

Координационная химия, 2020, T. 46, № 3, стр. 132-156
Комплексы металлов III группы на основе лигандов о-иминохинонового типа
И. В. Ершова 1, А. В. Пискунов 1, *
1 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
* E-mail: pial@iomc.ras.ru
Поступила в редакцию 27.09.2019
После доработки 30.10.2019
Принята к публикации 31.10.2019
Аннотация
Представлены и проанализированы известные к настоящему моменту литературные данные о синтезе, строении и химических свойствах комплексов металлов III группы на основе би-, три- и тетрадентатных редокс-активных лигандов о-иминохинонового типа.
Химия комплексов металлов с редокс-активными лигандами интенсивно развивается в последние несколько лет благодаря богатому разнообразию практически полезных свойств, приобретаемых соединениями этого типа за счет органического фрагмента [1–6]. Все больше необычных химических превращений становятся доступными за счет способности лигандов к обратимому окислительно-восстановительному превращению в координационной сфере металла [7–13]. Другим важным аспектом химии рассматриваемых соединений является возможность направленного дизайна магнитоактивных производных металлов с радикальными формами редокс-активных лигандов. Особенно важными эти свойства становятся при рассмотрении химии непереходных металлов, которые не обладают способностью легко изменять свою степень окисления и для которых не характерны парамагнитные формы устойчивых ионов. Недавно была опубликована работа, посвященная координационной химии непереходных элементов IV группы с редокс-активными о-иминохиноновыми лигандами [14]. В настоящем обзоре собраны литературные данные по синтезу, строению, а также химическим свойствам комплексов элементов III группы, включая редкоземельные металлы и актиноиды, с лигандами данного типа.
КОМПЛЕКСЫ МЕТАЛЛОВ III ГРУППЫ С БИДЕНТАТНЫМИ о-ИМИНОХИНОНОВЫМИ ЛИГАНДАМИ
В координационной сфере металла бидентатные ON-хелатирующие редокс-активные о-иминобензохиноновые лиганды могут находиться в одной из трех редокс-форм: в нейтральном (о‑иминобензохинон, imQ0), однократно восстановленном (о-иминобензосемихинолят, imSQ–), или дважды восстановленном (о-амидофенолят, AP2–) состоянии (схема 1 ).
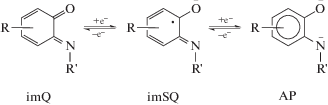
Схема 1 .
Основные методы синтеза о-иминобензохиноновых комплексов металлов III группы в целом схожи и включают прямое взаимодействие металлов (или их амальгам) с о-иминобензохинонами, обменные реакции между соответствующими производными щелочных металлов с галогенидами металлов III группы, а также реакции окислительного присоединения о-иминобензохинонов к соединениям металлов в низких степенях окисления. В подавляющем большинстве синтетических подходов к получению о-иминобензохиноновых производных Al, Ga, In и Tl стартовыми реагентами являются пространственно затрудненный 4,6-ди-трет-бутил-N-(2,6-ди-изо-пропилфенил)-о-иминобензохинон (DippimQ) и его моно- ((DippimSQ)Na) и динатриевая ((DippAP)Na2) соли (схема 2 ).
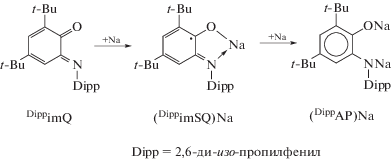
Схема 2 .
В ряде случаев, когда соответствующие о-иминобензохиноны неустойчивы в растворе, исходными соединениями для синтеза о-иминобензохиноновых производных металлов являются отвечающие им о-аминофенолы (дважды восстановленная протонированная форма о-иминобензохинона ((AP2–)H2).
Комплексы элементов подгруппы бора. Поскольку химия бора существенно отличается от других элементов III группы, при получении его о-иминохиноновых производных не используется ни один из рассмотренных выше наиболее общих способов. В качестве исходных соединений для синтеза большинства известных о-амидофенолятов бора выступают о-аминофенолы и производные борной кислоты [15, 16].
Взаимодействие двух эквивалентов фенилбороновой кислоты с бис-о-аминофенолами протекает в достаточно жестких условиях и приводит к образованию бис-оксазаборолидинов (1), содержащих два плоских пятичленных цикла CCNBO, в которых каждый атом бора имеет тригональную геометрию (cхема 3) [16]. Таким образом, несмотря на то, что в качестве исходных соединений были использованы формально тетрадентатные ONNO-лиганды, комплексы (1) представляют собой два о-амидофенолятных фрагмента ((AP)BPh), связанных алкильными мостиками. В растворе все соединения типа (1) симметричны за счет наличия поворотной оси C2, однако в кристалле данная симметрия нарушается. Наличие в одной молекуле сразу двух льюисовских кислотных центров делает (1) интересными объектами для бимолекулярного катализа.
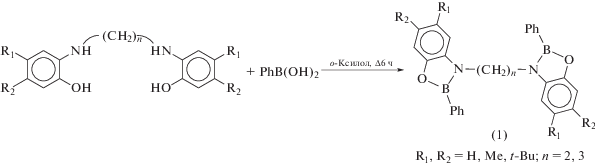
Схема 3 .
Иной тип комплекса, содержащего в одной молекуле два фрагмента (AP)B, получен с невысоким выходом при взаимодействии незамещенного о-аминофенола с дибораном B2(NMe2)4 (схема 4 ) [17]. Димерный комплекс (2) лежит на центре инверсии: атомы O и группы NH в двух фрагментах (AP)B располагаются в транс-положении относительно друг друга. Согласно РСА в кристаллической упаковке (2) наблюдается π-стэкинг с межплоскостным расстоянием 3.48 Å. Кроме того, каждая молекула в (2) связана с четырьмя другими молекулами в двух соседних колонках за счет контактов NH…O (2.32 Å), образуя бесконечные листы водородно-связанных молекул.

Схема 4 .
Борсодержащие ферроценильные комплексы (3), образующиеся при взаимодействии производных борной кислоты с о-аминофенолом, функционализированным бис(дифенилфосфин)ферроценильной группой (cхема 5) – перспективные лиганды для осуществления асимметричного катализа за счет возможности вторичных взаимодействий (secondary interactions) между боронатной группой, являющейся слабым льюисовским кислотным центром, и субстратом-основанием Льюиса [15]. Соединения (3) образуют устойчивые металлокомплексы (4) при взаимодействии с Me2Pt(COD) (COD = циклооктадиен) (cхема 5), а также стабильные аддукты с производными Rh(I), проявляющими каталитическую активность в реакциях гидрирования и гидроформилирования простых субстратов.
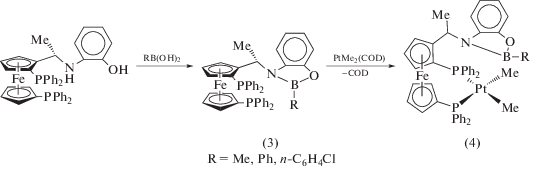
Схема 5 .
В то же время при взаимодействии незамещенного о-аминофенола с Me3Ga происходит депротонирование только гидроксильной группы лиганда и продуктом реакции является не о-амидофенолятный, а о-аминофенолятный комплекс (cхема 6) [18]. Анализ кристаллической структуры (5) показал, что молекулы комплекса не образуют димеров, но связаны с соседними молекулами за счет образования водородных связей NH···O.
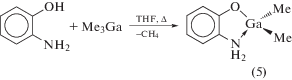
Схема 6 .
Металлический галлий не реагирует с DippimQ, однако в ходе реакции с амальгамой металла происходит неполное восстановление DippimQ с образованием смешанолигандного комплекса (6) (cхема 7) [19]. О наличии двух разнозаряженных о-иминохиноновых лигандов в (6) однозначно свидетельствуют данные спектроскопии ЭПР, указывающие на наличие быстрой (во временной шкале ЭПР) миграции неспаренного электрона между двумя редокс-активными лигандами. Декоординация молекулы THF при растворении (6) в гексане приводит к изменению геометрии в образующемся четырехкоординационном комплексе (7) на тетраэдрическую, при которой делокализация неспаренного электрона по обоим лигандам невозможна. Комплекс (8), образующийся при добавлении пиридина к раствору (7), выделен в индивидуальном состоянии. В растворе его структура аналогична структуре комплекса (6) и сохраняется в кристаллическом состоянии, о чем свидетельствует наличие полосы переноса заряда лиганд–лиганд (~2010 нм) в ближней ИК-области.
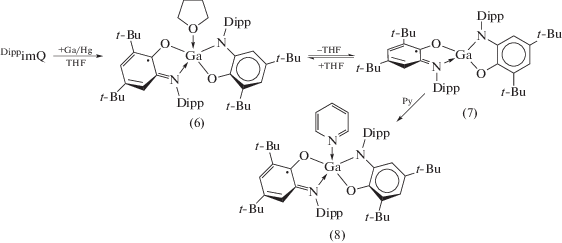
Схема 7 .
Одним из удобных методов синтеза металлокомплексов, содержащих редокс-активный лиганд в дианионном состоянии, является восстановление о-иминохинона галогенидами металлов в низких степенях окисления. Взаимодействие DippimQ с “GaI” приводит к образованию о-амидофенолята (9), который также можно синтезировать при взаимодействии DippimQ с металлическим галлием в присутствии стехиометрических количеств иода (схема 8 ) [19].
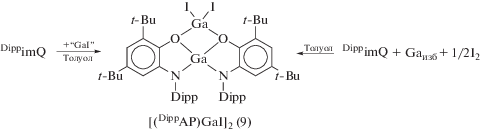
Схема 8 .
При использовании (DippAP)Na2 в качестве стартового соединения о-амидофенолятные про-изводные галлия образуются по его реакции обмена с галогенидами металла: димерный о-амидофенолят галлия (10) получали по реакции между (DippAP)Na2 и MeGaI2 (cхема 9) [20], тогда как образование (11) [21] либо (12) [19] в ходе взаимодействия (DippAP)Na2 с GaI3 зависит от соотношения исходных реагентов (2 : 1 или 3 : 2 соответственно) (cхема 10).
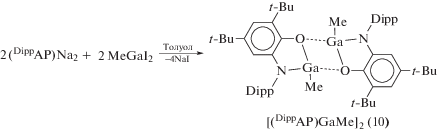
Схема 9 .
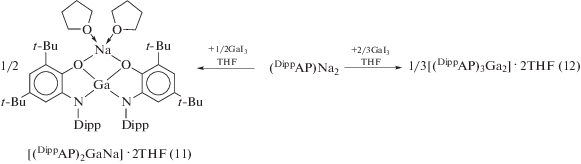
Схема 10 .
Взаимодействие DippimQ с иодидами индия в низких степенях окисления не приводит к образованию о-амидофенолятных производных металла: продуктом реакции DippimQ с InI является бирадикальный комплекс (13), а In2I4 в присутствии DippimQ подвергается диспропорционированию, в результате чего в ходе реакции образуется смесь продуктов (13) и (14) (cхема 11) [22]. Комплекс (14), содержащий координированный на металлоцентр нейтральный лиганд DippimQ, можно также получить при непосредственном взаимодействии DippimQ с InI3 в толуоле.
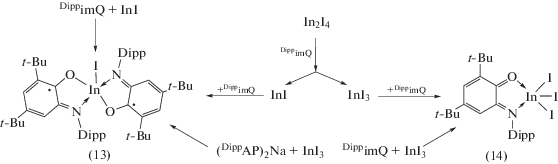
Схема 11 .
При взаимодействии (DippAP)Na2 с InI3 в неполярных растворителях происходит окисление о‑амидофенолятного лиганда ионом In3+, и продуктом реакции также является парамагнитный комплекс (13) (cхема 11). С целью исключить протекание редокс-процесса между (DippAP)2– и ионом In3+ авторы [23] использовали стратегию, позволяющую понизить электроноакцепторность иона In3+ за счет введения в координационную сферу металла нейтрального N-донорного лиганда, а также за счет замены электроноакцепторного атома иода на электронодонорную алкильную группу, что позволило получить о-амидофеноляты индия (15), (16) (cхема 12).
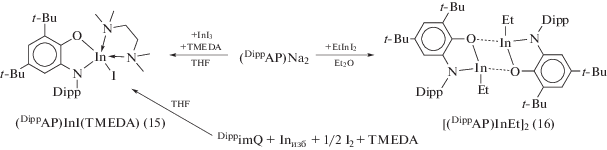
Схема 12 .
Полученный по обменной реакции (DippAP)Na2 с InI3 (2 : 1) в среде THF бис-о-амидофенолят индия, в отличие от галлиевого аналога (11), представляет собой ионный комплекс [(DippAP)2In]–[Na(DME)3]+ (17) (cхема 13) [24]. Комплекс (17) также образуется в результате восстановления анион-радикального лиганда (DippimSQ)– ионом одновалентного индия (cхема 13).
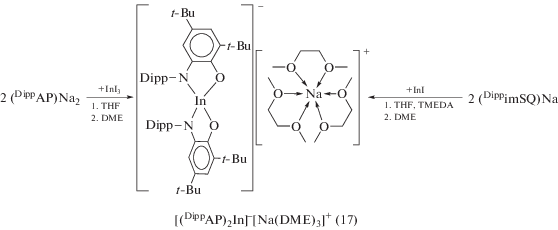
Схема 13 .
При взаимодействии бис-о-амидофенолятов галлия (11) и индия (17) с избытком алкил/аллил галогенидов (RHal) происходит окислительное присоединение двух молекул RHal к комплексам непереходных металлов, сопровождающееся образованием двух новых связей C–C [21, 24]. Реакции протекают в достаточно мягких условиях и приводят к образованию диамагнитных комплексов (18), содержащих лиганды иминоциклогекса-1,4-диенолятного типа (схема 14 ).
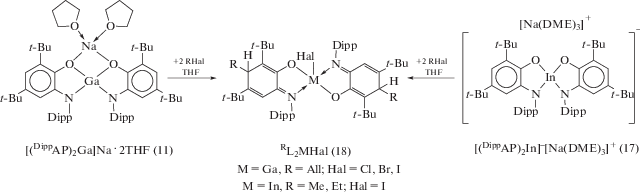
Схема 14 .
Взаимодействие о-амидофенолятных комплексов галлия (9, 10) с одноэлектронными окислителями приводит к окислению лиганда (DippAP)2– в исходных соединениях, в результате чего образуются новые парамагнитные производные металла (cхема 15) [25]. На первых этапах реакций происходит образование промежуточных монорадикальных продуктов, детектируемых методом спектроскопии ЭПР, которые подвергаются дальнейшей симметризации с образованием бирадикальных ме-таллокомплексов (19)–(22). Комплекс (19) также является основным продуктом окисления (10) кислородом [20].
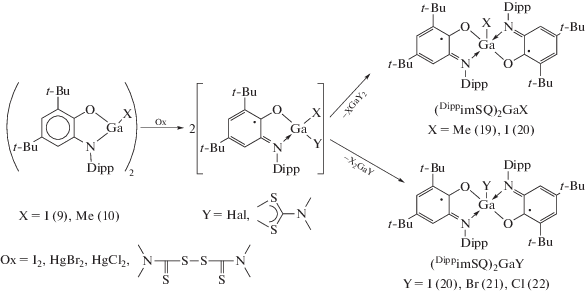
Схема 15 .
Аналогичным образом проходит окисление о‑амидофенолята индия [(DippAP)InEt]2 (16) одноэлектронными окислителями и кислородом. Однако при симметризации промежуточных монорадикальных комплексов во всех случаях происходит образование (DippimSQ)2InEt (23) [26]. Окисление (DippAP)InI-(TMEDA) (15) кислородом также приводит к образованию бирадикального комплекса (DippimSQ)2InI (13), в то время как продуктами его взаимодействия с I2 и HgCl2 являются монорадикальные комплексы (24), (25) (cхема 16) [26]. Устойчивость (24, 25) обусловлена координацией на металлоцентр нейтрального N-донорного лиганда, что предотвращает их последующую симметризацию [23].
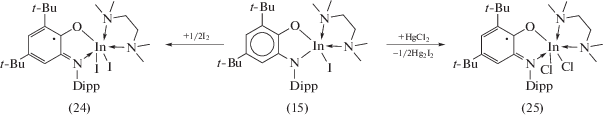
Схема 16 .
Направление симметризации монорадикальных комплексов индия определяется степенью заполнения координационной сферы металла в образующихся продуктах, ввиду этого образование шестикоординационного комплекса (26), а не пятикоординационного комплекса (13) при окислении (15) тетраметилтиурамдисульфидом вполне закономерно (cхема 17) [26].
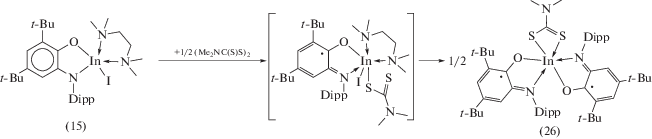
Схема 17 .
Окисление бис-о-амидофенолятного комплекса индия (17) галогенидами ртути(II) также приводит к образованию бис-о-иминобензосемихиноновых производных (27), (28) (cхема 18) [27].
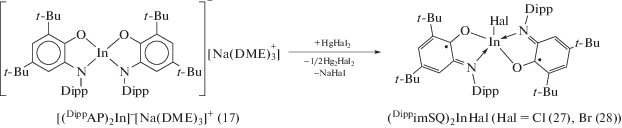
Схема 18 .
Наиболее удобным методом синтеза бирадикальных производных галлия [25, 28] и индия [22, 26] – конечных продуктов окисления о‑амидофенолятных комплексов (9), (10), (16), а также алюминия [29] является реакция обмена между (DippimSQ)Na с галогенидами/алкилгалогенидами соответствующих металлов (cхема 19).
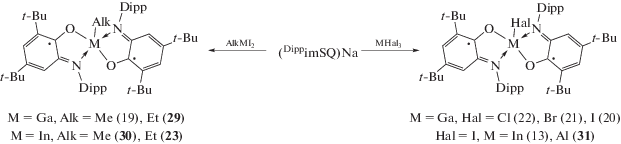
Схема 19 .
С использованием (DippimSQ)2MI (M = Al (31), Ga (20)) в качестве исходных соединений по реакции обмена с избытком неорганических солей (NaN3, KNCS, KNCO), а также со стехиометрическими количествами фенолята натрия были получены комплексы алюминия и галлия с различными неорганическими (а также феноксильным) заместителями в апикальном положении (32)–(38) (схема 20 ) [27–29].
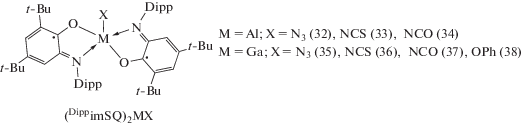
Схема 20 .
Магнитные свойства серии комплексов (Dip-pimSQ)2MX (13), (19)–(23), (27)–(38) сильно зависят от природы апикального заместителя X. В комплексах с алкильными заместителями имеет место слабое ферромагнитное взаимодействие между неспаренными электронами анион-радикальных лигандов, а для производных с неорганическими заместителями и комплекса с феноксильной группой наблюдается достаточно сильный антиферромагнитный обмен между спинами органических лигандов. При помощи DFT расчетов, выполненных для серии комплексов (DippimSQ)2GaX, был обнаружен косвенный канал антиферромагнитного обмена, формируемый за счет взаимодействия p-орбиталей атомов кислорода и азота о-иминобензохиноновых лигандов с р- или π-орбиталями гетероатомов апикального заместителя X [28]. При этом энергия магнитного обменного взаимодействия коррелирует с длиной связи Ga–X [27]. Координация нейтральных донорных лигандов на металлоцентр (схема 21 ) приводит к изменению геометрии комплексов с пяти- на шестикоординационную и нарушению косвенного канала магнитного обмена [27].
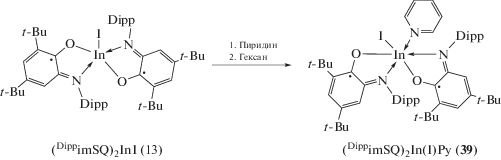
Схема 21 .
Трис-о-иминобензосемихиноновые комплексы галлия (imSQR)3Ga (40) образуются при взаимодействии соответствующих о-аминофенолов с GaCl3 в присутствии основания и кислорода воздуха [30]. Однако отсутствие в N-арильном фрагменте заместителей в орто-положениях по отношению к азоту может приводить к неустойчивости такого типа трирадикальных соединений и сопровождаться внутримолекулярными превращениями радикальных лигандов, что продемонстрировано на примере комплекса, содержащего метоксигруппы [31]. В результате реакции был получен монорадикальный комплекс галлия (41), содержащий гексадентатный лиганд – продукт внутримолекулярной перегруппировки с активацией связей C–H и образованием новых связей C–N (схема 22 ).
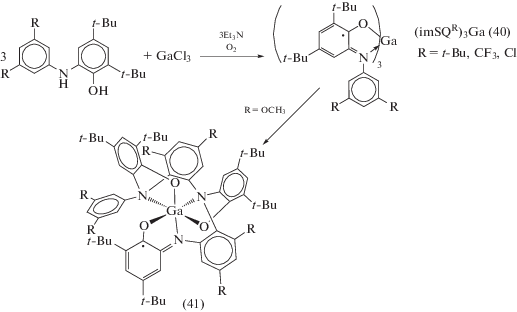
Схема 22 .
В качестве лиганда для синтеза о-иминохиноновых комплексов также используется N-(2,6-диметилфенил)фенантрен-о-иминохинон (DmpPhenimQ). Взаимодействие эквимолярной смеси DmpPhenimQ и B(C6F5)3 в присутствии водорода в кипящем толуоле в течение часа приводит к образованию смеси диамагнитного (42) и парамагнитного (43) продуктов, дальнейшее нагревание реакционной смеси сопровождается количественной конверсией (42) в о-амидофенолятный комплекс бора (44) (схема 23 ) [32]. Комплекс (43) – редкий пример структурно охарактеризованного полиароматического бороциклического радикала. Его устойчивость в аэробных условиях обусловлена делокализацией спиновой плотности по ароматическому каркасу редокс-активного лиганда, что подтверждается данными спектроскопии ЭПР.
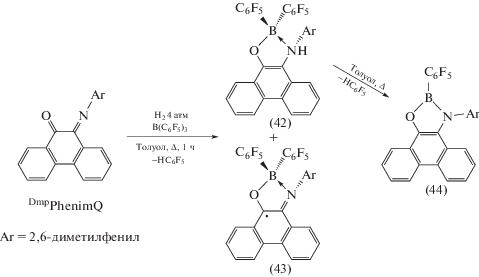
Схема 23 .
Поскольку в синтезе, представленном на схеме 23 , происходит образование трудноразделимой смести комплексов (43), (44), для их получения были предложены альтернативные синтетические процедуры: по реакции DmpPhenimQ с H2B(C6F5) · SMe2 (1 : 1) при комнатной температуре для комплекса (44) и по реакции DmpPhenimQ с HB(C6F5)2 для комплекса (43).
Продуктом взаимодействия эквимольных количеств DmpPhenimQ и Me3Al является диамагнитный биядерный комплекс (45), при образовании которого происходит нарушение сопряженной структуры лиганда в результате миграции одной из метильных групп AlMe3 к атому углерода, связанному с кислородным атомом лиганда, приводящей к образованию связи С–С (схема 24 ) [33].
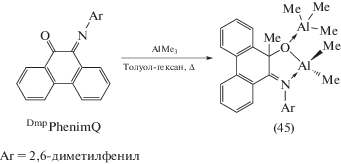
Схема 24 .
Комплекс галлия, содержащий координированный нейтральный о-иминохиноновый лиганд, получен при взаимодействии GaCl3 с аценафтен-1-имино-2-оном (DippMIAN) (cхема 25) [34]. Кристаллическая ячейка (46) содержит отдельные ионы [(DippMIAN)2GaCl2]+[GaCl4]–, предположительно образовавшиеся при диспропорционировании первоначально образующего комплекса (DippMIAN)GaCl3.
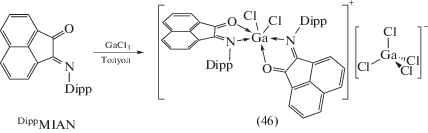
Схема 25 .
Комплексы галлия, содержащие DippMIAN в моно- и дивосстановленной формах, получены постадийным восстановлением лиганда металлическим галлием в присутствии стехиометрических количеств иода (cхема 26) [35]. Димерный диамагнитный комплекс (48) реагирует с фенилацетиленом с образованием продукта циклоприсоединения (49), а также проявляет каталитическую активность в реакциях гидроаминирования и гидроарилирования непредельных субстратов.
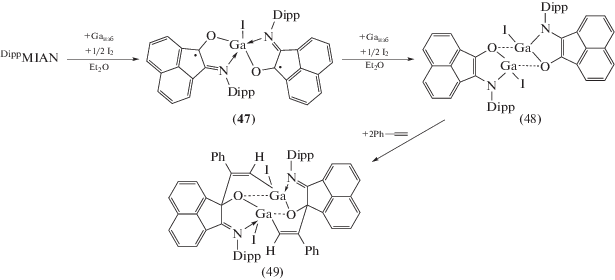
Схема 26 .
Единственным известным на данный момент о-иминобензохиноновым производным таллия является о-иминобензосемихинолят таллия(I) (DippimSQ)Tl (50), образующийся при восстановлении DippimQ амальгамой таллия [36].
Комплексы редкоземельных металлов и актинидов. Первые о-иминохиноновые комплексы редкоземельных металлов получены на основе N-(2,6-ди-изо-пропилфенил)фенантрен-о-иминохинона (DippPhenimQ) [37]. Продуктами окисления скандия и иттербия DippPhenimQ в бензоле являются трис-лигандные производные трехвалентных металлов (51), (52), содержащие эквивалентные анион-радикальные лиганды DippPhenimSQ. Oднако, согласно спектроскопии ЯМР, в среде THF полученные комплексы находятся в равновесии с о-амидофенолятной формой (cхема 27).
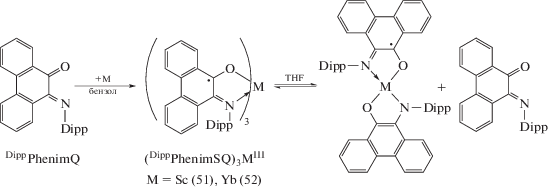
Схема 27 .
Взаимодействие DippPhenimQ с иодидами двухвалентных неодима и диспрозия также приводит к образованию металлокомплексов трехвалентных лантанидов, но координационная сфера металла в (53), (54), наряду с двумя атомами иода, содержит два разнозаряженных редокс-активных лиганда DippPhenimQ и DippPhenimSQ. При этом в обоих лигандах реализуется крайне нетипичный для о-иминохинонов η1-тип координации с металлом (схема 28 ). В ходе реакции происходит частичное диспропорционирование (53), (54) на трииодид лантанида и (DippPhenimSQ)3M, однако лишь комплекс диспрозия (DippPhenimSQ)3Dy (55) (строение аналогично 51 и 52) удалось выделить из реакционной смеси.
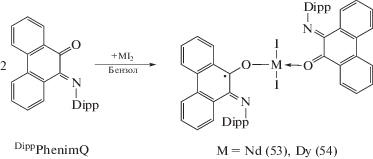
Схема 28 .
При окислении иттербия и европия DippimQ образуются производные не трех-, а двухвалентных лантанидов (56), (57), которые также можно получить по реакции обмена (DippAP)K2 с дииодидами соответствующих металлов (cхема 29) [38].
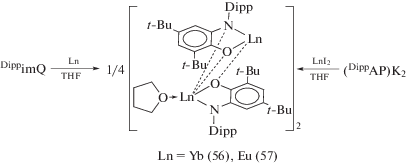
Схема 29 .
В результате окислительного присоединения эквивалента DippimQ к о-амидофенолятным производным Yb(II) (56) и Eu(II) (57) происходит окисление металлоцентра до Ln(III) (Ln = Yb, Eu) и образование смешанолигандных производных (58), (59), альтернативным способом получения которых является обменная реакция между (DippimSQ)K и LnI2 (2 : 1). При окислении (56) в среде некоординирующегося растворителя происходит образование димера (60), в котором о-амидофенолятные лиганды выступают в качестве мостиковых (cхема 30).
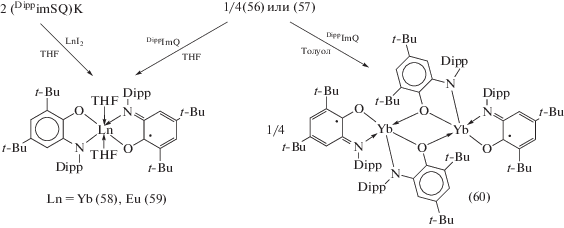
Cхема 30.
На основе DippimQ получена серия бис-о-иминохиноновых комплексов неодима(III), координированных лигандом DippimQ в трех различных редокс-состояниях [39]. Комплекс (DippimQ)2NdI3 (61), содержащий координированные нейтральные о-иминохиноновые лиганды, был получен при взаимодействии двух эквивалентов DippimQ с NdI3 · 3.5THF. Последовательное восстановление лигандов DippimQ в координационной сфере (61) KC8 приводит к образованию бис-о-иминобензосемихинонового (62) и бис-о-амидофенолятного (63) производных Nd(III). Комплекс (64), полученный при обработке (63) краун-эфиром, не содержит катион калия в координационной сфере неодима (cхема 31).
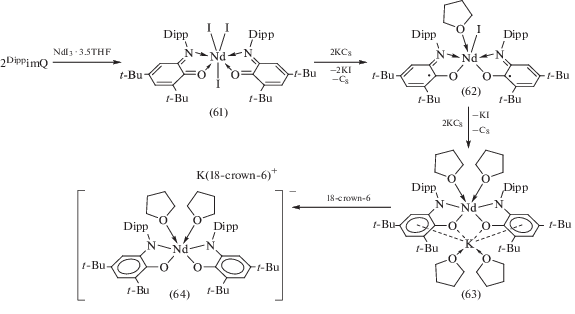
Схема 31 .
Комплекс (64) мгновенно окисляется элементарными серой и селеном до бис-о-иминобензосемихиноновых комплексов (65) и (66), соответственно, в которых атомы халькогена при координации атомом неодима образуют шестичленное кольцо (cхема 32). Эти реакции вызывают особый интерес ввиду того, что для редкоземельных элементов крайне нетипично участие в мультиэлектронных процессах. Комплекс (63) также реагирует с серой с образованием (65), в то же время его реакция с селеном не протекает по причине блокировки металлоцентра ионом калия, однако при добавлении к реакционной смеси краун-эфира образование (66) происходит мгновенно.
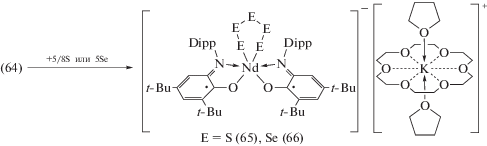
Схема 32 .
Комплексы актинидов с о-иминохиноновыми лигандами представлены соединениями урана и тория. Комплекс урана(IV) (67), содержащий о‑амидофенолятный лиганд DippAP, образуется из тетрабензилурана в результате последовательного восстановительного элиминирования бензильных радикалов под действием DippimQ [40]. Образование на первом этапе реакции моно о-иминосемихинонового интермедиата (68) было доказано при проведении эксперимента с U(CD2C6D5), однако при его целенаправленном синтезе из реакционной смеси был выделен комплекс (67) (cхема 33). Исходным соединением для целевого синтеза (68) служит (DippimSQ)UI3(THF)2 (69), образующийся в результате окислительного присоединения DippimQ к иодиду трехвалентного урана. При действии KC8 на (69) происходит восстановление лиганда (DippimSQ) и образуется о-амидофенолят урана(IV) (DippAP)UI2(THF)2 (70). Обменная реакция (70) с одним эквивалентом PhCH2K сопровождается образованием моноалкильного комплекса (DippAP)UI(CH2Ph)(THF)2 (71), тогда как попытка получить (67) по реакции обмена (70) с двумя эквивалентами PhCH2K приводит к разложению образующегося продукта.
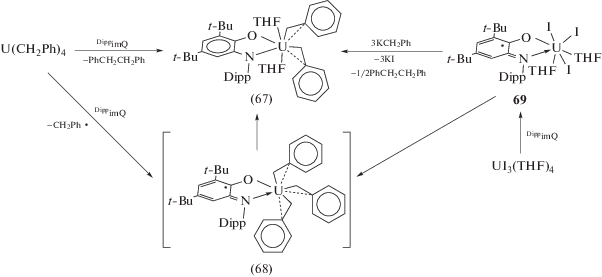
Схема 33 .
Обменная реакция межу N-трет-бутил-, N-адамантил- и N-dipp-замещенными о-амидофенолятами щелочных металлов и тетрахлоридом урана в соотношении 2 : 1 приводит к образованию бис-о-амидофенолятных комплексов урана(IV) (72)–(74) (схема 34 ) [41].
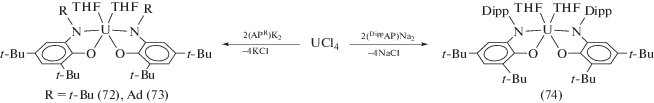
Схема 34 .
При действии на (72)–(74) эквивалента хлористого фенилиодония происходит окислительное присоединение хлора к исходным комплексам, и образуются бис-о-иминосемихиноновые производные (75)–(77) (схема 35 ). Бис-о-иминосемихиноновый комплекс (imSQt-Bu)2UI2(THF) (78) является продуктом окислительного присоединения иода к комплексу (72), тогда как реакция иода с комплексами (73), (74) приводит к их разложению.
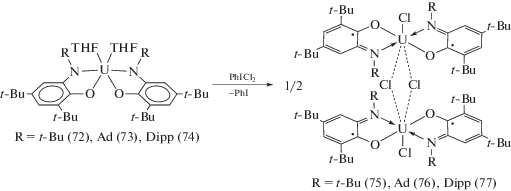
Схема 35 .
Первые биядерные комплексы актиноидов, в которых металлоцентры связаны посредством хинонового мостика, получены при взаимодействии дианионной формы 2,5-бис[2,6-ди-изо-пропил)анилид]-1,4-бензохинона и 2,5-бис[2-(метокси)анилид]-1,4-бензохинона с комплексами четырехвалентных урана и тория на основе триподального трис[2-амидо(2-пиридил)этил]амина (L) (схема 36 ) [42]. Наличие лиганда L в координационной сфере актиноида обуславливает кинетическую и термодинамическую стабильность образующихся биядерных комплексов (79)–(81). Комплексы тория (79, 80) диамагнитны, тогда как (81) содержит два магнитных центра U(IV). Однако, согласно данным магнетохимии, магнитное обменное взаимодействие между ними ничтожно слабое. Восстановленная форма (81) [K(18-c-6)(THF)2]+[81–] нестабильна и крайне чувствительна к кислороду и влаге воздуха, но данные РСА указывают, что она представляет собой новый тип комплекса UIVUIV, в котором металлоцентры связаны анион-радикальным пара-иминохиноновым мостиком.
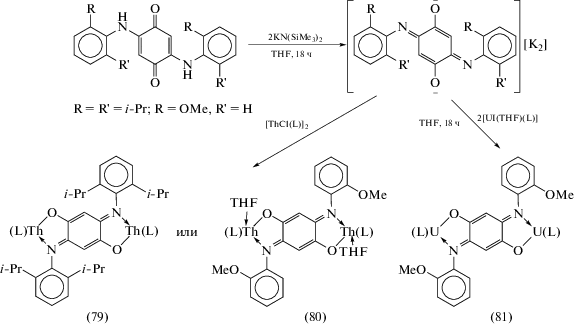
Схема 36 .
КОМПЛЕКСЫ МЕТАЛЛОВ III ГРУППЫ С ТРИДЕНТАТНЫМИ ЛИГАНДАМИ o-ИМИНОХИНОНОВОГО ТИПА
Комплексы элементов подгруппы бора. Диметильные производные металлов IIIA группы, содержащие тридентатный ONN-лиганд, образуются при взаимодействии енаминной формы N‑(1-(5,7-ди-трет-бутил-2-метил-2,3-дигидробензо[d]оксазол-2-ил)етилиден)-2,6-ди-изо-пропиланилина с Me3M (M = Al, Ga, In, Tl) (схема 37 ) [43]. Несмотря на наличие двух алкильных групп в координационной сфере металла, (82)–(85) в кристаллическом состоянии устойчивы по отношению к кислороду и влаге воздуха, однако в растворах различных растворителей в (82) наблюдается миграция одной из метильных групп от алюминия к группе C=N лиганда (схема 37 ). В отличие от производных алюминия и галлия, комплексы (84), (85) обладают интенсивной фотолюминесценцией при комнатной температуре, что делает их перспективными объектами для создания фотоактивных материалов.
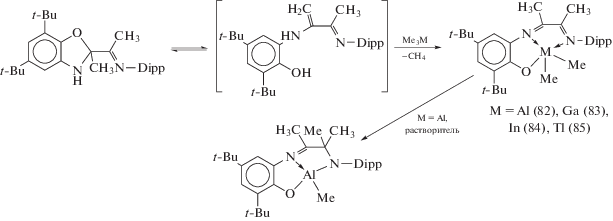
Схема 37 .
По реакции конденсации несимметричных амино-бис-фенолов с различными арилбороновыми кислотами получена богатая серия мономерных боронатов (86) (схема 38 ) [44]. Атом бора в (86) имеет тетраэдрическое окружение, образованное за счет ковалентного связывания с атомами кислорода тридентатного лиганда и донорно-акцепторной координации протонированного sp3-гибридизованного атома азота.
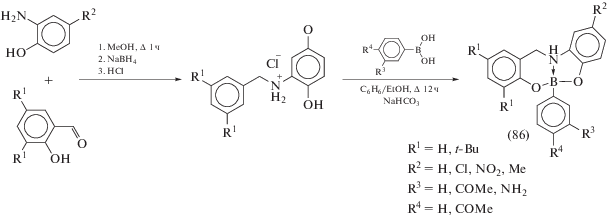
Схема 38 .
Аналогичное координационное окружение имеет атом бора в производных (87), полученных конденсацией незамещенных бис-феноламинов и фенилборной кислоты (схема 39 ) [45].
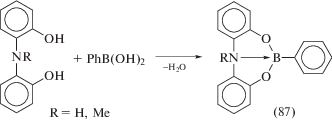
Схема 39 .
Большое количество комплексов металлов III группы получено на основе 3,5-ди-трет-бутил-1,2-хинон-1-(2-гидрокси-3,5-ди-трет-бутилфенил)имина (ONO). Указанный тридентатный лиганд содержит два кислородных атома в орто-положениях по отношению к атому азота. Будучи связанным в комплекс с металлом, он способен существовать в четырех различных редокс-состояниях (схема 40 ) [46]. Нейтральная (ONO0) и дважды восстановленная (ONO2) формы представляют собой радикалы, в то время как моно- (ONO1) и трианион (ONO3) диамагнитны.
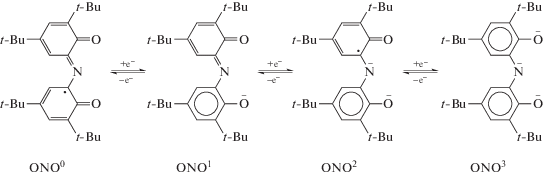
Схема 40 .
Геометрия координационного узла в комплексе бора (ON(H)O3)BCl (88), синтезированном по реакции трансметаллирования BCl3 цинкового комплекса Zn(ONO1)2 [47], аналогична соединениям (87) на основе незамещенных бис-феноламинов.
В связи с неустойчивостью свободного лиганда ONO основным методом получения металлокомплексов на его основе является темплатный синтез из 3,5-ди-трет-бутил-пирокатехина ((3,5-Cat)H2), аммиака и солей соответствующих металлов в спиртовой среде в присутствии кислорода воздуха. При взаимодействии (3,5-Cat)H2 c трихлоридами алюминия и галлия в мольном соотношении 4 : 1 в этаноле в присутствии кислорода воздуха образуются октаэдрические смешанолигандные комплексы состава (ONO1)(ONO2)MIII (M = Al (89), Ga (90)) [47]. Авторами [48] для синтеза смешанолигандного производного галлия (90) был использован электролиз раствора (3,5-Cat)H2 в жидком аммиаке с галлиевым анодом.
Диамагнитные комплексы таллия с общей формулой (ONO1)Tl(X)Ar (91) и (ONO1)TlAr2 (92), где X – галоген, Ar – фенил и его замещенные аналоги, также образуется в ходе темплатного синтеза при взаимодействии 3,5-ди-трет-бутил-аминофенола с моно- и диарильными комплексами таллия(III) (2 : 1) в среде полярных растворителей [49].
Серия комплексов алюминия, содержащих один лиганд ONO в различных степенях окисления, была синтезирована по реакции обмена галогенидов алюминия с солями щелочных металлов (ON(H)O3)M2 (M = Li, K) и (ONO1)K [50]. В ходе реакции галогенидов алюминия с (ON(H)O3)M2 (M = Li, K) при соотношении исходных компонентов 1 : 1 образуются пятикоординационные комплексы алюминия (93), (94), в которых атом металла находится в тригонально-бипирамидальном координационном окружении (cхема 41).
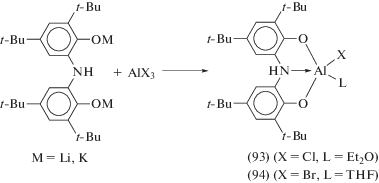
Схема 41 .
Взаимодействие AlCl3 c моноанионной солью (ONO1)K в присутствии стехиометрических количеств дифенилацетилацетоната $\left( {{\text{AcacPh}}_{{\text{2}}}^{--}} \right)$ либо 8-оксихинолина (QuinO–) приводит к образованию октаэдрических комплексов алюминия (95), (96), которые могут быть одноэлектронно восстановлены до монорадикальных производных (97), (98) (cхема 42).
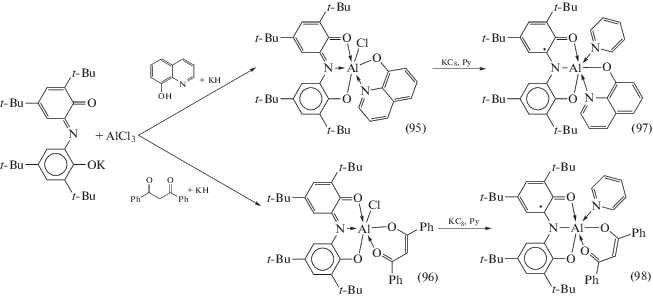
Схема 42 .
При окислении комплекса (93) различными о‑хинонами (Q) происходит вытеснение HCl и образуются комплексы алюминия с двумя редокс-активными лигандами различной природы (99)–(101) (cхема 43). Электронное строение (99)–(101) различно: в случае менее акцепторных о-фенантренхинона и пирен-4,5-диона образуются бирадикальные производные (ONO2)Al(SQ)(Py) (SQ = анион-радикал соответствующего о-хинона), в то время как анион-радикал сильно акцепторного тетрахлоро-о-хинона способен окислять лиганд ONO до моноаниона ONO1, в связи с чем электронное строение комплекса (99) следует рассматривать как совокупность двух форм – (ONO2)Al(SQ)(Py) и (ONO1)Al(Cat)(Py) (Cat = дианион о-хинона).
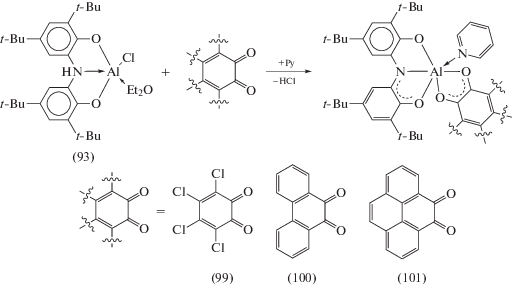
Схема 43 .
Биядерный дианионный комплекс галлия (102) был синтезирован при взаимодействии гексадентатного 1,2-бис(3,5-ди-трет-бутил-2-гидроксифенил)оксамида (H4(Bbpo)) с двумя эквивалентами хлорида галлия в присутствии аммонийной соли (cхема 44) [51]. Каждый атом галлия в (102) имеет искаженное тригонально-бипирамидальное окружение.
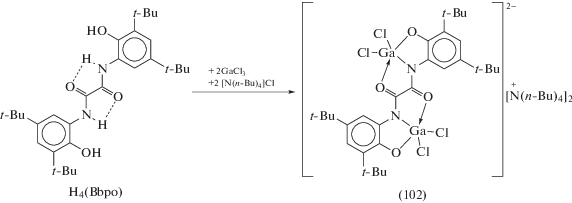
Схема 44 .
Электрохимические окисление диамагнитного дианиона (102) протекает обратимо в две одноэлектронные стадии с образованием моно- и бирадикальных производных соответственно (cхема 45).
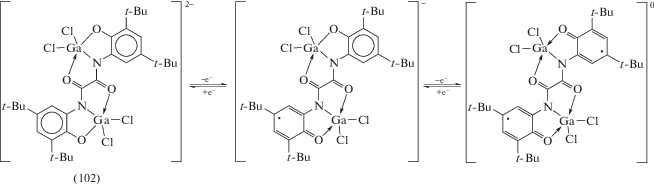
Схема 45 .
Комплексы редкоземельных металлов и актинидов. Трис-лигандные о-иминобензохиноновые комплексы редкоземельных элементов (ONO1)3MIII (M = Sc, Y, La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu) впервые были получены темплатным синтезом с использованием в качестве исходных соединений 3,5-ди-трет-бутил-пирокатехина и хлоридов или нитратов соответствующих металлов в присутствии аммиака в водно-спиртовой среде [52].
Комплексы (ONO1)3MIII (M = La (103), Sm (104), Yb (105)) также могут быть получены при взаимодействии бис-(2-гидрокси-3,5-ди-трет-бутилфенил)амина (ONO3H3) с гидратированными хлоридами металлов в присутствии амина и по реакции 3,5-ди-трет-бутил-1,2-хинон-1-(2-гидрокси-3,5-ди-трет-бутилфенил)имина (ONO1H) с силиламидами тех же металлов (3 : 1) [53]. Согласно РСА атом металла в (ONO1)3M (M = La (103) [53], Sm (104) [54]) находится в трехвалентном состоянии и координирован тремя эквивалентными лигандами ONO1, КЧ металла равно 9, а координационный полиэдр представляет собой трехшапочную тригональную призму. Комплексы (103, 104), а также комплекс алюминия (ONO1)(ONO2)Al (89) послужили основной для построения фотовольтаических ячеек, при этом наилучшие характеристики были зарегистрированы для полупроводниковых устройств с фотоактивным слоем на основе соединения алюминия [53].
Серия девятикоординационных трис-хелатных комплексов лантанидов(III) (106) синтезирована на основе восстановленной формы тридентатного 2,4,6,8-тетракис(трет-бутил)-9-гидроксифеноксазин-1-онового лиганда ((ONO')H3) по реакции с ацетатами (OAc)3Ln (Ln = La, Pr, Sm, Eu, Gd, Tb, Dy, Yb) (схема 46 ) [55, 56]. Координационный полиэдр в комплексах (ONO')3Ln (Ln = La, Pr, Sm, Eu, Gd, Tb, Dy) как и в комплексах (ONO)3Ln (Ln = La (103), Sm (104)) представляет собой трехшапочную тригональную призму, образованную шестью атомами кислорода и тремя атомами азота депротонированного лиганда ONO'.
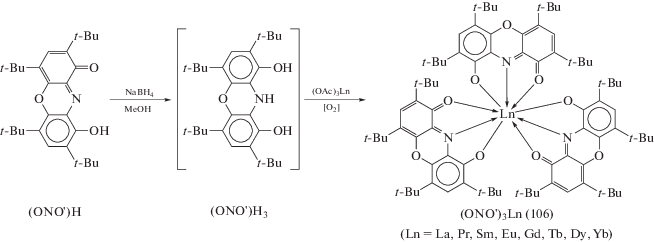
Схема 46 .
Комплекс урана (107), координированный лигандом ONO' в рассмотренной выше моноанионной форме, образуется при взаимодействии (ONO')H с бис-триметилсилиламидом уранила (схема 47 ) [57]. Возможность получения комплекса, координированного наиболее окисленной формой лиганда в данном случае обусловлена использованием в качестве стартового соединения производного урана(VI), что исключает протекание внутримолекулярного редокс-процесса, приводящего к восстановлению редокс-активного лиганда.
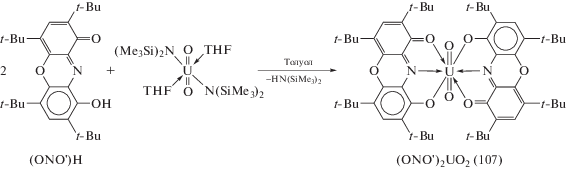
Схема 47 .
В то же время при взаимодействии иодида урана(III) с монокалиевой солью (ONO')K, как и в случае образования о-иминохинонового комплекса (69), происходит перенос электрона с металла на лиганд и образуется комплекс урана(IV) (108), координированный лигандом в дважды восстановленной форме (схема 48 ). Проведение данной реакции в присутствии эквивалента KC8 приводит к восстановлению редокс-активного лиганда и образующийся комплекс (109) содержит ONO' в трианионном состоянии. Соединение (109) также генерируется при непосредственном восстановлении (108) карбидом калия (схема 48 ).
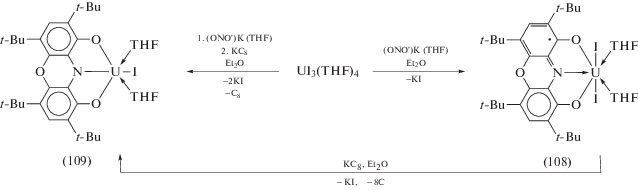
Схема 48 .
Пентаметилциклопентадиенильный аналог (109) – (ONO')UIV(Cp*)(THF)2 (110) – синтезирован по аналогичной схеме с использованием в качестве исходного соединения урана комплекс (Cp*)UIIII2(THF)3, генерируемый in situ из UI3(THF)4 и K(Cp*).
КОМПЛЕКСЫ МЕТАЛЛОВ III ГРУППЫ С ТЕТРАДЕНТАТНЫМИ ЛИГАНДАМИ о-ИМИНОХИНОНОВОГО ТИПА
Химия комплексов металлов III группы с тетрадентатными о-иминохиноновыми лигандами развита в гораздо меньшей степени, чем для элементов подгруппы кремния [14]. Серия шестикоординационных комплексов алюминия (111)–(114), стабилизированных фосфиноксидными донорными лигандами, синтезирована при взаимодействии потенциальнo тетрадентатных ${{{\text{N}}}_{{\text{2}}}}{\text{O}}_{{\text{2}}}^{{3 - }}$ формазанатных лигандов (FormR) с Al(OiPr)3 в присутствии двух эквивалентов фосфиноксидов различной природы (схема 49 ) [58]. Атом алюминия в (111)–(114) находится в октаэдрическом окружении, сформированном O,N,N,O-атомами формазанатного лиганда в экваториальных положениях и двумя фосфиноксидными донорами в аксиальных положениях. Комплекс (112) в комбинации с (n-Pr)3N является электролюминесцентным эмиттером с максимумом интенсивности электролюминесценции при 735 нм, что указывает на потенциальную возможность использования шестикоординационных формазанатных комплексов алюминия в качестве функциональных материалов.
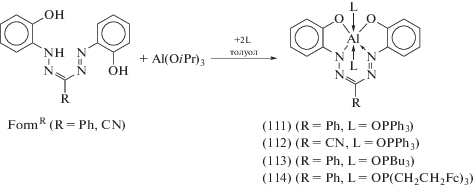
Схема 49 .
Взаимодействие FormCN с BF3 · OEt2 в присутствии триэтиламина не приводит к образованию четырехкоординационного комплекса бора, координированного O,N,N,O-атомами формазанатного лиганда, как в случае производных алюминия (111)–(114), поскольку в данном случае потенциально способные сформироваться пятичленные хелатные циклы были бы слишком напряженными, вместо этого в ходе реакции образуется смесь из менее стерически загруженных соединений (115)–(119) (схема 50 ) [59].
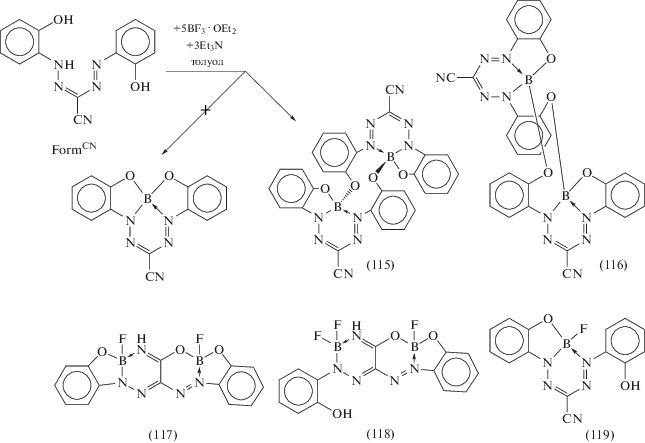
Схема 50 .
Тетрадентатный ONNO-лиганд, образующийся в ходе конденсации 3,5-ди-трет-бутилпирокатехина и этилендиамина, реагирует с одним эквивалентом AlEt3 в тетрагидрофуране с образованием соответствующего этильного комплекса алюминия (120), в то время как добавление неполярного растворителя (пентана) к реакционной смеси сопровождается образованием димера (121) (схема 51 ) [60]. Геометрия координационного окружения каждого пентакоординированного металлоцентра в (121) представляет собой практически идеальную квадратную пирамиду, основание которой образовано планарной координацией тетрадентатного ONNO-лиганда, а мостиковый атом азота занимает апикальное положение. Димер (121) при комнатной температуре реагирует с двумя эквивалентами бензилового спирта с образованием алкоксидного производного (122). Комплексы (120)–(122) при комнатной температуре проявляют высокую каталитическую активность в полимеризации rac-лактида с раскрытием цикла, причем каталитическая система может генерироваться in situ из ONNO и AlEt3 в THF при последующем (спустя 15 мин) добавлении смеси бензиловый спирт–rac-лактид (1 : 100).
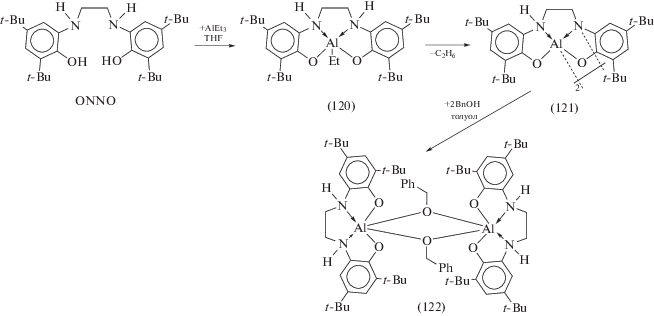
Схема 51 .
При взаимодействии тетрадентатного N,N'-бис-(иминофенол)аценафтена (Phen-BIAN) с UO2(OAc)2 в спиртовой среде образуется диамагнитный димерный комплекс (123), содержащий два уранильных металлоцентра, тетрадентатно координированных лигандом Phen-BIAN и связанных оксидными мостиками, в результате чего каждый UO2-центр находится в пентагонально-бипирамидальном координационном окружении (схема 52 ) [61]. Электрохимическое восстановление комплекса указывает на широкий диапазон достижимых окислительных состояний урана за счет образования смешановалентных производных U(VI)/U(V) и U(V)/U(IV) в растворе.
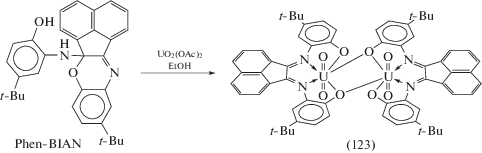
Схема 52 .
В заключение необходимо отметить, что область химии, затрагивающая изучение комплексов металлов с различными редокс-активными лигандами интенсивно развивается в последние годы и подключает к себе все большее число объектов. Синтезируются новые типы органических лигандов, способных изменять свою степень окисления в координационной сфере металла, направленно модифицируются уже известные редокс-активные системы. о-Иминохиноновые лиганды обладают большим потенциалом для такой модификации, что достигается путем введения различных функциональных заместителей в азотсодержащий фрагмент [62–74]. Другой перспективной ветвью развития данного направления химии стали первые работы по исследованию о-иминохиноновых комплексов редкоземельных элементов и актинидов [37–42, 53, 55–57, 61].
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Tezgerevska T., Alley K.G., Boskovic C. // Coord. Chem. Rev. 2014. V. 268. P. 23.
Broere D.L.J., Plessius R., van der Vlugt J.I. // Chem. Soc. Rev. 2015. V. 44. P. 6886.
Jacquet J., Cheaib K., Ren Y. et al. // Chem. Eur. J. 2017. V. 23. P. 1530.
Kaim W., Paretzki A. // Coord. Chem. Rev. 2017. V. 344. P. 345.
Abakumov G.A., Piskunov A.V., Cherkasov V.K. et al. // Russ. Chem. Rev. 2018. V. 87. P. 393.
Van der Vlugt J.I. // Chem. Eur. J. 2019. V. 25. P. 2651.
Broere D.L.J., van Leest N.P., de Bruin B. et al. // Inorg. Chem. 2016. V. 55. P. 8603.
Bagh B., Broere D.L.J., Sinha V. et al. // J. Am. Chem. Soc. 2017. V. 139. P. 5117.
Paul G.C., Ghorai S., Mukherjee C. // Chem. Commun. 2017. V. 53. P. 8022.
Leconte N., d’Hardemare A.M., Philouze C., Thomas F. // Chem. Commun. 2018. V. 54. P. 8241.
Yakub A.M., Moskalev M.V., Bazyakina N.L., Fedushkin I.L. // Russ. Chem. Bull. 2018. V. 67. P. 473.
Zhang W., Dodonov V.A., Chen W. et al. // Chem. Eur. J. 2018. V. 24. P. 14994.
Dodonov V.A., Chen W., Zhao Y. et al. // Chem. Eur. J. 2019. V. 25. P. 8259.
Чегерев М.Г., Пискунов А.В. // Коорд. химия. 2018. Т. 44. С. 109. (Chegerev M.G., Piskunov A.V. // Russ. J. Coord. Chem. 2018. V. 44. P. 258. https://doi.org/10.1134/S1070328418040036).
Kimmich B.F.M., Landis C.R., Powell D.R. // Organometallics. 1996. V. 15. P. 4141.
Barba V., Rodriguez A., Ochoa M.E. et al. // Inorg. Chim. Acta. 2004. V. 357. P. 2593.
Alibadi M.A.M., Batsanov A.S., Bramham G. et al. // Dalton Trans. 2009. P. 5348.
Gracey G.D., Rettig S.J., Storr A., Trotter J. // Can. J. Chem. 1987. V. 65. P. 2469.
Piskunov A.V., Maleeva A.V., Mescheryakova I.N., Fukin G.K. // Eur. J. Inorg. Chem. 2012. V. 2012. P. 4318.
Piskunov A.V., Mescheryakova I.N., Ershova I.V., Fukin G.K. // Inorg. Chem. Commun. 2012. V. 24. P. 227.
Piskunov A.V., Ershova I.V., Fukin G.K., Shavyrin A.S. // Inorg. Chem. Commun. 2013. V. 38. P. 127.
Piskunov A.V., Mescheryakova I.N., Bogomyakov A.S. et al. // Inorg. Chem. Commun. 2009. V. 12. P. 1067.
Piskunov A.V., Mescheryakova I.N., Fukin G.K. et al. // New J. Chem. 2010. V. 34. P. 1746.
Piskunov A.V., Meshcheryakova I.N., Fukin G.K. et al. // Dalton Trans. 2013. V. 42. P. 10533.
Пискунов А.В., Ершова И.В., Фукин Г.К. // Изв. АН. Сер. хим. 2014. № 4. С. 916.
Piskunov A.V., Meshcheryakova I.N., Ershova I.V. et al. // RSC Adv. 2014. V. 4. P. 42494.
Ershova I.V., Bogomyakov A.S., Fukin G.K., Piskunov A.V. // Eur. J. Inorg. Chem. 2019. V. 2019. P. 938.
Piskunov A.V., Ershova I.V., Bogomyakov A.S. et al. // Inorg. Chem. 2015. V. 54. P. 6090.
Piskunov A.V., Ershova I.V., Bogomyakov A.S., Fukin G.K. // Inorg. Chem. Commun. 2016. V. 66. P. 94.
Chaudhuri P., Wagner R., Pieper U. et al. // Dalton Trans. 2008. P. 1286.
Chaudhuri P., Bill E., Wagner R. et al. // Inorg. Chem. 2008. V. 47. P. 5549.
Bamford K.L., Longobardi L.E., Liu L. et al. // Dalton Trans. 2017. V. 46. P. 5308.
Gao B., Su Q., Gao W., Mu Y. // Acta Crystallogr. E. 2011. V. 67. P. m1374.
Razborov D.A., Lukoyanov A.N., Makarov V.M. et al. // Russ. Chem. Bull. 2015. V. 64. P. 2377.
Razborov D.A., Lukoyanov A.N., Moskalev M.V. et al. // Russ. J. Coord. Chem. 2018. V. 44. P. 380. https://doi.org/10.1134/S1070328418060040
Poddel’sky A.I., Abakumov G.A., Bubnov M.P. et al. // Russ. Chem. Bull. Int. Ed. 2004. V. 53. P. 1189.
Bochkarev M.N., Fagin A.A., Druzhkov N.O. et al. // J. Organomet. Chem. 2010. V. 695. P. 2774.
Klementyeva S.V., Lukoyanov A.N., Afonin M.Yu. et al. // Dalton Trans. 2019. V. 48. P. 3338.
Coughlin E.J., Zeller M., Bart S.C. // Angew. Chem. Int. Ed. 2017. V. 56. P. 12142.
Matson E.M., Franke S.M., Anderson N.H. et al. // Organometallics. 2014. V. 33. P. 1964.
Matson E.M., Opperwall S.R., Fanwick P.E., Bart S.C. // Inorg. Chem. 2013. V. 52. P. 7295.
Hohloch S., Pankhurst J.R., Jaekel E.E. et al. // Dalton Trans. 2017. V. 46. P. 11615.
Egorova E.N., Druzhkov N.O., Shavyrin A.S. et al. // RSC Adv. 2015. V. 5. P. 19362.
Abreu A., Alas S.J., Beltran H.I. et al. // J. Organomet. Chem. 2006. V. 691. P. 337.
Farfan N., Joseph-Nathan P., Chiquete L.M., Contreras R. // J. Organomet. Chem. 1988. V. 348. P. 149.
Chaudhuri P., Hess M., Hildenbrand K. et al. // Inorg. Chem. 1999. V. 38. P. 2781.
Camacho-Camacho C., Merino G., Martinez-Martinez F.J. et al. // Eur. J. Inorg. Chem. 1999. P. 1021.
Brown M.A., Castro J.A., McGarvey B.R., Tuck D.G. // Can. J. Chem. 1999. V. 77. P. 502.
Stegmann H.B., Ulmschneider K.B., Scheffler K. // J. Organomet. Chem. 1974. V. 72. P. 41.
Szigethy G., Heyduk A.F. // Dalton Trans. 2012. V. 41. P. 8144.
Beckmann U., Bill E., Weyhermüller T., Wieghardt K. // Eur. J. Inorg. Chem. 2003. V. 2003. P. 1768.
Lyubchenko S.N., Kogan V.A., Olekhnovich L.P. // Russ. J. Coord. Chem. 1996. V. 22. P. 534.
Малеев А.А., Трофимова О.Ю., Пушкарев А.П. и др. // Росс. нанотехнологии. 2015. Т. 10. № 7–8. С. 88.
Furmanova N.G., Lyubchenko S.N., Kogan V.A., Olekhovich L.P. // Crystallogr. Rep. 2000. V. 45. P. 439.
Ivakhnenko E.P., Simakov V.I., Knyazev P.A. et al. // Mendeleev Commun. 2016. V. 26. P. 49.
Ivakhnenko E.P., Romanenko G.V., Simakov V.I. et al. // Inorg. Chim. Acta. 2017. V. 458. P. 116.
Pattenaude S.A., Kuehner C.S., Dorfner W.L. et al. // Inorg. Chem. 2015. V. 54. P. 6520.
Maar R.R., Kenaree A.R., Zhang R. et al. // Inorg. Chem. 2017. V. 56. P. 12436.
Barbon S.M., Staroverov V.N., Gilroy J.B. // Angew. Chem. Int. Ed. 2017. V. 56. P. 8173.
Gesslbauer S., Cheek H., White A.J.P., Romain C. // Dalton Trans. 2018. V. 47. P. 10410.
Niklas J.E., Farnum B.H., Gorden J.D., Gorden A.E.V. // Organometallics. 2017. V. 36. P. 4626.
Broere D.L.J., Metz L.L., de Bruin B. et al. // Angew. Chem. Int. Ed. 2015. V. 54. P. 1516.
Paretzki A., Hübner R., Ye S. et al. // J. Mater. Chem. C. 2015. V. 3. P. 4801.
Piskunov A.V., Ershova I.V., Gulenova M.V. et al. // Russ. Chem. Bull. 2015. V. 64. P. 642.
Ali A., Dhar D., Barman S.K. et al. // Inorg. Chem. 2016. V. 55. P. 5759.
Broere D.L.J., Plessius R., Tory J. et al. // Chem. Eur. J. 2016. V. 22. P. 13965.
Maity S., Kundu S., Bera S. et al. // Eur. J. Inorg. Chem. 2016. V. 2016. P. 3691.
Bubrin M., Paretzki A., Hübner R. et al. // Z. Anorg. Allg. Chem. 2017. V. 643. P. 1621.
Maity S., Kundu S., Mondal S. et al. // Inorg. Chem. 2017. V. 56. P. 3363.
Paul G.C., Banerjee S., Mukherjee C. // Inorg. Chem. 2017. V. 56. P. 729.
Safaei E., Bahrami H., Wojtczak A. et al. // Polyhedron. 2017. V. 122. P. 219.
Piskunov A.V., Pashanova K.I., Bogomyakov A.S. et al. // Dalton Trans. 2018. V. 47. P. 15049.
Ershova I.V., Smolyaninov I.V., Bogomyakov A.S. et al. // Dalton Trans. 2019. V. 48. P. 10723.
Пискунов А.В., Пашанова К.И., Ершова И.В. и др. // Изв. АН. Сер. хим. 2019. № 4. С. 757.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия