Изв. АН. Сер. хим. 2010. С. 355. (Piskunov A.V., Meshcheryakova I.N., Baranov E.V. et al. // Russ. Chem. Bull. 2010. V. 59. P. 361).
Пискунов А.В., Чегерев М.Г., Ваганова Л.Б. и др. // Коорд. химия. 2015. Т. 41. С. 394 (Piskunov A.V., Chegerev M.G., Vaganova L.B. et al. // Russ. J. Coord. Chem. 2015. V. 41. P. 428). https://doi.org/10.1134/S1070328415070076
Гордон А., Форд Р. // Спутник химика. М: Мир, 1976. С. 541 (Gordon A., Ford R., The Chemist’s Companion: A Handbook of Practical Data, Techniques, and References, New York: Wiley, 1972).
Stoll S., Schweiger A.J. // J. Magn. Reson. 2006. V. 178. P. 42.
Ивахненко Е.П., Карсанов И.В., Хандкарова В.С. и др. // Изв. АН СССР. Сер. хим. 1986. С. 2755 (Ivakhnenko E.P., Karsanov I.V., Khandrakova V.S. et al. // Russ. Chem. Bull. 1986. V. 35. P. 2526).
Вольева В.Б., Прокофьева Т.И., Прокофьев А.И. и др. // Изв. АН. Сер. хим. 1995. V. 9. P. 1789 (Vol’eva V.B., Prokofeva T.I., Prokofev A.I. et al. // Russ. Chem. Bull. 1995. V. 44. P. 1720).
Stegmann H.B., Scheffler K. // Chem. Ber. 1968. V. 101. P. 262.
Frisch M.J., Trucks G.W., Schlegel H.B. et al. Gaussian 09. Revision D.01. Wallingford (CT, USA): Gaussian, Inc., 2013.
Becke A.D. // J. Chem. Phys. 1993. V. 98. P. 5648.
Rigaku Oxford Diffraction. CrysAlisPro software system. Version 1.171.35.19. Wroclaw (Poland): Rigaku Corporation, 2011.
Sheldrick G.M. // Acta Crystallogr. C. 2015. V. 71. P. 3.
Spek A.L. // Acta Crystallogr. C. 2015. V. 71. P. 9.
Romanenko G.V., Ivakhnenko E.P., Minkin V.I. et al. // Inorg. Chim. Acta. 2014. V. 418. P. 66.
Ivakhnenko E.P., Starikov A.G., Lyssenko K.A. et al // Inorg. Chim. Acta. 2014. V. 410. P. 144.
Piskunov A.V., Mescheryakova I.N., Fukin G.K. et al. // Heteroatom Chem. 2009. V. 20. P. 332.
Piskunov A.V., Meshcheryakova I.N., Piskunova M.S. et al. // J. Mol. Struct. 2019. V. 1195. P. 417.
Пискунов А.В., Мещерякова И.Н., Смолянинов И.В. и др. // Изв. АН. Сер. хим. 2013. С. 149 (Piskunov A.V., Meshcheryakova I.N., Smolyaninov I.V. et al. // Russ. Chem. Bull. 2013. V. 62. P. 147).
Абакумов Г.А., Черкасов В.К., Пискунов А.В. и др. // Изв. АН. Сер. хим. 2006. С. 1103 (Abakumov G.A., Cherkasov V.K., Piskunov A.V. et al. // Russ. Chem. Bull. 2006. V. 55. P. 1146).
Gillespie R.J. // J. Chem. Soc. 1963. P. 4672.
Пискунов А.В., Сухошкина О.Ю., Смолянинов С.В. // Журн. oбщ. химии. 2010. Т. 80. С. 629 (Piskunov A.V., Sukhoshkina O.Y., Smolyaninov I.V. // Russ. J. Gen. Chem. 2010. V. 80. P. 790).
Ивахненко Е.П., Карсанов И.В., Хандкарова В.С. и др. // Изв. АН СССР Сер. хим. 1987. С. 68 (Karsanov I.V., Ivakhnenko E.P., Khandkarova V.S. et al. // Russ. Chem. Bull. 1987. V. 36. P. 56).
Addison A.W., Rao T.N., Reedijk J. et al. // Dalton Trans. 1984. P. 1349.
Пискунов А.В., Айвазьян И.А., Абакумов Г.А. и др. // Изв. АН. Сер. хим. 2007. С. 253 (Piskunov A.V., Aivaz’yan I.A., Abakumov G.A. et al. // Russ. Chem. Bull. 2007. V. 56. P. 261).
Brown S. N. // Inorg. Chem. 2012. V. 51. P. 1251.
Chegerev M.G., Piskunov A.V., Starikova A.A. et al. // Eur. J. Inorg. Chem. 2018. P. 1087.
Бацанов С.С. // Журн. неорган. химии. 1991. Т. 36. № 12. С. 3015 (Batsanov S.S. // Russ. J. Inorg. Chem. 1991. V. 36. P. 1694).
Ивахненко Е.П., Кощиенко Ю.В., Чернышев А.В. и др. // Журн. oбщ. химии. 2016. Т. 86. № 7. С. 1188 (Ivakhnenko E.P., Koshchienko Y.V., Chernyshev A.V. et al. // Russ. J. Gen. Chem. 2016. V. 86. P. 1664).
Ивахненко Е.П., Кощиенко Ю.В., Князев П.А. и др. // Коорд. химия. 2016. Т. 42. № 4. С. 221 (Ivakhnenko E.P., Koshchienko Yu.V., Knyazev P.A. et al // Russ. J. Coord. Chem. 2016. V. 42. P. 509). https://doi.org/10.1134/S1070328416040011
Антипин М.Ю., Ивахненко Е.П., Кощиенко Ю.В. и др. // Изв. АН. Сер. хим. 2013. № 8. С. 1744 (Antipin M.Yu., Ivakhnenko E.P., Koshchienko Yu.V. et al. // Russ. Chem. Bull. 2013. V. 62. P. 1744).
Speier G., Whalen A.M., Csihony J., Pierpont C.G. // Inorg. Chem. 1995. V. 34. P. 1355.
Bhattacharya S., Pierpont C.G. // Inorg. Chem. 1994. V. 33. P. 6038.
Whalen A.M., Bhattacharya S., Pierpont C.G. // Inorg. Chem. 1994. V. 33. P. 347.
Пискунов А.В., Цыс К.В., Чегерев М.Г. и др. // Коорд. химия. 2019. Т. 45. № 9. С. 527 (Piskunov A.V., Tsys K.V., Chegerev M.G. et al. // Russ. J. Coord. Chem. 2019. V. 45. P. 626). https://doi.org/10.1134/S1070328419090069
Tsys K.V., Chegerev M.G., Fukin G.K. et al. // Mendeleev Commun. 2018. V. 28. P. 527.
Абакумов Г.А., Черкасов В.К., Пискунов А.В. // Доклады АН. 2005. Т. 404. №. 4. С. 496 (Abakumov G.A., Cherkasov V.K., Piskunov A.V. et al. // Dokl. Chem. 2005. V. 404. P. 189).
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия