

Координационная химия, 2020, T. 46, № 3, стр. 157-171
N-гетероаналоги 9,10-фенантренхинона – иминохиноны и диимины в координационной химии
Г. Г. Казаков 1, 2, *, Н. О. Дружков 1, В. К. Черкасов 1, 2
1 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
2 Нижегородский государственный университет им. Н.И. Лобачевского
Нижний Новгород, Россия
* E-mail: gkazakov@iomc.ras.ru
Поступила в редакцию 09.10.2019
После доработки 18.10.2019
Принята к публикации 31.10.2019
Аннотация
Обсуждаются особенности синтеза N-гетероаналогов 9,10-фенантренхинона. Рассмотрены особенности их строения. Обобщены сведения об использовании этих соединений в координационной химии как нейтральных, моно- и дианионных лигандов.
В последние десятилетия в координационной химии проявляется большой интерес к лигандам переменной валентности, так называемым редокс-активным лигандам. Такие лиганды способны обратимо принимать один или два электрона с образованием анион-радикальной или дианионной формы, находясь в координационной сфере металла. Основными представителями данного класса лигандов являются о-хиноны, о-иминохиноны, α-дитиолены и α-диимины [1]. Наличие подобных лигандов в составе металлокомплекса может оказывать существенное влияние на их реакционную способность. Один из интенсивно изучаемых редокс-активных лигандов – 1,2-бис(имино)аценафтен (BIAN). При использовании этого лиганда удалось получить соединения непереходных металлов со связью металл–металл, способных обратимо присоединять алкины [2], подобно комплексам переходных металлов. В случае магниевого комплекса с моно(имино)аценафтенхиноном такое присоединениe оказывается необратимым [3]. На примере дигаллана на основе бис(имино)аценафтена была показана активация малых молекул [4] и присоединения гетерокумуленов [5, 6].
Комплексы BIAN с переходными металлами широко известны и зарекомендовали себя эффективными катализаторами циклоизомеризации [7], полимеризации алкенов [8, 9], сополимеризации CO и стирола [10], CO2 и метиленциклопропена [11], гидрирования алкинов [12].
Особенностью ближайших структурных аналогов BIAN – 9,10-фенантрен-моно- и дииминов, является сочетание конденсированной ароматической системы фенантрена и возможности регулирования стерических и электронных эффектов путем изменения заместителей при атомах азота или введения дополнительных групп в периферические части молекулы. Такая вариативность позволяет сделать предположение об их потенциальной способности образовывать широкий спектр соединений с различными элементами, где диимин будет находиться в нейтральном, анион-радикальном или дианионном состоянии.
ФЕНАНТРЕНХИНОНИМИНЫ
В координационной химии фенантренхинон используется как нейтральный, анион-радикальный и дианионный лиганды [13, 14]. Однако получение и использование моно- и дииминовых производных 9,10-фенантренхинона, содержащих заместители при атоме азота, было недостаточно изучено до недавнего времени.
Первое упоминание фенантренхинонимина приходится на конец XIX в. [15]. Позднее он нашел применение как прекурсор фенантрендиамина [16] и только в конце XX в. появилась первая работа, посвященная изучению хинониминовых комплексов меди(I) и их окисления кислородом [17].
В начале XXI в. был получен фенантренхинонимин, содержащий заместители при атомах азота, по реакции 1,2-нуклеофильного присоединения амина к карбонильной группе о-хинона получили замещенные о-иминофенантренхиноны [18]. В качестве амина использовали о-толуидин, 2,6-диметиланилин и 2,6-ди-изо-пропиланилин. Реакции проводили в метаноле при нагревании в присутствии каталитических количеств муравьиной кислоты. Выделить продукты в препаративных количествах удалось лишь в случае стерически загруженных дизамещенных анилинов.
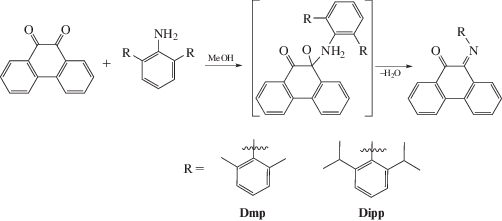
Позднее появилось сообщение группы авторов [19] о синтезе фенантренхинониминов, содержащих 2,6-ди-изо-пропилфенильный заместитель с атомом галогена в пара-положении, синтез которых осуществлен по такой же методике. Описанные соединения были выбраны в качестве модели для изучения нековалентных взаимодействий неподеленных электронных пар галогенов и атомов водорода с сопряженными системами.
В результате попыток заменить второй атом кислорода повышением жесткости условий реакции в [20] наряду с хинонимином получен продукт взаимодействия изо-пропильной группы в заместителе при атоме азота с фенантреновой частью.
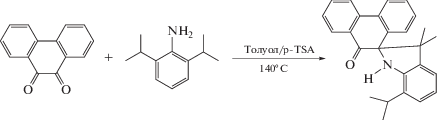
Таким образом, получить фенантрендиимины простой конденсацией в присутствии кислот Бренстеда оказалось невозможно.
ФЕНАНТРЕНДИИМИНЫ
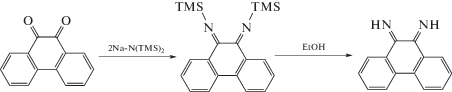
Впервые незамещенный 9,10-фенантрендиимин получил G. Tuchtenhagen в 1968 г. по реакции бис(триметилсилил)амида натрия с 9,10-фенантренхиноном [21]. Однако введение заместителей у атомов азота этим методом невозможно, что ограничивает применение этих дииминов в координационной химии.
Замещенный фенантрендиимин удалось получить в 1971 г. путем восстановительного циклодегидрирования N,N'-(1,2-дифенилэтан-1,2-диилиден)дианилина [22]:
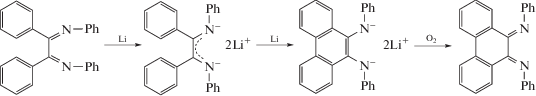
Позднее в [23] было предложено несколько новых методов получения α-дииминовых производных фенантренхинона: из 2-гидрокси-1,2-дифенилэтанона и из дибензоила.
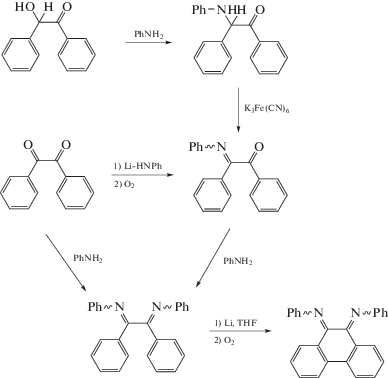
Взаимодействие иминокетона с другим анилином дает возможность получить 9,10-фенантрендиимины с различными заместителями при атомах азота. Реакция дибензоила с избытком анилина приводит к диимину только в случае анилинов с небольшими заместителями в ароматическом кольце: толил-, метоксифенил- или 2-изо-пропиланилин. Получение фенантрендииминов с алкильными заместителями по данной методике вообще невозможно.
Кроме этого, в [23] была обнаружена необычность строения N,N'-дифенил-9,10-фенантренди-имина, имеющего Z,Z-конфигурацию в твердом состоянии и в растворе, что несвойственно дииминам с жесткой структурой. На рис. 1 представлено его молекулярное строение в двух разных проекциях.
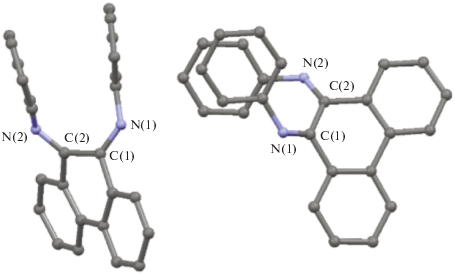
В случае более стерически затрудненного заместителя – о-изо-пропилфенила – конфигурация меняется на Z,E, что авторы обнаружили по наличию сигналов от неэквивалентных протонов изо-пропильных групп в спектре ЯМР.
Синтез нового N,N'-дизамещенного 9,10-фенантрендиимина был описан в [24] по реакции 9,10-фенантренхинона с 2,6-диметиланилином в присутствии системы TiCl4/1,4-диазабицикло[2.2.2]октан (DABCO) при 140°С с выходом продукта 50%.
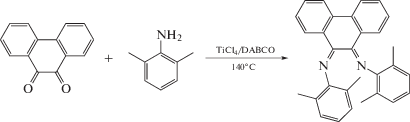
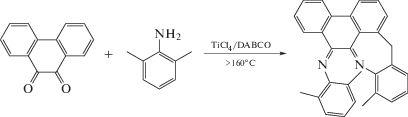
Эта реакция чувствительна к температуре и при проведении ее с повышением температуры до 160°С образуется продукт взаимодействия метильной группы заместителя в Е-кофигурации и фенантреновой части с выходом до 35%.
В 2011 г. в лаборатории химии элементоорганических соединений ИМХ РАН модифицировали методику получения дииминов в присутствии тетрахлорида титана. Так, применяя шестикратный избыток первичных аминов в присутствии 2 экв. TiCl4 в толуоле в более мягких условиях, при температуре от комнатной до 70°С, получили фенантрендиимины с заместителями при атомах азота различными по природе и электронным свойствам [25].
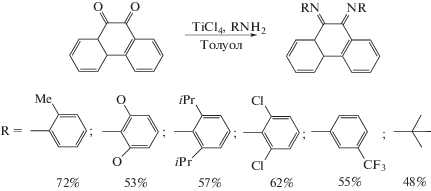
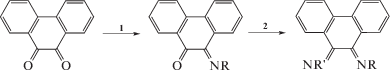
На основании того, что взаимодействие 9,10-фенантренхинона с первичными аминами в кипящем метаноле в присутствии муравьиной кислоты останавливается на образовании фенантренхинонимина, авторы предложили методику получения N,N'-дизамещенных 9,10-фенантрендииминов с различными заместителями при атомах азота в две стадии: 1) взаимодействие 9,10-фенантренхинона с избытком 2,6-диизопропиланилина в метаноле в присутствии муравьиной кислоты с образованием хинонимина. Выход 85%; 2) реакция 10-((2,6-ди-изо-пропилфенил)имино)фенантрен-9(10Н)-она в толуоле с ароматическим или алифатическим амином (например, с трет-бутиламином) и TiCl4 при комнатной температуре с образованием ди-имина. Выход 75% (для t-BuNH2) и 70% (для 2,6-диметиланилина).
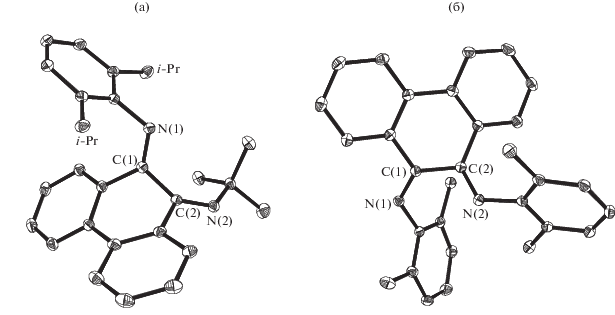
Методом РСА авторы обнаружили, что в кристаллическом состоянии диимины с Dmp, Dipp и t‑Bu-заместителями, а также диимин с алкильным и арильным заместителем, находятся в Z,E-форме, причем иминная группа с арильным заместителем находится в Е-форме, а с алкильной – в Z-форме (рис. 2).
Рис. 2.
Молекулярное строение фенантрендииминов со стерически затрудненными заместителями: N-трет-бутил-N'-(2,6-ди-изо-прoпилфенил) (а), N,N'-бис(2,6-диметилфенил) – замещенные диимины (б).
Также было обнаружено, что N,N'-ди-трет-бутил-9,10-фенантрендиимин сохраняет Z,E-конфигурацию в растворе, о чем свидетельствуют сигналы от неэквивалентных трет-бутильных групп в спектре ЯМР. Аналогично ведут себя фенантрендиимины со стерически затрудненными арильными заместителями.
В литературе имеется один пример [26] получения фенантрендиимина без использования тетрахлорида титана. Применяемый метод заключается в кипячении этанольных растворов п-нитроанилина и фенантренхинона в присутствии каталитических количеств ледяной уксусной кислоты. Одновременно с исследованиями, проводимыми в ИМХ РАН, в 2012 г. появилось сообщение группы китайских ученых [27], посвященное синтезу N‑гетероаналогов 9,10-фенантренхинона, также основанное на получении фенантрендииминов как из фенантренхинона, так и из фенантренхинониминов с применением тетрахлорида титана. Основное отличие этой методики заключается в использовании DABCO и более высокой температуры (140°С).
СВОЙСТВА N-ГЕТЕРОАНАЛОГОВ 9,10-ФЕНАНТРЕНХИНОНА
Свойства фенантренхинониминов. Комплексы переходных металлов с хинонимином представляют интерес в качестве катализаторов полимеризации и различных органических реакций. Роль фенантренхинонимина в этих случаях заключается в стабилизации различных состояний металлов благодаря возможности принимать несколько восстановленных состояний.
Известно, что восстановление о-иминобензохинонов и бензохинонов металлическим калием идет в две стадии и приводит к дианионной форме через стадию образования устойчивого анион-радикала. Авторами [18] было обнаружено, что фенантренхинонимины при восстановлении щелочными металлами ведут себя аналогично.
В спектре ЭПР анион-радикальной формы фенантренхинонимина наблюдается триплет (1 : 1 : 1) от сверхтонкого взаимодействия неспаренного электрона с атомом азота и мультиплет от расщепления на протонах в фенантреновой части (aN = 6.6 Э, gi = 2.0041).
Комплексы на основе фенантренхинониминов можно получать реакциями окислительного присоединения к нульвалентными комплексам переходных металлов. При взаимодействии тетракарбонила никеля с 2,6-ди-изо-пропил замещенным фенантренхинонимином образуется анион-радикальный комплекс [18].
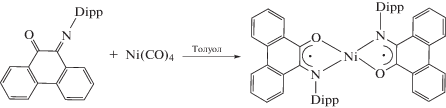
При использовании Ni(COD)2 и нейтрального лиганда удалось получить дианионный комплекс [28].
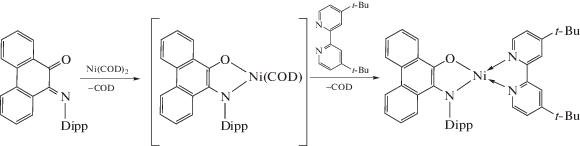
Металлы, такие как железо [29], магний [30], скандий и иттербий [31], способны восстанавливать фенантренхинонимины до анион-радикальной формы.
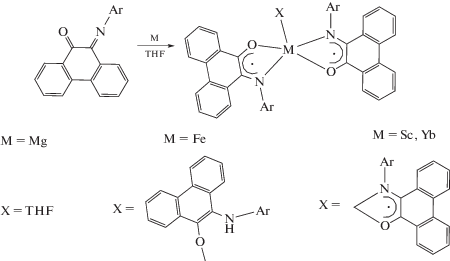
В случае солей железа [29] и хрома [27] комплексы возможно получать окислением M(II) до M(III).
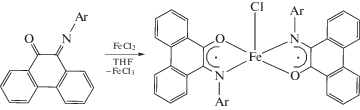
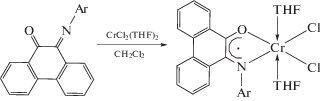
Однако при изменении растворителя на дихлорметан в случае железа окисление не происходит, вместо этого образуется комплекс с фенантренхинонимином в нейтральном состоянии [29].
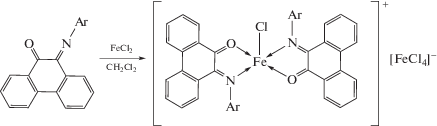
Похожим образом фенантренхинонимины взаимодействуют с солями кобальта(II) [32].
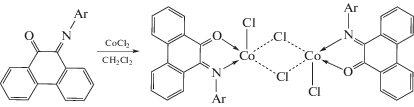
Анион-радикальные комплексы хрома и комплексы кобальта c нейтральным фенантренхинонимином показывают хорошую селективность при полимеризации диенов с образованием цис-1,4-продукта.
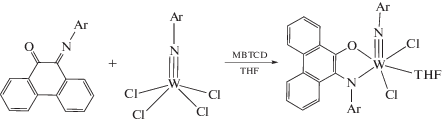
При восстановлении высоковалентного хлорида вольфрама в присутствии фенантренхинонимина с помощью 1-метил-3,6-бис(триметилсилил)-1,4-циклогексадиена (MBTCD) образуется дианионный комплекс. Замена атомов хлора на алкильные группы позволяет получить прекурсор катализатора метатезисной полимеризации с раскрытием цикла нонборнена, однако его активность невысока по сравнению с диазадиеновым аналогом из-за высокой термической стабильности [33].
Восстановление хинониминов иодидами диспрозия(II) и неодима(II) приводит к образованию анион-радикальных комплексов необычного строения. В них присутствуют два лиганда, нейтральный и одновосстановленный, связанные с металлом не хелатно, а посредством атомов кислорода [31].
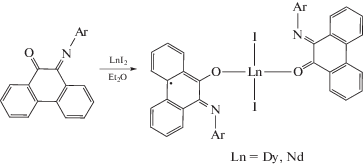
При восстановлении фенантренхинониминов гидридами осмия(II) [34] и рутения(II) [35] возможно получить анион-радикальные комплексы, в которых металл сохраняет изначальную степень окисления.
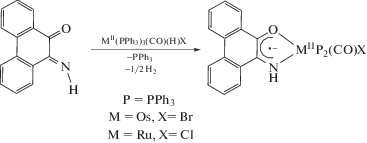
Интересную особенность проявляют анион-радикальные комплексы осмия(III). Взаимодействие таких соединений с галогенами (I2, Cl2, Br2) сопровождается изменением N,O-хелатирования на N,C и анион-радикального состояния лиганда [34].
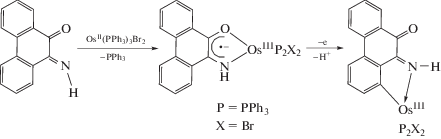
Подобные комплексы рутения такого свойства не проявляют, их окисление приводит к биядерному комплексу в случае иода или к комплексу с лигандом в нейтральной форме в случае брома [35].
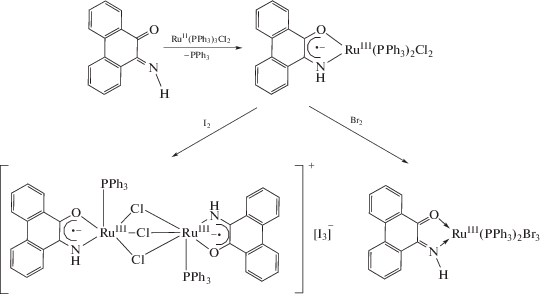
Другим прекурсором для получения анион-радикальных комплексов рутения может послужить ацетилацетонат рутения [36].
Особенностью всех вышеперечисленных соединений является высокая чувствительность к следам влаги и воздуха. Однако фенантренхинонимин можно использовать для получения стабильных на воздухе анион-радикальных соединений, что было продемонстрировано на примере борциклических радикалов [37].
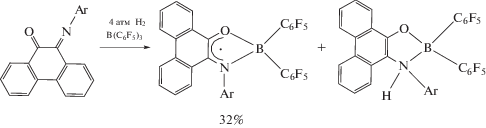
В литературе представлены разрозненные сообщения о комплексах фенантренхинониминов с переходными металлами (Ni, Cr, Co, Fe, Cu, Ru, Os), магнием и бором, где лиганд находится в нейтральной, анион-радикальной или дианионной форме. Основным состоянием фенантренхинониминов в комплексах можно считать одновосстановленную (анион-радикальную) форму. Анализ структурных данных позволяет заметить, что длины связей в хелатном цикле для всех форм лиганда лежат в узком диапазоне (табл. 1).
Таблица 1.
Длины связей в хелатном цикле для комплексов на основе фенантренхинониминов
| Форма | Связь, Å | ||
|---|---|---|---|
| С–С | С–N | C–O | |
| Нейтральная | 1.46–1.48 | 1.27–1.30 | 1.25–1.27 |
| Анион-радикальная | 1.42–1.45 | 1.30–1.35 | 1.27–1.29 |
| Дианионная | 1.38–1.39 | 1.40–1.42 | 1.30–1.34 |
Свойства фенантрендииминов. Структурные аналоги 9,10-фенантрендииминов, такие как о‑хиноны, иминохиноны, 1,4-диазадиены (DAD) и BIAN, образуют стабильные анион-радикальные комплексы при взаимодействии со щелочными металлами, которые легко удается зафиксировать с помощью ЭПР. Однако, несмотря на родственность фенантрендииминов этим соединениям, анион-радикальная форма лиганда при восстановлении калием методом ЭПР не регистрируется. Для оценки возможности фенантрендииминовых лигандов участвовать в редокс-превращениях методом ЦВА авторами [38] были изучены их электрохимические свойства. Было проведено восстановление ряда фенантрендииминов, протекающее в этом случае в одну необратимую двухэлектронную стадию с образованием дианиона.
Волны, соответствующей одноэлектронному восстановлению нейтральных дииминов до их анион-радикальной формы, на кривой ЦВА не наблюдалось, что говорит о малой устойчивости свободной анион-радикальной формы в растворе. Было определенно, что анион-радикальная форма быстро диспропорционирует на дианион и исходный диимин.
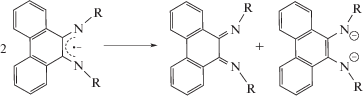
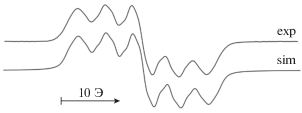
Несмотря на нестабильность анион-радикальной формы большинства фенантрендииминов, при восстановлении литием удалось получить их одноэлектронно-восстановленную форму. На спектрах ЭПР в этом случае наблюдается накопление сигнала анион-радикального производного фенантрендиимина, интенсивность которого в дальнейшем уменьшается вплоть до исчезновения (рис. 3).
Рис. 3.
Спектр ЭПР литиевого комплекса с N,N-бис(2,6-ди-изо-пропилфенил)-9,10-фенантрендиимином, gi = 2.0030, ${{a}_{{{\text{N1}}}}}$ = ${{a}_{{{\text{N2}}}}}$ = 4.5 Э, ${{a}_{{{\text{H1}}}}}$ = ${{a}_{{{\text{H2}}}}}$ = = 1.45 Э, ${{a}_{{{\text{H3}}}}}$ = ${{a}_{{{\text{H4}}}}}$ = 0.85 Э, aLi = 1.05 Э.
В случае алкил-алкильного и алкил-арильного фенантрендииминов, даже в случае восстановления литием, зафиксировать одноэлектронно-восстановленную форму методом ЭПР не удается.
Такое поведение фенантрендииминов авторы объясняют тем, что при восстановлении их калием образуется сольватно-разделенная электронная пара и получающийся анион радикал диспропорционирует. В реакции с литием получается контактная ионная пара, что стабилизирует образующийся анион-радикал.
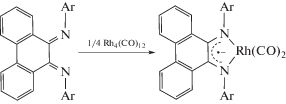
При переходе от ионно-построенной частицы к комплексу, где металл образует с анион-радикалом хелатный цикл, стабилизация анион-радикала возрастатает, что продемонстрировано в [38] в реакции с карбонилом родия и дихлоридом хрома [27]:
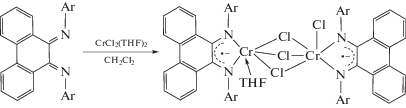
Интересны примеры анион-радикальных комплексов с незамещенным фенантрендиимином, где в качестве исходного соединения применяется фенантрендиамин. В литературе известны примеры получения двухпалубных комплексов кобальта [39] и многопалубных комплексов родия [40].
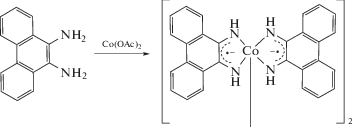
Несмотря на нестабильность анион-радикальной формы в растворе, координация на металл может стабилизировать это состояние лиганда. На данный момент в литературе имеются лишь приведенные выше соединения.
В применении фенантрендииминов как нейтральных лигандов для получения координационных соединений можно выделить два периода: работы с фенантрендиимином без заместителей при атомах азота и работы после разработки методов получения замещенных дииминов. На базе простого фенантрендиимина получены комплексы с переходными [41] и благородными металлами [36, 42–45]. Их синтез осуществлялся из диаминофенантрена или бис(триметилсилил)фенантрендиимина.
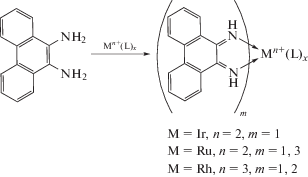
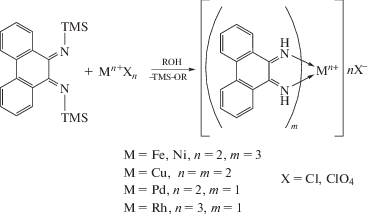
Однако в случае кобальта происходит образование необычного комплекса, где один лиганд образует амидную связь с металлом, меняющим свою степень окисления.
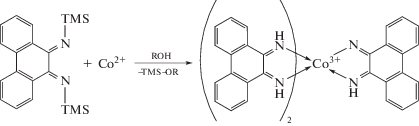
При построении металлокомплексов на базе замещенных фенантрендииминов применяется простое взаимодействие соли металла с исходным диимином. В литературе имеется несколько сообщений о производных переходных металлов (Zn [38, 46], Co [46], Cu [46], Ni [24, 46, 47]).
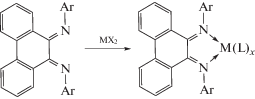
Комплексы NiBr2 c фенантрендииминами в нейтральной форме показали себя хорошими катализаторами полимеризации алкенов [24, 48–50].
Комплексы с благородными металлами освещены намного меньше – имеется лишь несколько сообщений одной группы исследователей [51, 52]. Обнаружено, что палладациклопентадиеновые комплексы, содержащие нейтральный фенантрендииминовый лиганд способны выступать в качестве катализаторов синтеза сопряженных диенов из алкинов.
Фенантрендиимины способны легко восстанавливаться до дианионной формы с помощью магния [30] или окислительного присоединения к диоксанату дихлорида германия(II) [38]. Обнаружено, что магниевый комплекс способен катализировать стереоселективную полимеризацию с раскрытием цикла рацемической смеси лактидов [53].
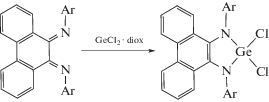
Известно, что структурный аналог 9,10-фенантрендииминов BIAN ведет себя в аналогичных условиях по-другому, образуя анион-радикальное производное [54]:
Диамидный комплекс титана(IV) можно получить взаимодействием диамина с тетрахлоридом титана в присутствии трет-бутиламина [55].
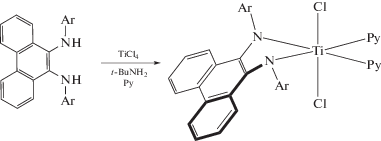
Искажение геометрии координационного узла авторы объясняют перекрытием неподеленных электронных пар азота со свободной dxy-орбиталью титана.
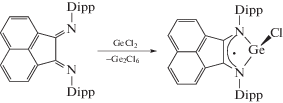
Благодаря сочетанию расширенной π-системы фенантрена и возможности регулирования стерической загруженности координационного узла фенантрендиимины представляют интерес для стабилизации низковалентных соединений элементов 13–15 групп, так называемых аналогов карбена Ардуэнго. В литературе имеются лишь несколько сообщений на данную тему. N-гетероциклический карбен был получен обработкой гидридом калия имидазолиевой соли [56].
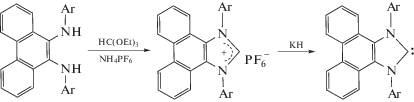
В случае необъемного заместителя при азоте (п-Tol) карбен выделяется в виде димера, однако уже в случае o-Tol возможно выделить мономерный продукт.
N-гетероциклические гермилены [56, 57] и станнилены [57] были получены двумя путями: взаимодействием дилитиевой соли диамида с дихлоридом металла или реакцией диамина с силиламидом металла в низкой степени окисления.
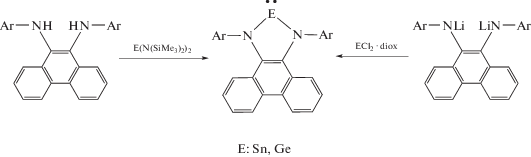
Попытка получения плюмбилена по реакции литиевой соли диимина с дииодидом свинца оказалась неудачной и привела к выделению металла.
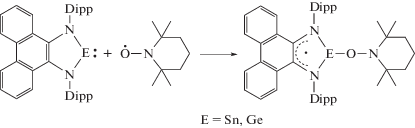
Интересная особенность N-гетероциклических аналогов карбена – способность вступать в окислительно-восстановительные превращения за счет изменения степени окисления лиганда. Известно, что металлокомплексы, содержащие дианионные лиганды на основе о-хинонов и α‑дииминов при действии на них стабильных радикалов превращаются в анион-радикальные производные, которые хорошо регистрируются методом ЭПР [58]. Для исследования возможности образования и устойчивости парамагнитных производных гермиленов и станниленов на основе N,N'-бис(2,6-ди-изо-пропилфенил)-9,10-фенантрендиимина авторами [57] были проведены реакции с кислородцентрированными органическими радикалами: 2,2,6,6-тетраметилпиперидин-1-ил)оксилом (ТЕМПО) и 3,6-ди-трет-бутил-2-этоксифеноксильным радикалом.
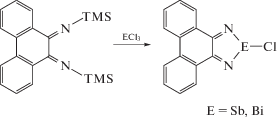
Первые известные 1,3,2-диазастиболы и 1,3,2-диазабисмолы были синтезированы с использованием фенантрендиимина. В случае сурьмы стабилизация гетероцикла также возможна с применением бензилдиимина, однако в случае висмута соединение будет стабильным только при использовании фенантрендиимина [59].
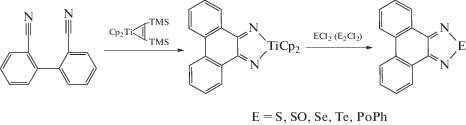
Определенный интерес для получения N-гетероциклических соединений, аннелированных фенантреном, представляют реакции с диазатитанациклопентадиеном. Основными особенностями данного пути является отсутствие фенантренхинона в схеме превращений и заместителей при атомах азота, что может повлиять на стабильность гетероцикла [60].
Таким образом, анализ известных литературных источников показывает, что N-гетероаналоги фенантренхинона способны образовывать комплексы во всех трех редокc-формах: нейтральной, анион-радикальной и дианионной. Примечательно, что, несмотря на структурную близость фенантрендииминов и BIAN, свойства и поведение комплексных соединений на их основе различаются. Интересные открытия в случае BIAN сделаны на примерах комплексов элементов 13 группы, однако сообщений о подобных работах с фенантрендииминами в литературе не наблюдается. Изучение соединений элементов 14 группы на основе фенантрендииминов показало их способность присоединять свободные радикалы с сохранением валентного состояния металлоцентра за счет участия в этом превращении редокc-активного лиганда. Подобное поведение комплексов непереходных металлов может оказаться перспективным в плане их использования в каталитических процессах. Однако химия N-гетероаналогов фенантренхинона находится в самом начале своего развития. Основным фактором, сдерживающим развитие этой области исследований, являлся недостаток рациональных и удобных методов синтеза 9,10-фенантрен-моно- и -дииминов. Такие методики появились только в начале XXI века, что открывает простор для дальнейших исследований металлокомплексных соединений на основе этих объектов.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Kaim W. // Dalton Trans. 2019. V. 48. P. 8521.
Fedushkin I.L., Nikipelov A.S., Morozov A.G. et al. // Chem. Eur. J. 2012. V. 18. P. 255.
Razborov D., Lukoyanov A., Baranov E. et al. // Dalton Trans. 2015. V. 44. P. 20532.
Fedushkin I.L., Skatova A.A., Dodonov V.A. et al. // Inorg. Chem. 2016. V. 55. P. 9047.
Zhang W., Dodonov V.A., Chen W. et al. // Chem. Eur. J. 2018. V. 24. P. 14994.
Dodonov V.A., Chen W., Zhao Y. et al. // Chem. Eur. J. 2019. V. 25. P. 8259.
Heumann A., Giordano L., Tenaglia A. // Tetrahedron Lett. 2003. V. 44. P. 1515.
Cherian A.E., Lobkovsky E.B., Coates G.W. // Chem. Commun. 2003. V. P. 2566.
Leatherman M.D., Svejda S.A., Johnson L.K. et al. // J. Am. Chem. Soc. 2003. V. 125. P. 3068.
Binotti B., Carfagna C., Zuccaccia C. et al. // Chem. Commun. 2005. № 1. P. 92.
Takeuchi D., Yasuda A., Osakada K. // Dalton Trans. 2003. P. 2029.
van Laren M.W., Elsevier C.J. // Angew. Chem. Int. Ed. 1999. V. 38. P. 3715.
Климов Е.С., Абакумов Г.А., Гладышев Е.Н. // Докл. АН СССР. 1974. Т. 218. С. 844.
Климов Е.С., Лобанов А.В., Абакумов Г.А. // Изв. АН СССР. Сер. хим. 1981. Т. 4. С. 2028.
Anschütz R., Schultz G. // Liebigs Annalen. 1879. V. 196. P. 32.
Pschorr R. // Ber. Dtsch. Chem. Ges. 1902. V. 35. P. 2729.
Balogh-Hergovich É., Speier G. // Inorg. Chim. Acta 1984. V. 84. P. 129.
Abakumov G.A., Cherkasov V.K., Druzhkov N.O. et al. // Synth. Commun. 2006. V. 36. P. 3241.
Farrell D., Kingston S.J., Tungulin D. et al. // Eur. J. Org. Chem. 2017. V. 2017. P. 5597.
Li L., Gomes C.S.B., Gomes P.T. et al. // ARKIVOC: Online J. Org. Chem. 2009. V. 2009. P. 95.
Tuchtenhagen G., Ruhlmann K. // Liebigs Annalen. 1968. V. 711. P. 174.
MacPherson E., Smith J. // Tetrahedron. 1971. V. 27. P. 2645.
van Belzen R., Klein R.A., Smeets W.J.J. et al. // Recl. Trav. Chim. Pays-Bas. 1996. V. 115. P. 275.
Li L., Jeon M., Kim S.Y. // J. Mol. Catal. A: Chem. 2009. V. 303. P. 110.
Cherkasov V.K., Druzhkov N.O., Kocherova T.N. et al. // Tetrahedron. 2012. V. 68. P. 1422.
Arun T.R., Raman N. // Spectrochim. Acta. A. 2014. V. 127. P. 292.
Gao B., Luo X., Gao W. et al. // Dalton Trans. 2012. V. 41. P. 2755.
Cameron L.A., Ziller J.W., Heyduk A.F. // Chem. Sci. 2016. V. 7. P. 1807.
Xu B., Ma A., Jia T. et al. // Dalton Trans. 2016. V. 45. P. 17966.
Gao B., Xin L., Gao W. et al. // Polyhedron. 2013. V. 63. P. 91.
Bochkarev M.N., Fagin A.A., Druzhkov N.O. et al. // J. Organomet. Chem. 2010. V. 695. P. 2774.
Gao B., Li D., Duan Q. // Polyhedron. 2019. V. 169. P. 287.
Tanahashi H., Tsurugi H., Mashima K. // Organometallics. 2015. V. 34. P. 731.
Bera S., Mondal S., Maity S. et al. // Inorg. Chem. 2016. V. 55. P. 4746.
Bera S., Maity S., Weyhermueller T. et al. // Dalton Trans. 2016. V. 45. P. 8236.
Mandal A., Kundu T., Ehret F. et al. // Dalton Trans. 2014. V. 43. P. 2473.
Bamford K.L., Longobardi L.E., Liu L. et al. // Dalton Trans. 2017. V. 46. P. 5308.
Абакумов Г.А., Дружков Н.О., Кочерова Т.Н. и др. // Докл. РАН. 2016. Т. 467. С. 418.
Chern S.-S., Liaw M.-C., Peng S.-M. // J. Chem. Soc., Dalton Trans. 1993. P. 359.
Chern S.-S., Lee G.-H., Peng S.-M. // J. Chem. Soc., Dalton Trans. 1994. P. 1645.
Schlosser K., Hoyer E. // Z. Anorg. Allg. Chem. 1972. V. 387. P. 91.
Shaffer D.W., Ryken S.A., Zarkesh R.A. et al. // Inorg. Chem. 2011. V. 50. P. 13.
Pyle A.M., Chiang M.Y., Barton J.K. // Inorg. Chem. 1990. V. 29. P. 4487.
Dapporto P., Denti G., Dolcetti G. et al. // J. Chem. Soc., Dalton Trans. 1983. P. 779.
Krotz A.H., Kuo L.Y., Barton J.K. // Inorg. Chem. 1993. V. 32. P. 5963.
Raman N., Arun T.R., Mahalakshmi R. et al. // Inorg. Chem. Commun. 2014. V. 46. P. 263.
Kramer W.W., Cameron L.A., Zarkesh R.A. et al. // Inorg. Chem. 2014. V. 53. P. 8825.
Pourtaghi-Zahed H., Zohuri G.H. // Polymer Bulletin. 2013. V. 70. P. 1769.
Pourtaghi-Zahed H., Zohuri G.H., Ahmadjo S. // J. Polym. Res. 2014. V. 21. P. 365.
Sa S., Jeon M., Kim S.Y. // J. Mol. Catal. A: Chem. 2014. V. 393. P. 263.
van Belzen R., Hoffmann H., Elsevier C.J. // Angew. Chem. Int. Ed. Engl. 1997. V. 36. P. 1743.
van Belzen R., Elsevier C.J., Dedieu A. et al. // Organometallics. 2003. V. 22. P. 722.
Gao B., Zhao D., Li X. et al. // RSC Adv. 2015. V. 5. P. 440.
Fedushkin I.L., Khvoinova N.M., Baurin A.Y. et al. // Inorg. Chem. 2004. V. 43. P. 7807.
Ketterer N.A., Ziller J.W., Rheingold A.L. et al. // Organometallics. 2007. V. 26. P. 5330.
Ullah F., Kindermann M.K., Jones P.G. et al. // Organometallics. 2009. V. 28. P. 2441.
Druzhkov N.O., Kazakov G.G., Shavyrin A.S. et al. // Inorg. Chem. Commun. 2018. V. 90. P. 92.
Naka A., Hill N.J., West R. // Organometallics. 2004. V. 23. P. 6330.
Diel B.N., Huber T.L., Ambacher W.G. // Heteroat. Chem. 1999. V. 10. P. 423.
Kiel G.R., Samkian A.E., Nicolay A.l. et al. // J. Am. Chem. Soc. 2018. V. 140. P. 2450.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия