

Координационная химия, 2020, T. 46, № 3, стр. 172-192
Моноядерные о-хиноновые комплексы кобальта и железа с тетрадентатными N-донорными основаниями: строение и свойства
А. А. Старикова 1, *, М. Г. Чегерев 1, А. Г. Стариков 1
1 Научно-исследовательский институт физической и органической химии
Южного федерального университета
Ростов-на-Дону, Россия
* E-mail: alstar@ipoc.sfedu.ru
Поступила в редакцию 25.09.2019
После доработки 28.10.2019
Принята к публикации 31.10.2019
Аннотация
Рассмотрены накопленные в литературе данные об электронном строении и магнитном поведении моноядерных о-бензохиноновых комплексов кобальта и железа с тетрадентатными азотсодержащими основаниями. Значительный интерес к выбранным объектам исследования вызван их способностью проявлять магнитную бистабильность, что указывает на широкие перспективы практического применения данного класса соединений в молекулярной электронике и спинтронике. На основании результатов квантовохимических расчетов обсуждено влияние структурных особенностей таких комплексов на их магнитные свойства.
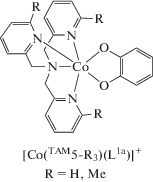
В последние годы объектами интенсивных исследований являются комплексы переходных металлов с редокс-активными лигандами [1–20]. Повышенное внимание к таким системам обусловлено их способностью демонстрировать механизмы магнитной бистабильности (спин-кроссовер (СКО) [21–24], валентную таутомерию (ВТ) [25–27] и др.) и, как следствие, возможностью их использования в качестве молекулярных магнитов, молекулярных переключателей и спиновых кубитов [28–37]. Распространенным структурным мотивом магнитно-активных соединений являются катионные комплексы металлов (М), включающие один редокс-активный лиганд (Ln) и тетрадентатное азотсодержащее макроциклическое основание (TAMn, n = 1–5) (схема 1 ). Сконструированные таким образом молекулы в зависимости от природы тетраазамакроцикла и металла (железо или кобальт) могут проявлять спиновые переходы в результате ВТ [38] или СКО [39, 40], а также демонстрировать каталитическую активность и имитировать катехол-диоксигеназу [41, 42]. Настоящий обзор посвящен систематическому анализу электронного строения и магнитных свойств соединений указанного типа. Первая часть обзора содержит данные экспериментальных исследований, а во второй – рассмотрены результаты квантовохимического изучения полученных ранее и новых о‑бензохиноновых комплексов кобальта и железа с тетраазамакроциклическими лигандами.
СТРОЕНИЕ И МАГНИТНОЕ ПОВЕДЕНИЕ o-ХИНОНОВЫХ КОМПЛЕКСОВ КОБАЛЬТА И ЖЕЛЕЗА С ТЕТРААЗАМАКРОЦИКЛИЧЕСКИМИ ЛИГАНДАМИ
Производные пиридинофана. Тетраазамакроциклические N,N'-диалкил-2,11-диаза[3.3]-(2,6)пиридинофаны (пиридинофаны) (TAM1-R2) способны координироваться к металлоцентрам, оставляя вакантными два цис-положения в координационной сфере металла для формирования октаэдрического окружения [43]. В соединениях кобальта включение в эти позиции о-хинона (диоксолена) приводит к образованию потенциального валентно-таутомерного фрагмента [27]. Однако вопреки ожиданиям комплекс [Co(TAM1-t-Bu2)- (L1b)]B(п-C6H4Cl)4 демонстрирует высокотемпературный (>400 K) спин-кроссовер [40]. Это соединение – единственный пример о-хинонового комплекса кобальта, проявляющего спиновые переходы по механизму СКО, а не ВТ.
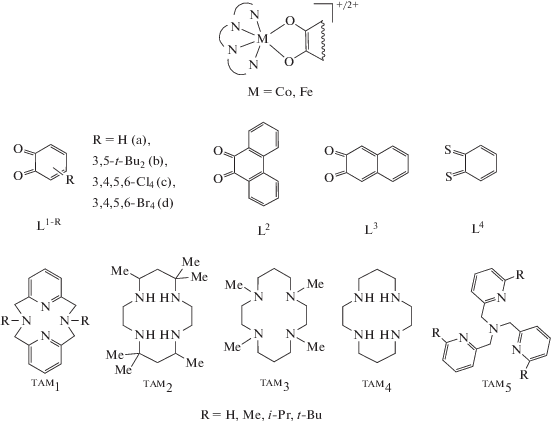
Схема 1 .
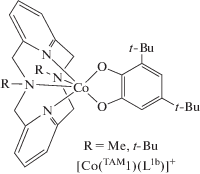
Согласно магнетохимическим измерениям, при комнатной температуре между низкоспиновым ионом кобальта(II) и анион-радикальной семихиноновой (SQ) формой редокс-активного лиганда наблюдается ферромагнитное связывание; незавершенность спинового перехода не позволила изучить обменные взаимодействия между металлоцентром и органическим радикалом [40]. Исследование этого соединения с использованием фемтосекундной электронной и ИК-колебательной нестационарной спектроскопии поглощения показало, что селективное фотовозбуждение раствора комплекса при комнатной температуре приводит к сверхбыстрому превращению низкоспиновой фракции в высокоспиновую [44]. В противоположность этому, использование макроцикла с метильными заместителями при атомах азота способствует формированию комплекса [Co(TAM1-Me2)(L1b)]+, который находится в LSСoIIICat (Cat = дианионная катехолатная форма лиганда) состоянии и не демонстрирует спиновые переходы.
С целью тонкой настройки лигандной системы данного типа магнитно-активных соединений недавно была синтезирована серия комплексов [Co(TAM1-R2)(L1b)]BPh4 (R = Me, Et, i-Pr, t-Bu), детальное исследование которых позволило установить роль алкильных заместителей пиридинофанового лиганда в реализации механизма магнитной бистабильности и его природу (СКО или ВТ) [45]. Показано, что производное [Co(TAM1-i-Pr2)(L1b)]BPh4 претерпевает термически индуцированную редокс-изомерную перегруппировку [LSCoIIICat]+ ⇄ [HSCoIISQ]+, в то время как аналогичный комплекс с TAM1-Et2 преимущественно находится в диамагнитном состоянии. Результаты квантовохимического моделирования этих систем обсуждены во второй части данного обзора.
Комплекс железа с изо-пропильным производным пиридинофана [Fe(TAM1-i-Pr2)(L1b)]BPh4 в широком диапазоне температур представлен изомером, включающим высокоспиновый трехвалентный ион металла [45]. Аналогичные координационные соединения с метилзамещенным пиридинофановым макроциклом [41, 42] существуют либо в виде дикатиона, которому на основании данных мёссбауэровской спектроскопии, РСА и магнетохимических измерений приписана структура [LSFeIII(TAM1-Me2)(SQ)]2+ [42], либо в катионной форме [HSFeIII(TAM1-Me2)(Cat)]+ [41].
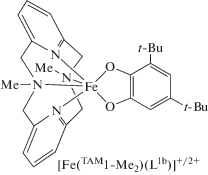
В комплексе [LSFeIII(TAM1-Me2)(SQ)]2+ неспаренные электроны низкоспинового иона металла и семихинона характеризуются сильным антиферромагнитным связыванием, приводящим к диамагнетизму. Это соединение представляет собой первый пример выделенного и структурно охарактеризованного комплекса, включающего низкоспиновый трехвалентный ион железа и SQ‑анион-радикал. В [41, 42] обсуждается каталитическая активность синтезированных систем, их мимикрия под диоксигеназу.
В комплексах железа(II) и железа(III) с TAM1-Me2 и в соединениях железа(II) и кобальта(II) с трет-бутилпроизводным пиридинофана TAM1-t-Bu2, включающих N-донорные вспомогательные лиганды (бипиридил (Вipy), 1,10-фенантролин (Рhen), бипиримидин (Вpym)), наблюдался СКО [46].
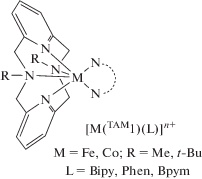
Производные циклама. Другим хорошо изученным классом тетраазамакроциклов являются цикламовые производные: 5,7,7,12,14,14-гексаметил-1,4,8,11-тетраазациклотетрадекан (TAM2), 1,4,8,11-тетраметил-1,4,8,11-тетраазациклотетрадекан (TAM3) и 1,4,8,11-тетраазациклотетрадекан (TAM4).
3,5-Ди-трет-бутилкатехолатные комплексы железа(III) с TAM2, TAM4 и триазациклононаном описаны в [47]. Магнитные и ЭПР измерения указывают на присутствие во всех соединениях трехвалентного иона металла. С целью выявления условий, при которых стабилизируются различные окислительные состояния металлов и лигандов, синтезированы и охарактеризованы семихиноновый и катехолатный комплексы кобальта(II) и кобальта(III) с TAM2 [48].
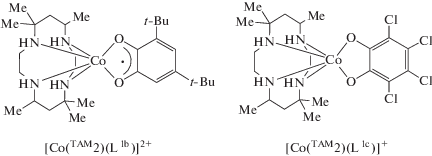
В [49] изучено переключение электронных состояний LSСoIIICat и HSCoIISQ хиноновых соединений с общей формулой [Co(TAM2)(L2)]Y (Y = PF6, BPh4, I). Такие комплексы претерпевают ВТ-переходы [LSCoIII(TAM2)(Cat)]+ ⇄ [HSCoII(TAM2)(SQ)]+, температура которых варьируется в зависимости от противоионов. Повышение давления приводит к смещению равновесия в направлении низкоспиновой структуры, имеющей меньший объем.
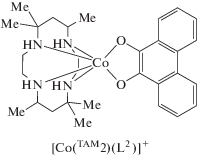
Изучение комплекса [Co(TAM2)(L2)]PF6 [49] продолжено в [50]. Рассмотренные в ней сольваты демонстрируют фотоиндуцированную ВТ.
В продолжение исследований комплексов с цикламовыми производными синтезированы соединения кобальта [Сo(TAM3)(L1b)]PF6 и [Сo(TAM3)- (L2)]PF6 [51]. Оба комплекса существуют в форме, включающей высокоспиновый ион кобальта(II) и SQ, в отличие от рассмотренного выше производного с ТАМ2. Основное триплетное состояние этих соединений формируется в результате сильного антиферромагнитного обмена между HSСoII и SQ, обусловленного неортогональностью магнитных орбиталей.
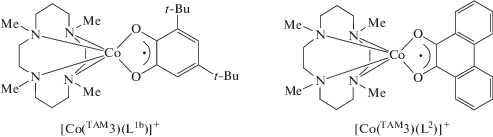
В результате экспериментов с использованием спектроскопии с временным разрешением охарактеризованы фотоиндуцированные превращения комплекса кобальта [Сo(TAM3)(L1b)]PF6 [52]. Показано вовлечение в переход HSCoIISQ → LSCoIIICat не менее трех различных состояний (HSCoIISQ, LSCoIIICat и LSCoIISQ/ISCoIIICat).
В [53] обобщены полученные ранее данные по о-бензохиноновым и 9,10-фенантренхиноновым комплексам кобальта с производными ТАМ2–4, претерпевающим ВТ переходы, где противоионы – PF6, I, BPh4; растворители – H2O, CH2Cl2, C6H5CH3. Обнаружено существование зависимости температуры перехода от природы противоиона и присутствия в кристалле молекул растворителя. Важным участником фотоуправляемой спиновой перегруппировки, помимо изомеров LSCoIIICat и HSCoIISQ, является электромер LSCoIISQ. Между парамагнитными центрами выявлены антиферромагнитные обменные взаимодействия. Позже этой группой авторов было установлено, что замена дихлорметана его дейтероаналогом в соединениях [Co(TAM2)(L2)]+ сопровождается резким изменением свойств сольватов и приводит к появлению гистерезиса [54]. Впоследствии синтезирован и структурно охарактеризован аналогичный комплекс, содержащий анион трополона [55].
Производные трис(2-пиридилметил)амина. Формально трис(2-пиридилметил)амин (TAM5) не имеет макроцикла, однако его поведение в составе координационных соединений с переходными металлами близко к обсуждаемому классу органических N-донорных лигандов, с ним получено большое число комплексов кобальта и железа, демонстрирующих магнитно-активные свойства.
Комплексы кобальта. В [56] описан фотоиндуцированный (а также термически управляемый) внутримолекулярный перенос электрона в диоксоленовых комплексах кобальта на основе бис(6-метил-(2-пиридилметил))(2-пиридилметил)амина (TAM5-Me2), характеризующийся долгим временем жизни метастабильных электронных состояний. Позже показано [57], что соединение [Co(TAM5-Me2)- (L1b)]PF6 претерпевает ВТ с критической температурой 370 K.
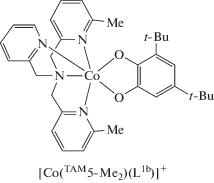
При изучении серии о-хиноновых комплексов кобальта на основе трис(2-пиридилметил)аминовых лигандов [Co(TAM5-Men)(L1b)]PF6 · Solv (n = = 0–3) установлено, что стерические препятствия, создаваемые метильными заместителями, регулируют редокс-свойства металла, определяя распределение заряда в фрагменте Со–L1b при комнатной температуре [57]. В твердом состоянии комплексы с n = 0, 1 представляют собой диамагнитные LSCoIIIСat производные, в то время как в системе, содержащей три метильные группы в тетраазамакроцикле, приписано электронное строение [HSCoII(TAM5-Me3)(SQ)]+. Соединение [Co(TAM5-Me2)(L1b)]PF6 · С2H5OH претерпевает энтропийно-управляемую перегруппировку ВТ при комнатной температуре. При охлаждении наблюдалась фотоиндуцированная ВТ в комплексах с n = 0–2. Полученные результаты демонстрируют эффективность молекулярного дизайна для контроля релаксационных свойств фотоиндуцированных метастабильных состояний.
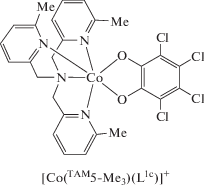
В продолжение работ [56, 57] группой Dei синтезирован и исследован с использованием метода РСА, магнитных измерений и электронной спектроскопии аналогичный комплекс кобальта с тетрахлорированным о-бензохиноном L1c и TAM5-Me3 [58]. Показано, что данная система в кристаллическом состоянии претерпевает ВТ при 300 K (СoIIICat → CoIISQ), в растворе процесс наблюдается при 250 K. Впервые определены термодинамические параметры ВТ в катионных диоксоленовых комплексах кобальта (1 : 1), которые подтвердили энтропийно-управляемый характер этого процесса.
В [56–58] продемонстрирована чрезвычайная чувствительность ВТ процесса к энергии кристаллической решетки. Для изучения этого эффекта получены различные сольваты (этанол, толуол) синтезированного ранее комплекса ВТ [Co(TAM5-Men)(L1b)]PF6 [59]. Установлено влияние растворителя, из которого осуществляется перекристаллизация, на термически индуцированную и фотоуправляемую ВТ. С использованием спектров поглощения рентгеновского излучения оценена роль иона кобальта в энтропийно- и фотоиндуцированной перегруппировке ВТ, проявляемой [Co(TAM5-Me2)(L1b)](PF6) · C6H5CH3 [60]. Высокая устойчивость и фотостабильность делают данное соединение хорошим кандидатом для применения в молекулярных устройствах. Влияние допирования [Co(TAM5-Me2)(L1b)]+ изоморфным цинковым комплексом [Zn(TAM5-Me2)- (L1b)](PF6) · C6H5CH3 на энтропийно- и фотоиндуцируемые переходы ВТ изучено в [61]. Показано, что разбавление комплексом диамагнитного металла практически не влияет на скорость релаксации оптически индуцированного метастабильного высокоспинового состояния.
Данные по диоксоленовым комплексам кобальта с тетраазамакроциклическими лигандами, демонстрирующими ВТ, обобщены в обзоре [29]. Показано, что относительная устойчивость редокс-изомеров определяется температурой, а облучение светом соответствующей длины волны способствует трансформации стабильного изомера в метастабильное состояние.
Воздействие электростатического поля на критическую температуру перегруппировки ВТ в комплексе [Co(TAM5-Me2)(L1b)]PF6 изучено в [62]. Полученные результаты демонстрируют новый способ управления магнетизмом молекул и открывают перспективы использования соединений ВТ для разработки устройств молекулярной спинтроники. В [63] посредством разностной ИК-спектроскопии исследована фотоиндуцированная валентная таутомерия в комплексах [Co(TAM5-Men)(L1b)]+.
Посредством квантовохимического моделирования серии соединений [Co(TAM5-Me3)(L1a–d)]+ выявлен комплекс [Co(TAM5-Me3)(L1d)]+, способный демонстрировать равновесие [LSCoIIICat]+⇄ ⇄ [HSCoIISQ]+ [38]. Сделанное на основании результатов расчетов предположение подтверждено последующим синтезом и экспериментальными исследованиями (электронная спектроскопия, изучение магнитной восприимчивости в твердом состоянии и в растворе) соединений [Co(TAM5-Me3)(L1d)]PF6 и [Co(TAM5-Me3)(L1d)]BPh4. Установлено, что в растворе комплекс [Co(TAM5-Me3)- (L1d)]+ претерпевает термически управляемую ВТ с критическими температурами в диапазоне 291–359 K, зависящими от растворителя. В твердом состоянии комплексы [Co(TAM5-Me3)(L1d)]PF6 и [Co(TAM5-Me3)(L1d)]BPh4 проявляют незавершенные переходы ВТ.
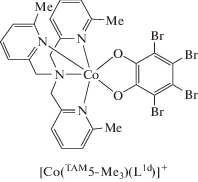
Авторы [64, 65] на основе TAM5 получили катионные (противоионы ${\text{PF}}_{{\text{6}}}^{--}$ и ${\text{BPh}}_{{\text{4}}}^{--}$) комплексы кобальта с 2,3-дигидроксинафталином (L3). Исследования показали, что обсуждаемые системы содержат низкоспиновый трехвалентный ион кобальта и катехолатную форму редокс-лиганда: в диапазоне 2–380 K комплексы диамагнитны [66].
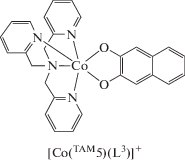
Авторы [67] получили три 3,5-ди-трет-бутил-о-хиноновых комплекса кобальта с пиразолил замещенным и трис(2-пиридилметил)аминами: TAM6 (PzPy2) = (3,5-диметил-1H-пиразолил)-N,N-бис(2-пиридилметил)амином, TAM7 (Pz2Py) = N,N-бис((3,5-диметил-1H-пиразолил)метил)-N-(2-пиридилметил)амином и TAM8 (Pz3) = трис((3,5-диметил-1H-пиразолил)метил)амином. Соединение [Co(TAM6)(L1b)]PF6 диамагнитнo, в то время как комплексы с TAM7 и TAM8 характеризуются основным высокоспиновым состоянием [HSCoII(TAM7, 8)(SQ)]PF6.
В [68] сообщается о синтезе, кристаллической структуре и электрохимическом поведении девяти новых комплексов кобальта(III) с тетрабромкатехолом L1d. В качестве азотсодержащих лигандов использованы различные бидентатные основания (этилендиамин, пропилендиамин, 2-аминометилпиридин, 8-аминохинолин и др.), а также структурный аналог TAM5 – трис(2-аминоэтил)амин (TAM9). Все соединения в основном состоянии диамагнитны, за исключением производного с 8‑аминохинолином, способного претерпевать в растворе термически индуцированную ВТ, которая сопровождается резким изменением электронных спектров.
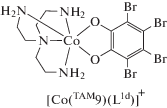
Комплексы железа. Первые работы по о-хиноновым производным железа с лигандом TAM5 появились в начале 1990-х гг. [69, 70]. Соединения [Fe(TAM5)(L1b)]BPh4 [69] и [Fe(TAM5-Me)(L1b)]СlO4 [70], как и большинство комплексов данного металла, включающих редокс-лиганды (L1,L4) и различные тетраазамакроциклы (TAM5-R (R = H, Men, Br, ((4-MeO)2, 4-Cl)2, (4-NO2), (4-NO2)2), TAM10–17) (схема 2 ), рассматриваются в качестве функциональной модели фермента катехол-диоксигеназы [71–78].
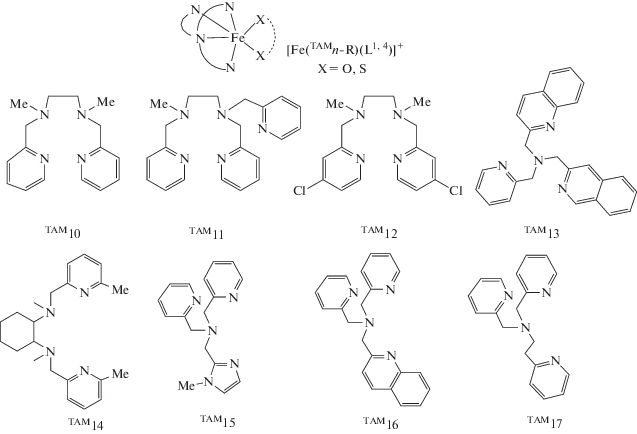
Схема 2 .
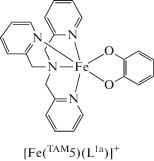
В [79] сообщается о синтезе [Fe(TAM5)- (L1a)]BPh4 – близкого аналога соединения, описанного ранее [69]. При изучении магнитной восприимчивости в твердом состоянии обнаружен СКО ионов трехвалентного железа S = 1/2 ⇌ S = = 5/2. Детальное исследование этого комплекса проведено позже [39]. С учетом полученных экспериментальных и теоретических данных обсуждается возможность существования в основном и возбужденном состояниях эквивалентной смеси FeIISQ и FeIIICat конфигураций.
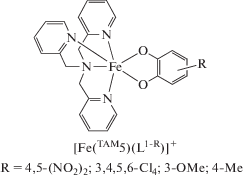
Этой же группой исследователей получен ряд соединений с общей формулой [Fe(TAM5)(L1-R)]X (R = 4,5-(NO2)2; 3,4,5,6-Cl4; 3-OMe; 4-Me; X = = ${\text{BPh}}_{{\text{4}}}^{ - };$ ${\text{NO}}_{{\text{3}}}^{ - };$ ${\text{PF}}_{{\text{6}}}^{ - };$ ${\text{ClO}}_{{\text{4}}}^{ - }$) [80]. Их магнитное поведение в твердом состоянии показывает общие черты: протекание термически управляемого СКО на трехвалентном ионе железе. Установлено, что электронная конфигурация низкоспиновых состояний во многом определяется химическим давлением, испытываемым комплексом под влиянием решетки, которое способствует смешиванию LSFeIIICat и LSFeIISQ, позволяющих ожидать как СКО, так и ВТ.
В работе [81] изучены фотофизические свойства синтезированных ранее [39, 80] катехолатных комплексов железа(III) в твердом состоянии. Проанализирован процесс фотовозбуждения, инициирующего переход LS → HS при низких температурах. Впоследствии были проведены ЭПР исследования электронной структуры этих соединений [82]. Установлено, что введение более электроноакцепторных заместителей в хиноновый лиганд приводит к повышению анизотропии g-тензора, что обусловлено увеличением вклада формы Fe-Cat [83].
Идентифицированы два полиформа комплекса с тетрахлоркатехолом [Fe(TAM5)(L1c)]PF6: моноклинный и орторомбический [84]. С понижением температуры до 210 K оба претерпевают спиновый переход между высоко- и низкоспиновым состояниями.
Позднее с использованием оптической фемтосекундной спектроскопии в режиме накачка–зондирование и методов пикосекундной рентгеновской дифракции изучена динамика фотопереключения спинового состояния в катионе [Fe(TAM5)(L1c)]+ с анионами ${\text{SbF}}_{{\text{6}}}^{--}$ и ${\text{РF}}_{{\text{6}}}^{--}$ [85, 86]. В обоих случаях СКО протекает в три стадии: сначала происходит возбуждение в состояние с переносом заряда лиганд–металл, далее следует релаксация в метастабильное состояние HS за 300 нс. Этот процесс сопровождается увеличением размера молекул, приводящим к ударной волне через кристалл, результатом чего является его расширение в период времени 10–10 000 нс. В заключение оптическая энергия переходит в тепловую, способствуя конверсии в состояние HS за 50 мкс. Комплекс [Fe(TAM5)(L1c)]SbF6 проявляет неполный термический спин-кроссовер S = 1/2 ⇌ S = 5/2 при 250 K [87]. При наложении давления температура перехода увеличивается почти на 50 K. Выделенные тетрахлоркатехолатные комплексы железа c производными диазепана охарактеризованы с помощью спектральных методов и электрохимии и, как и многие из рассмотренных выше, исследованы на способность к имитации катехол-диоксигеназы [88].
Синтезирован и структурно охарактеризован моноядерный катехолатный комплекс железа(III) [Fe(TAM17)(L1а)]PF6 [89], претерпевающий неполный СКО.
Моноядерные комплексы Fe(II) и Co(II) с редокс-активным лигандом на основе хлораниловой кислоты также демонстрируют магнитно-активные свойства [90]. Показано, что спиновым состоянием комплекса железа(II) можно управлять, варьируя температуру (СКО при 251 K), давление и облучение светом (LIESST при 42 K), а комплекс кобальта(II) претерпевает редокс-индуцированное переключение спинового состояния с образованием комплекса LSCoIII, при этом окислительное состояние редокс-активного лиганда остается неизменным.
о-Xиноновые металлокомплексы, функционализированные радикальными фрагментами. В настоящее время этот тип металлокомплексов привлекает особое внимание благодаря присутствию в них трех носителей спина: ион металла, семихинон и парамагнитный заместитель. Группой исследователей под руководством Shultz опубликован цикл работ [91–96], посвященных синтезу и изучению комплексов первого переходного ряда с о-хиноновыми лигандами, включающими нитронилнитроксильный (NN) радикал, связанный с о-бензохиноновым кольцом непосредственно либо через мостиковые группы. Большая часть таких соединений в качестве вспомогательных лигандов содержит гидро-трис(3-куменил-5-метилпиразолил)-борат) [92, 94–96].
Установлено, что бис-о-NN-семихиноновый комплекс кобальта [Co(L1b-Ph-NN)2(Bipy)], координационная сфера которого достроена до октаэдра бипиридилом, способен претерпевать ВТ [93].
Работа [94] посвящена исследованию соединений кобальта(III) Co(L1b-NN)2(Py)2 и Co(L1b-Ph-NN)2-(Py)2. В ней подчеркивается, что во всех изученных системах [91–96] в паре SQ–NN наблюдаются сильные ферромагнитные обменные взаимодействия.
В 2015 г. появилось сообщение [97] о комплексе железа(III) с NN-катехолом, содержащим трет-бутильный заместитель в третьем положении, и тетраазамакроциклом TAM5. Соединение [FeIII(TAM5)(L1b-NN)]BPh4, строение которого установлено методами РСА и мёссбауэровской спектроскопии, проявляет слабые ферромагнитные обменные взаимодействия Fe(III)–NN и претерпевает спин-кроссовер.
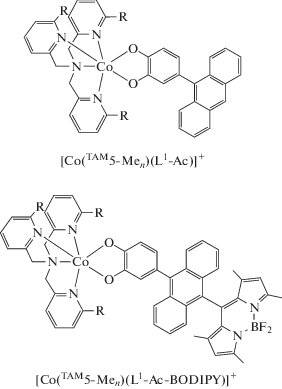
Получены комплексы кобальта с хинонами, функционализированными антраценом, и трис(2-пиридилметил)аминовыми производными [CoTAM5-Men)(L1-Ac)]PF6 (n = 0–3) [98]. В твердом состоянии соединение [Co(TAM5)(L1-Ac)]PF6 находится в диамагнитной форме (LSCoIIICat), в то время как комплекс [Co(TAM5-Men)(L1-Ac)]PF6 (n = 3), включающий три метильные группы при атомах азота тетраазамакроциклического лиганда, содержит высокоспиновый ион кобальта(II) и семихиноновую форму редокс-активного лиганда (HSCoIISQ). Системы с n = 1, 2 демонстрируют ВТ, в том числе индуцированную светом (при 5 K). В развитие этого направления получены аналогичные комплексы [Co(TAM5-Men)(L1-Ac-BODIPY)]PF6 (n = 0, 3), содержащие в редокс-активном лиганде бордипиррометан (BODIPY) [99]. Показано, что BODIPY не оказывает влияния на спиновое состояние комплекса.
Таким образом, о-хиноновые комплексы металлов с тетраазамакроциклическими лигандами являются объектами разносторонних экспериментальных исследований. В то же время сложности в определении электронных и спиновых состояний изомеров, выявление закономерностей “структура–свойства” требуют использования методов квантовой химии.
ТЕОРЕТИЧЕСКОЕ МОДЕЛИРОВАНИЕ ЭЛЕКТРОННОГО СТРОЕНИЯ И МАГНИТНЫХ СВОЙСТВ о-БЕНЗОХИНОНОВЫХ КОМПЛЕКСОВ КОБАЛЬТА И ЖЕЛЕЗА С ТЕТРААЗАМАКРОЦИКЛИЧЕСКИМИ ЛИГАНДАМИ
С целью изучения магнитного поведения синтезированных ранее соединений [40, 45, 49, 53–59] и выявления структурных параметров, ответственных за возможность протекания в рассматриваемых системах механизмов переключения спиновых состояний, выполнено квантовохимическое моделирование моноядерных о-хиноновых комплексов кобальта и железа с тетрадентатными макроциклическими азотсодержащими основаниями. Все расчеты проводили с помощью программы Gaussian 09 [100] методом теории функционала плотности (DFT) с использованием функционала UTPSSh [101, 102] и расширенного базиса 6-311++G(d,p), сочетание которых корректно воспроизводит энергетические и магнитные характеристики катионных комплексов, проявляющиx механизмы магнитной бистабильности [12, 45, 103–108]. Согласно полученным в данном приближении результатам расчетов экспериментально изученных систем, СКО на ионе железа реализуется при ΔEHS-LS < 11 ккал/моль [103, 104], СКО на ионе кобальта при ΔEHS-LS < 5 ккал/моль, а ВТ в комплексах с ионом кобальта при ΔEHS-LS < 10 ккал/моль [109, 110]. Локализацию стационарных точек на поверхности потенциальной энергии (ППЭ) осуществляли путем полной оптимизации геометрии молекулярных структур с проверкой стабильности DFT волновой функции и расчетом силовых констант. Поскольку ни малликеновские, ни NBO заряды не дают правильного представления о степени окисления центрального иона в комплексах с редокс-активными лигандами, значение этого параметра наиболее целесообразно оценивать по количеству неспаренных электронов на атоме металла и длинам характеристических связей лигандов. Вычисление параметров обменного взаимодействия (J, см–1) осуществляли в рамках формализма “нарушенной симметрии” (broken symmetry, BS) [111] с использованием метода обобщенной проекции спина, предложенного Yamaguchi [112]. Подробная информация о методике проведения DFT исследований комплексов переходных металлов с редокс-активными лигандами содержится в [113].
Комплексы кобальта с производными пиридинофана. Согласно расчетам, основным состоянием катионного комплекса [Co(TAM1-t-Bu2)(L1a)]+ на основе о-бензохинона и ди-трет-бутильного производного пиридинофана является электронный изомер (электромер [114]) LSCoIISQ на триплетной поверхности ППЭ [109].
Спиновая плотность присутствует на металле, а также на атомах кислорода и прилегающиx к ним атомах углерода (рис. 1), что указывает на низкоспиновое состояние двухзарядного иона кобальта и семихиноновую форму лиганда. Геометрические характеристики структуры LSCoIISQ [109] хорошо согласуются с данными РСА комплекса при 200 K [40, 44]. В изомере LSCoIISQ предсказано ферромагнитное связывание неспаренных электронов (J = 579 см–1) [109], что подтверждает предположение [40, 44] о наличии в этой форме комплекса сильных обменных взаимодействий ферромагнитного типа.
Рис. 1.
Вычисленное распределение спиновой плотности в электромерах LSCoIISQ и HSCoIISQ комплекса [Co(TAM1-t-Bu2)(L1a)]+. Здесь и на рис. 3–7 атомы водорода опущены для ясности.
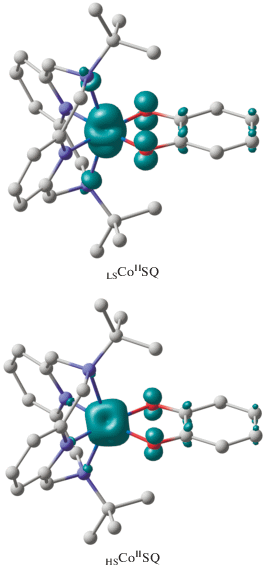
Высокоспиновый изомер HSCoIISQ на квинтетной ППЭ дестабилизирован относительно LS-CoIISQ на 6.2 ккал/моль [109], что позволяет ожидать неполный переход [Co(TAM1-t-Bu2)(L1a)]+ в высокоспиновое состояние. Этот вывод согласуется с результатами магнетохимических измерений комплекса [40, 44]. Расчет состояния BS указывает на антиферромагнитный обмен (J = –46 см–1) [109] между неспаренными электронами семихинона и высокоспинового иона кобальта. Результат коррелирует с умеренными антиферромагнитными взаимодействиями, наблюдаемыми в комплексах высокоспинового Co(II) с семихиноновыми лигандами [51].
Дестабилизация структуры LSCoIIICat относительно LSCoIISQ, являющейся основным состоянием комплекса, делает невозможным термически инициированный внутримолекулярный редокс-процесс.
Включение в расчетную схему тетра(п-хлорфенил)боратного противоиона привело к понижению разности энергий между низкоспиновыми изомерами LSCoIIICat и LSCoIISQ; последний, как и в рассмотренном выше катионном комплексе, является основным состоянием и характеризуется сильными ферромагнитными взаимодействиями спинов неспаренных электронов [110]. Оптимальная разность энергий между структурами LSCoIISQ и HSCoIISQ позволяет ожидать переключение магнитных свойств соединения [Co(TAM1-t-Bu2)(L1a)]B(п-C6H4Cl)4 посредством спин-кроссовера. Дестабилизация изомеров, включающих трехзарядный ион кобальта и катехолатную форму о-хинона, обусловлена невозможностью формирования коротких координационных связей Со–N вследствие стерических препятствий, создаваемых трет-бутильными группами азотсодержащего макроциклического лиганда.
Замена в пиридинофановом лиганде объемистых заместителей на метильные группы и учет противоиона стабилизирует изомер LSCoIIICat на синглетной ППЭ [109]. Следовательно, комплекс [Co(TAM1-Me2)(L1a)]BF4 будет находиться в диамагнитном состоянии, что полностью согласуется с экспериментом [40, 44].
Для выяснения влияния алкильных заместителей на магнитные свойства рассматриваемого типа комплексов методом DFT изучены соединения, включающие этильные и изопропильные группы [45]. Расчеты комплекса [Co(TAM1-Et2)- (L1b)]BPh4 указывают на энергетическую предпочтительность низкоспиновой формы LSCoIIICat. Электромер HSCoIISQ дестабилизирован относительно основного состояния на 10.9 ккал/моль, что позволяет предположить неполный переход ВТ. Этот вывод подтверждается увеличением эффективного магнитного момента в обсуждаемом соединении при температуре выше 340 K [45]. Как и в рассмотренных выше комплексах с TAM1-t-Bu2 и TAM1-Me2, высокоспиновый электромер характеризуется антиферромагнитным связыванием спинов неспаренных электронов иона металла и SQ.
В комплексе [Co(TAM1-i-Pr2)(L1b)]BPh4 предсказан иной результат. Разность энергий между основным состоянием LSCoIIICat и высокоспиновым изомером HSCoIISQ (4.6 ккал/моль) благоприятствует протеканию ВТ, наблюдаемой в эксперименте [45].
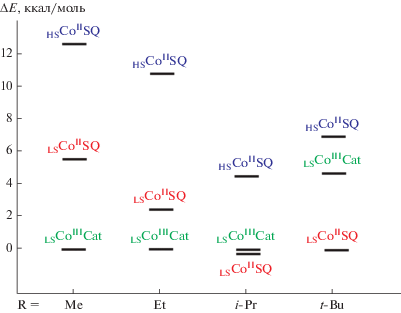
Сравнение энергетических характеристик электромеров рассмотренных выше комплексов кобальта показало (рис. 2), что увеличение размера алкильного заместителя в тетраазамакроцикле (Me → Et → i-Pr) способствует сближению энергий изомеров HSCoIISQ и LSCoIIICat и приводит к благоприятным условиям для протекания перегруппировок ВТ в комплексе [Co(TAM1-i-Pr2)- (L1b)]BPh4. При дальнейшем увеличении объемности заместителя (соединение [Co(TAM1-t-Bu2)- (L1f)]B(п-C6H4Cl)4) происходит инверсия относительных энергий: электромер LSCoIISQ стабилизируется относительно LSCoIIICat на 4.7 ккал/моль, создавая условия для перехода СКО, а не ВТ. Таким образом, варьирование алкильных заместителей в TAM1 позволяет управлять относительной устойчивостью электромерных форм комплексов и, как следствие, типом механизма переключения спиновых состояний.
Рис. 2.
График относительных энергий электромеров о-хиноновых комплексов кобальта с N-диалкилпроизводными TAM1.
Согласно результатам расчетов, электроноакцепторные заместители в о-бензохиноне повышают устойчивость состояния LSCoIIICat, способствуя облегчению внутримолекулярного переноса электрона: энергетические характеристики электромеров комплекса с тетрахлор-о-бензохиноном [Co(TAM1-Me2)(L1c)]+ позволяют ожидать протекания перегруппировки ВТ [110]. Как и в рассмотренных выше соединениях, обмен между спинами неспаренных электронов семихинонового радикала и высокоспинового иона металла изомера HSCoIISQ носит антиферромагнитный характер (J = –62 см–1) [110].
Наиболее устойчивой формой [Co(TAM1-t-Bu)- (L1c)]+ является электромер LSCoIISQ, включающий лиганд в форме SQ и низкоспиновый двухвалентный ион кобальта, между неспаренными электронами которых предсказаны сильные ферромагнитные обменные взаимодействия. Высокоспиновая структура HSCoIISQ дестабилизирована относительно основного состояния на 5.8 ккал/моль, что дает основаниe ожидать в обсуждаемом комплексе термически управляемый СКО, осложненный антиферромагнитным обменом в высокоспиновом изомере (J = –22 см–1) [110].
Таким образом, характер обменных взаимодействий в рассмотренных о-бензохиноновых комплексах кобальта с производными TAM1 определяется электронной конфигурацией атома металла и не зависит от заместителей в лигандах: в изомерах с низкоспиновым Co(II) предсказан сильный ферромагнитный обмен, а в структурах с высокоспиновым Co(II) – антиферромагнитный обмен умеренной силы.
Комплексы кобальта с трис(2-пиридилметил)аминами. В катионном комплексе кобальта [Co(TAM5)(L1a)]+ разность энергий между изомерами LSCoIIICat и HSCoIISQ позволяет ожидать редокс-изомерную перегруппировку, что не согласуется с экспериментальными данными [57], указывающими на превалирование диамагнитного состояния в широком диапазоне температур. Включение в расчетную схему гексафторфосфат-аниона приводит к дестабилизации высокоспинового электромера HSCoIISQ и блокированию валентной таутомерии в соединении [Co(TAM5)- (L1a)]PF6 [110, 113]. Вне зависимости от спинового состояния металла (LSCoIISQ и HSCoIISQ) и присутствия противоиона обмен между SQ и ионом кобальта носит сильный ферромагнитный характер.
Результаты расчетов электромеров комплекса кобальта [Co(TAM5-Me2)(L1a)]PF6 с трис(2-пиридилметил)аминовым лигандом, содержащим две метильные группы в положении 6 пиридиновых колец, хорошо воспроизводят экспериментальные данные [57], указывающие на протекание перегруппировки ВТ LSCoIIICat ⇄ HSCoIISQ при комнатной температуре. Заместители в азотсодержащем лиганде не оказывают существенного влияния на силу ферромагнитных обменных взаимодействий [110].
Введение дополнительной метильной группы способствует стабилизации парамагнитной формы HSCoIISQ комплекса [Co(TAM5-Me3)(L1a)]PF6, включающей высокоспиновый ион кобальта и семихинон, обмен между которыми также носит ферромагнитный характер (J = 197 см–1).
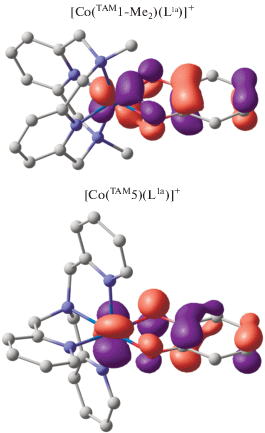
Различный характер обменных взаимодействий в высокоспиновых состояниях комплексов с пиридинофановым и трис(2-пиридилметил)аминовым лигандами обусловлен ориентацией магнитных орбиталей. Как видно из рис. 3, в соединении [Co(TAM1-Me2)(L1a)]+ имеет место значительное перекрывание, способствующее появлению канала антиферромагнитного обмена; ортогональность магнитных орбиталей комплекса [Co(TAM5)(L1a)]+ приводит к ферромагнитному связыванию спинов неспаренных электронов металла и SQ анион-радикала.
Рис. 3.
Магнитные орбитали электромеров HSCoIISQ комплексов [Co(TAM1-Me2)(L1a)]+ и [Co(TAM5)-(L1a)]+.
Таким образом, в исследованных о-бензохиноновых комплексах кобальта с производными TAM1 и TAM5 возможность переключения спинового состояния, тип механизма этого процесса, а также характер обменных взаимодействий определяются природой тетраазамакроциклического лиганда и заместителями в о-хиноне. Введение алкильных групп, создающих стерические препятствия в координационном узле, позволяет управлять редокс-состояниями комплексов. Учет противоионов при проведении расчетов, а также электроноакцепторные заместители приводят к повышению устойчивости низкоспиновых изомеров, включающих трехзарядный ион металла и катехолатную форму лиганда. Рассмотренные системы демонстрируют важность включения в расчетную схему внешнесферных противоионов при изучении координационных соединений переходных металлов с редокс-активными лигандами.
Комплексы железа с производными пиридинофана. Выполненные ранее экспериментальные исследования о-хиноновых комплексов железа с пиридинофановым макроциклом посвящены установлению их каталитической активности и способности имитировать поведение фермента диоксигеназы [41, 42]; магнитные свойства этих соединений не изучались. С целью выяснения возможности реализации механизмов магнитной бистабильности в подобных системах выполнены расчеты монокатионного о-хинонового комплекса железа с N,N'-диметил-2,11-диаза[3.3]-(2,6)пиридинофаном [Fe(TAM1-Me2)(L1a)]+ [115].
Анализ вычисленных геометрических характеристик и распределения спиновой плотности указывают на одновременное присутствие двух низкоспиновых форм – LSFeIIICat и LSFeIISQ. Наличие четырех неспаренных электронов на металле и значительное количество спиновой плотности на атомах кислорода и углерода о-бензохинона позволяет приписать электромеру на секстетной ППЭ состояние HSFeIISQ, включающее двухзарядный ион железа в высокоспиновом состоянии и семихиноновый радикал, между которыми предсказаны сильные ферромагнитные обменные взаимодействия (J = 185 см–1). Вычисленная разность энергий между изомерами комплекса [Fe(TAM1-Me2)(L1а)]+ составляет 10.6 ккал/моль, что дает основание ожидать в нем термически инициированный спиновый переход [115].
Расчет нейтрального комплекса [Fe(TAM1-Me2)- (L1а)]PF6 свидетельствует о включении в низко- и высокоспиновые электромеры катехолатной формы редокс-лиганда и трехзарядных ионов железа. Учeт противоиона не сопровождается изменением энергетических характеристик изомеров LSFeIIICat и HSFeIIICat (ΔE = 10.6 ккал/моль), что позволяет предположить протекание термически управляемого спин-кроссовера, в который может быть вовлечен электромер ISFeIIICat с промежуточным спиновым состоянием металла (ΔE = = 8.5 ккал/моль) [115]. Таким образом, результаты квантовохимических расчетов комплекса [Fe(TAM1-Me2)(L1а)]PF6 указывают на проявление им магнитно-активных свойств, что может послужить стимулом к экспериментальному изучению этого соединения.
Далее были рассмотрены дикатионные о-бензохиноновые комплексы железа с N-диалкилпроизводными пиридинофана [116].
Согласно расчетам [116], основным состоянием соединения [Fe(TAM1-Me2)(L1а)]2+ является низкоспиновый изомер LSFeIIISQ на триплетной ППЭ, включающий трехвалентный ион металла и SQ-форму редокс-активного лиганда (рис. 4). Вычисленные длины связей в координационном узле и бензохиноновом кольце хорошо коррелируют со значениями, установленными при помощи РСА [42]. В обсуждаемом электромере предсказаны сильные обменные взаимодействия антиферромагнитного характера [116], что также находится в согласии с экспериментальными данными, указывающими на диамагнетизм комплекса [42]. На септетной ППЭ локализован изомер HSFeIIISQ, содержащий высокоспиновый трехвалентный ион железа и семихинон, между которыми предсказано сильное антиферромагнитное связывание. Относительная энергия электромеров (ΔE = 14.4 ккал/моль) не позволяет ожидать протекания в комплексе [Fe(TAM1-Me2)- (L1а)]2+ термически инициированного спинового перехода.
Рис. 4.
Вычисленное распределение спиновой плотности в электромерах дикатионных комплексов [Fe(TAM1-Me2)(L1a)]2+, [Fe(TAM2-Me2)(L1c)]2+ и [Fe(TAM1-t-Bu2)(L1a)]2+.
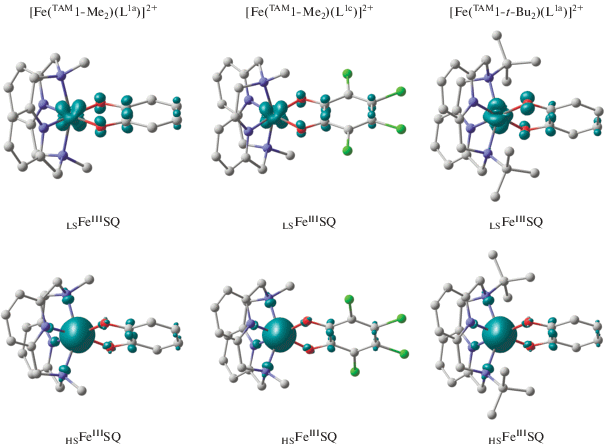
Аналогичные результаты получены для производного [Fe(TAM1-Me2)(L1c)]2+. Расчеты показали, что хлорные заместители в радикальном лиганде не оказывают существенного влияния на энергетические и магнитные характеристики электромеров данного комплекса [116]. Дестабилизация изомера HSFeIIISQ по отношению к низкоспиновой структуре LSFeIIISQ на 14.1 ккал/моль свидетельствует о термической недостижимости высокоспинового состояния. В электромере на дублетной ППЭ сильные антиферромагнитные обменные взаимодействия будут способствовать диамагнетизму комплекса в широком диапазоне температур.
Как было показано ранее [40, 45, 109, 110], объемистые заместители при донорных атомах тетраазамакроциклических лигандов затрудняют формирование низкоспинового изомера, что приводит к сокращению разности энергий между электромерами до значений, благоприятствующих протеканию термически управляемого СКО. Расчеты комплекса [Fe(TAM1-t-Bu2)(L1а)]2+ показали, что замена метильных заместителей при атомах азота пиридинофанового лиганда на трет-бутильные группы сопровождается изменениями геометрических и энергетических характеристик электромеров [116]. Предсказан-ное сужение энергетической щели между электромерами LSFeIIISQ и HSFeIIISQ до 7.7 ккал/моль делает возможным реализацию спинового перехода на ионе железа. Таким образом, посредством квантовохимического моделирования дикатионных о‑семихиноновых комплексов железа с производными пиридинофана установлено, что электроноакцепторные группы в радикальном лиганде не оказывают значи-тельного влияния на магнитные свойства рассмотренных координационных соединений, в то время как варьирование заместителей при атомах азота тетрадентатного основания сопровождается изменением геометрических и энергетических характеристик изомеров. Это позволяет ожидать в дикатионном о-бензохиноновом комплексе железа с N,N'-ди-трет-бутил-2,11-диаза[3.3]-(2,6)пиридинофаном термически инициированный СКО.
Комплексы железа с трис(2-пиридилметил)аминами. Согласно расчетам катионного и электронейтрального комплексов железа с трис(2-пиридилметил)амином [Fe(TAM5)(L1а)]+ и [Fe(TAM5)(L1а)]PF6, основное состояние представлено структурами на дублетной ППЭ, в которых ожидается смесь конфигураций LSFeIIICat + LSFeIISQ, включающих низкоспиновые ионы металла в различных окислительных состояниях [115], что согласуется с предположениями, сделанными в [39]. Минимумы на секстетной ППЭ соответствуют электромерам HSFeIISQ, содержащим двухзарядный ион железа в высокоспиновом состоянии и семихиноновый радикал, между которыми предсказаны сильные ферромагнитные обменные взаимодействия. Энергетические характеристики изомеров комплексов [Fe(TAM5)(L1а)]+ и [Fe(TAM5)(L1а)]PF6 позволяют ожидать термически инициированный спиновый переход.
Рассмотрение комплекса [Fe(TAM5)(L1c)]PF6 с о-хлоранилом показало, что присутствие хлорзаместителей приводит к стабилизации структур, включающих катехолатную форму редокс-активного лиганда и трехзарядный ион железа [115]. Вычисленная разность энергий между низко- и высокоспиновым изомерами согласуется с протеканием СКО в этом соединении [80]. Следовательно, присутствие в о-бензохиноне галогенов не влияет на реализацию спиновых переходов комплексов [Fe(TAM5)(L1c)]+ и [Fe(TAM5)(L1c)]PF6.
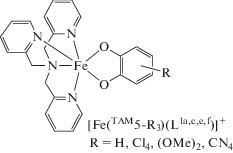
На рис. 5 приведено распределение спиновой плотности в низкоспиновых изомерах комплексов Fe(TAM5) с о-бензохинонами, содержащими электроноактивные заместители. Две мощные электронодонорные OMe-группы (комплекс [Fe(TAM5)(L1e)]+) стабилизируют двухвалентное состояние железа, на что указывает практически полное отсутствие спиновой плотности на ионе металла; в противоположность этому четыре электроноакцепторные цианогруппы (соединение [Fe(TAM5)(L1f)]+) переводят железо в окислительное состояние +3. Предсказанная расчетами делокализация спиновой плотности в экспериментально изученных системах [Fe(TAM5)(L1a)]+ и [Fe(TAM5)(L1c)]+ имеет промежуточное положение между рассмотренными выше случаями. С одной стороны, этот результат полностью согласуется с данными магнетохимических и мёссбауэровских исследований, свидетельствующих о высокой лабильности электронных состояний данного типа комплексов, а с другой – демонстрирует влияние модификации о-бензохинонового лиганда на реализацию ВТ или СКО.
Рис. 5.
Вычисленное распределение спиновой плотности в низкоспиновых электромерах комплексов [Fe(TAM5)(L1e, a, c, f)]+.
Таким образом, квантовохимическое изучение диоксоленовых комплексов железа с трис(2-пиридилметил)аминовыми лигандами показало способность этих соединений демонстрировать спин-кроссовер. Установлено, что заместители в о-бензохиноне не оказывают существенного влияния на разность энергий между низко- и высокоспиновыми состояниями комплекса, однако играют определяющую роль в стабилизации электронных конфигураций. Присутствие в бензохиноновом кольце заместителей, обладающих ярко выраженными электроноакцепторными свойствами, способствует смещению электронной плотности с иона железа и повышению устойчивости структур, включающих трехзарядный ион металла и дианионную форму редокс-активного лиганда. В то же время электронодонорные группы в о-хиноне стабилизируют электромеры FeIISQ.
о-Xиноновые комплексы кобальта и железа, функционализированные радикальными фрагментами. Взаимное влияние аценовых и о-бензохиноновых групп изучено в [99], посвященной синтезу и исследованию моноядерных комплексов кобальта с антраценовым фрагментом. Полициклическая группа в обсуждаемых системах диамагнитна вследствие малого времени жизни возбужденного триплетного состояния. Увеличение числа сопряженных колец до пяти способствует облегчению перехода аценов в парамагнитную форму.
С целью объединения в одной молекуле металлсодержащего центра, потенциально способного претерпевать термически инициированный спиновый переход, и полиаценовой линкерной группы, синглетно-триплетные переходы которой управляются облучением светом, выполнено компьютерное моделирование соединений [M(TAM1-t-Bu2)(L5)]+ и [M(TAM1-t-Bu2)(L6)]+ (M = Co, Fe). Координационная сфера металла в этих комплексах достроена N,N'-ди-трет-бутил-2,11-диаза[3.3]-(2,6)пиридинофаном. Радикальная группа в соединениях [M(TAM1-t-Bu2)(L5)]+ представлена линейным гептаценом. Как следует из полученных ранее результатов расчетов [117], энергетическая щель между синглетной (бирадикальной) и триплетной формами гептацена не превышает 7 ккал/моль, что благоприятствует его переходу в высокоспиновое состояние при облучении светом. Особенностью соединений [M(TAM1-t-Bu2)(L6)]+ является присутствие в полициклическом углеводороде концевых пятичленных колец, способствующих, согласно экспериментальным и теоретическим данным [117–119], стабилизации триплетного состояния такого ацена уже при пяти конденсированных циклах.
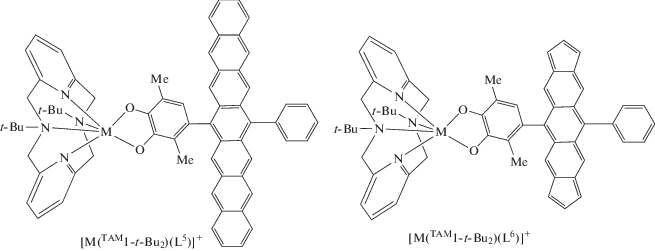
В соответствии с полученными результатами независимо от типа терминальных колец и иона металла аценовый линкер стабилизируется в виде синглетного бирадикала, характеризующегося сильным антиферромагнитным связыванием [120]. Спиновая плотность в гептацене (соединения [M(TAM1-t-Bu2)(L5)]+ (M = Co, Fe)) сконцентрирована в центральной части линкера на противолежащих атомах углерода полиароматической цепи (рис. 6, 7). Иное распределение спиновой плотности найдено в комплексах [Co(TAM1-t-Bu2)(L6)]+ и [Fe(TAM1-t-Bu2)(L6)]+: парамагнитные центры полициклического фрагмента этих соединений расположены на атомах углерода терминальных пятичленных циклов.
Рис. 6.
Вычисленное распределение спиновой плотности в электромерах комплексов [Co(TAM1-t-Bu2)(L5)]+ и [Co(TAM1-t-Bu2)(L6)]+.
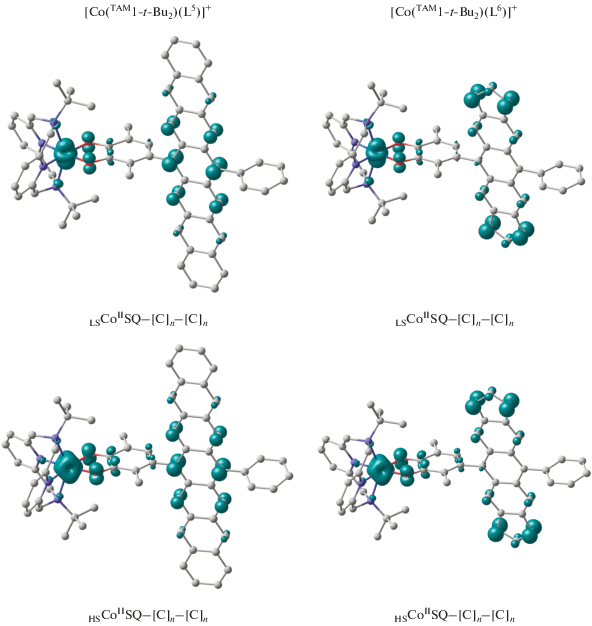
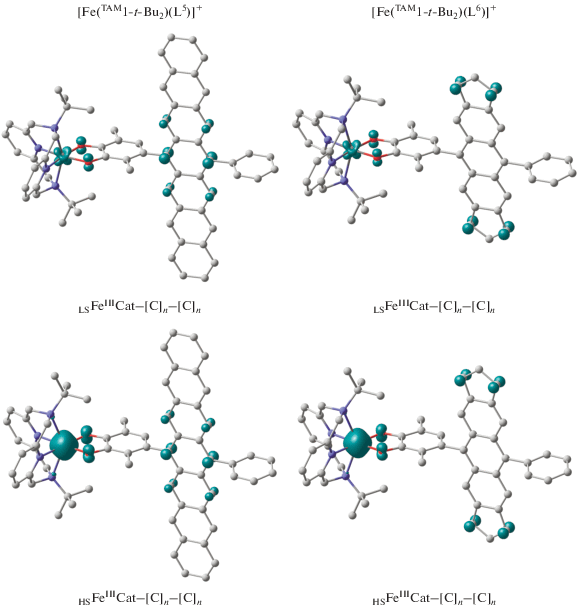
Рис. 7.
Вычисленное распределение спиновой плотности в электромерах комплексов [Fe(TAM1-t-Bu2)(L5)]+ и [Fe(TAM1-t-Bu2)(L6)]+.
В комплексах кобальта основным состоянием является электромер LSCoIISQ–[С]n–[С]n, включающий низкоспиновый двухзарядный ион металла и семихинон, что согласуется с проведенными ранее исследованиями моноядерных о-бензохиноновых соединений кобальта с TAM1-t-Bu2 [40, 45, 110]. На септетной ППЭ локализованы высокоспиновые изомеры с электронной конфигурацией HSCoIISQ–[С]n–[С]n. Энергетическая щель между низко- и высокоспиновыми состояниями комплексов [Co(TAM1-t-Bu2)(L5)]+ и [Co(TAM1-t-Bu2)- (L6)]+ составляет 6.3 и 6.2 ккал/моль соответственно, что указывает на возможность реализации термически инициированного СКО [120]. Обменные взаимодействия между неспаренными электронами семихинонов и ионов металла носят такой же характер, как и в рассмотренных ранее моноядерных соединениях с пиридинофановыми лигандами [40, 45, 110]: в низкоспиновом фрагменте LSCoII–SQ предсказано сильное ферромагнитное связывание, а спины неспаренных электронов в паре HSCoII–SQ взаимодействуют антиферромагнитно.
На основании полученных данных можно заключить, что длина и структура аценового фрагмента комплексов [Co(TAM1-t-Bu2)(L5)]+ и [Co(TAM1-t-Bu2)(L6)]+ не оказывают существенного влияния на возможность проявления последними перегруппировок, сопровождающихся переключением спиновых состояний. В то же время бирадикальный линкер способствует появлению дополнительных каналов обмена LSCoII–[С]n, HSCoII–[С]n и SQ–[C]n [120], характеристиками которых можно управлять, варьируя угол поворота хинонового кольца относительно полициклической цепи [121].
В построенных аналогичным способом соединениях железа [Fe(TAM1-t-Bu2)(L5)]+ и [Fe(TAM1-t-Bu2)(L6)]+ найденные на квартетной и октетной ППЭ электромеры включают трехвалентные ио-ны металла и катехолатную форму редокс-активного лиганда (рис. 7). Как и в рассмотренном выше дикатионном производном [Fe(TAM1-t-Bu2)- (L1a)]2+ [116], трет-бутильные группы при атомах азота тетрадентатного макроцикла создают оптимальные условия для стабилизации обеих электромерных форм. Разности энергий между состояниями LSFeIIICat–[С]n–[C]n и HSFeIIICat–[С]n–[C]n не превышают 5 ккал/моль, что позволяет ожидать реализацию СКО в комплексах [Fe(TAM1-t-Bu2)- (L5)]+ и [Fe(TAM1-t-Bu2)(L6)]+. Замена трет-бутильных заместителей на метильные приводит к стабилизации низкоспинового состояния и блокированию спинового перехода. Полученный результат подтверждает существенное влияние алкильных групп при атомах азота тетраазамакроциклического лиганда на энергетические характеристики данных систем.
Согласно вычисленным параметрам обмена, между парамагнитными центрами, локализованными на ионах железа (как в низко-, так и в высокоспиновом состояниях), и полиароматическом углеводороде ожидаются антиферромагнитные взаимодействия средней силы.
Таким образом, посредством квантовохимических расчетов показано, что комплексообразование ионов кобальта и железа с о-бензохиноновыми лигандами, включающими аценовые группы, может приводить к системам, проявляющим термически переключаемые спиновые состояния металлоцентрированных фрагментов и фотоуправляемые синглетно-триплетные переходы полициклического радикала. Механизм магнитной бистабильности таких комплексов будет определяться природой тетраазамакроциклического лиганда, в то время как наличие дополнительных каналов обмена зависит от структуры аценовой цепи.
В заключение отметим, что приведенные в обзоре результаты экспериментальных и теоретических исследований о-хиноновых комплексов металлов с тетраазамакроциклическими лигандами указывают на широкие перспективы практического применения данного класса магнитно-активных соединений в молекулярной электронике и спинтронике. Посредством выполненных нами квантовохимических расчетов объяснено магнитное поведение экспериментально изученных систем, однозначная интерпретация данных магнетохимических исследований которых осложнена комбинацией спиновых переходов и обменных взаимодействий. В изученной серии о-хиноновых комплексов кобальта и железа с тетраазамакроциклическими лигандами установлена зависимость реализации спиновых переходов, типа механизма таких перегруппировок, а также характера обменных взаимодействий от природы тетрадентатного азотсодержащего основания и заместителей в редокс-активном фрагменте. Функционализация структурного мотива [M(TAMn)(Ln)]+/2+, например, путем введения радикальных групп в о‑хиноновый фрагмент или конструирования на их основе биядерных структур открывает широкие перспективы получения новых магнитно-активных систем. Систематическое компьютерное моделирование электронного строения и магнитных свойств рассматриваемого типа соединений позволило предложить новые комплексы, потенциально способные проявлять механизмы магнитной бистабильности. Использование при конструировании новых молекул строительных блоков, представляющих собой известные соединения, позволяет ожидать успешный синтез предсказанных систем.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Poddel’sky A.I., Cherkasov V.K., Abakumov G.A. // Coord. Chem. Rev. 2009. V. 253. № 3–4. P. 291.
Teki Y., Shirokoshi M., Kanegawa S., Sato O. // Eur. J. Inorg. Chem. 2011. № 25. P. 3761.
Alley K.G., Poneti G., Aitken J.B. et al. // Inorg. Chem. 2012. V. 51. № 7. P. 3944.
Dai J., Kanegawa S., Li Z. et al. // Eur. J. Inorg. Chem. 2013. № 24. P. 4150.
Witt A., Heinemann F.W., Sproules S., Khusniyarov M.M. // Chem. Eur. J. 2014. V. 20. № 35. P. 11149.
Madadi, A., Itazaki, M., Gable R.W. et al.// Eur. J. Inorg. Chem. 2015. № 30. P. 4991.
Witt A., Heinemann F.W., Khusniyarov M.M. // Chem. Sci. 2015. V. 6. № 8. P. 4599.
Drath O., Gable R.W., Moubaraki B. et al. // Inorg. Chem. 2016. V. 55. № 9. P. 4141.
Zolotukhin A.A., Bubnov M.P., Bogomyakov A.S. et al. // Inorg. Chim. Acta. 2016. V. 440. P. 16.
Drath O., Gable R.W., Poneti G. et al. // Cryst. Growth Des. 2017. V. 17. № 6. P. 3156.
Zolotukhin A.A., Bubnov M.P., Arapova A.V. et al.// Inorg. Chem. 2017. V. 56. № 24. P. 14751.
Piskunov A.V., Pashanova K.I., Ershova I.V. et al. // J. Mol. Struct. 2018. V. 1165. P. 51.
Старикова А.А., Минкин В.И. // Успехи химии. 2018. Т. 87. № 11. С. 1049 (Starikova A.A., Minkin V.I. // Russ. Chem. Rev. 2018. V. 87. № 11. Р. 1049).
Piskunov A.V., Pashanova K.I., Bogomyakov A.S. et al. // Dalton Trans. 2018. V. 47. № 42. P. 15049.
Золотухин А.А., Бубнов М.П., Черкасов В.К., Абакумов Г.А. // Коорд. химия. 2018. Т. 44. № 2. С. 123 (Zolotukhin A.A., Bubnov M.P., Cherkasov V.K. et al. // Russ. J. Coord. Chem. 2018. V. 44. № 4. Р. 272). https://doi.org/10.1134/S1070328418040085
Пискунов А.В., Пашанова К.И., Ершова И.В. и др.// Изв. АН. Сер. хим. 2019. № 4. С. 757 (Piskunov A.V., Pashanova K.I., Ershova I.V. et al. // Russ. Chem. Bull. 2019. V. 68. № 4. P. 757).
Protasenko N.A., Poddel’sky A.I., Bogomyakov A.S. et al. // Inorg. Chim. Acta. 2019. V. 489. P. 1.
Черкасова А.В., Кожанов К.А., Золотухин А.А. и др. // Коорд. химия. 2019. Т. 45. № 7. С. 404 (Cherkasova A.V., Kozhanov K.A, Zolotukhin A.A. et al. // Russ. J. Coord. Chem. 2019. V. 45. № 7. P. 489). https://doi.org/10.1134/S1070328419070029
Ershova I.V., Smolyaninov I.V., Bogomyakov A.S. et al. // Dalton Trans. 2019. V. 48. № 28. P. 10723.
Zolotukhin A.A., Bubnov M.P., Bogomyakov A.S. et al. // Inorg. Chim. Acta. 2019. V. 488. P. 278.
Hauser A. // Coord. Chem. Rev. 1991. V. 111. P. 275.
Spin Crossover in Transition Metal Compounds. I–III / Topics in Curr. Chem. V. 233–235 / Eds. Gütlich P., Goodwin H.A. Berlin–Heidelberg: Springer-Verlag, 2004.
Spin-Crossover Materials: Properties and Applications / Ed. Halcrow M.A. Chichester: John Wiley & Sons, 2013. 564 p.
Минкин В.И., Стариков А.Г. // Изв. АН. Сер. хим. 2015. № 3. С. 475 (Minkin V.I., Starikov A.G. // Russ. Chem. Bull. 2015. V. 64. № 3. P. 475).
Buchanan R.M., Pierpont C.G. // J. Am. Chem. Soc. 1980. V. 102. № 15. P. 4951.
Evangelio E., Ruiz-Molina D. // Eur. J. Inorg. Chem. 2005. № 15. P. 2957.
Tezgerevska T., Alley K.G., Boskovic C. // Coord. Chem. Rev. 2014. V. 268. P. 23.
Минкин В.И. // Изв. АН. Сер. хим. 2008. № 4. С. 673 (Minkin V.I. // Russ. Chem. Bull. 2008. V. 57. № 4. P. 687).
Dei A., Sorace L. // Appl. Magn. Reson. 2010. V. 38. № 2. P. 139.
Molecular Switches / Eds. Feringa B.L., Browne W.R. Weinheim: Wiley-VCH, 2011. 824 p.
Troiani F., Affronte M. // Chem. Soc. Rev. 2011. V. 40. № 6. P. 3119.
Aromí G., Aguilà D., Gamez P. et al. // Chem. Soc. Rev. 2012. V. 41. № 2. P. 537.
Sato O. // Nature Chem. 2016. V. 8. № 7. P. 644.
Khusniyarov M.M. // Chem. Eur. J. 2016. V. 22. № 43. P. 15178.
Demir S., Jeon I.-R., Long J.R., Harris T.D. // Coord. Chem. Rev. 2015. V. 289–290. P. 149.
Senthil Kumar K., Ruben M. // Coord. Chem. Rev. 2017. V. 346. P. 176.
Drath O., Boskovic C. // Coord. Chem. Rev. 2018. V. 375. P. 256.
Gransbury G.K., Boulon M.-E., Petrie S. et al. // Inorg. Chem. 2019. V. 58. № 7. P. 4230.
Simaan A.J., Boillot M.-L., Carrasco R. et al. // Chem. Eur. J. 2005. V. 11. № 6. P. 1779.
Graf M., Wolmershäuser G., Kelm H. et al. // Angew. Chem. Int. Ed. 2010. V. 49. № 5. P. 950.
Koch W.O., Krüger H.-J. // Angew. Chem. Int. Ed. 1995. V. 34. № 23–24. P. 2671.
Koch W.O., Schünemann V., Gerdan M. et al. // Chem. Eur. J. 1998. V. 4. № 7. P. 1255.
Krüger H.J. // Chem. Ber. 1995. V. 128. № 6. P. 531.
Rupp F., Chevalier K., Graf M. et al. // Chem. Eur. J. 2017. V. 23. № 9. P. 2119.
Tezgerevska T., Rousset E., Gable R.W. et al. // Dalton Trans. 2019. V. 48. № 31. P. 11674.
Krüger H.-J. // Coord. Chem. Rev. 2009. V. 253. № 19–20. P. 2450.
Dei A., Gatteschi D., Pardi L. // Inorg. Chem. 1993. V. 32. № 8. P. 1389.
Benelli C., Dei A., Gatteschi D., Pardi L. // Inorg. Chim. Acta. 1989. V. 163. № 1. P. 99.
Caneschi A., Dei A., Fabrizi De Biani F. et al. // Chem. Eur. J. 2001. V. 7. № 18. P. 3926.
Carbonera C., Dei A., Sangregorio C., Letard J.-F. // Chem. Phys. Lett. 2004. V. 396. № 1–3. P. 198.
Caneschi A., Dei A., Gatteschi D., Tangoulis V. // Inorg. Chem. 2002. V. 41. № 13. P. 3508.
Neuwahl F.V.R., Righini R., Dei A. // Chem. Phys. Lett. 2002. V. 352. № 5–6. P. 408.
Bencini A., Caneschi A., Carbonera C. et al. // J. Mol. Struct. 2003. V. 656. № 1–3. P. 141.
Cador O., Dei A., Sangregorio C. // Chem. Commun. 2004. № 6. P. 652.
Bencini A., Beni A., Costantino F. et al. // Dalton Trans. 2006. № 5. P. 722.
Beni A., Dei A., Rizzitano M., Sorace L. // Chem. Commun. 2007. № 21. P. 2160.
Beni A., Dei A., Laschi S. et al. // Chem. Eur. J. 2008. V. 14. № 6. P. 1804.
Dei A., Feis A., Poneti G., Sorace L. // Inorg. Chim. Acta. 2008. V. 361. P. 3842.
Dapporto P., Dei A., Poneti G., Sorace L. // Chem. Eur. J. 2008. V. 14. № 35. P. 10915.
Poneti G., Mannini M., Sorace L. et al. // Chem. Phys. Chem. 2009. V. 10. № 12. P. 2090.
Dei A., Poneti G., Sorace L. // Inorg. Chem. 2010. V. 49. № 7. P. 3271.
Droghetti A., Sanvito S. // Phys. Rev. Lett. 2011. V. 107. № 4. P. 47.
Patricia T.T., Sandra M.V., Manuela L. et al. // Phys. Chem. Chem. Phys. 2012. V. 14. № 2. P. 1038.
Zhang Y.-M., Li A.-H., Yu F. // Acta Crystallogr. E. 2011. V. 67. № 7. P. m966.
Yu F. // Acta Crystallogr. E. 2012. V.68. № 10. P. m1248.
Bonnitcha P.D., Kim B.J., Hocking R.K. et al. // Dalton Trans. 2012. V. 41. № 37. P. 11293.
Yu F., Li B. // Inorg. Chim. Acta. 2012. V. 392. P. 199.
Panja A. // RSC Advances. 2013. V. 3. № 15. P. 4954.
Jang H.G., Cox D.D., Que L., Jr. // J. Am. Chem. Soc. 1991. V. 113. № 24. P. 9200.
Chiou Y.-M., Que, L., Jr. // Inorg. Chem. 1995. V. 34. № 14. P. 3577.
Mialane P., Tchertanov L., Banse F. et al. // Inorg. Chem. 2000. V. 39. № 12. P. 2440.
Jo D.-H., Chiou Y.-M., Que L., Jr. // Inorg. Chem. 2001. V. 40. № 13. P. 3181.
Pascaly M., Duda M., Schweppe F. et al. // Dalton Trans. 2001. № 6. P. 828.
Merkel M., Pascaly M., Wieting M. et al. // Z. Anorg. Allg. Chem. 2003. V. 629. № 12–13. P. 2216.
Merkel M., Schnieders D., Baldeau S.M., Krebs B. // Eur. J. Inorg. Chem. 2004. № 4. P. 783.
Merkel M., Pascaly M., Krebs B. et al.// Inorg. Chem. 2005. V. 44. № 21. P. 7582.
Hitomi Y., Yoshida M., Higuchi M. et al. // J. Inorg. Biochem. 2005. V. 99. P. 755.
Higuchi M., Hitomi Y., Minami H. et al. // Inorg. Chem. 2005. V. 44. № 24. P. 8810.
Simaan A.J., Boillot M.-L., Rivire E. et al. // Angew. Chem. Int. Ed. 2000. V. 39. № 1. P. 196.
Floquet S., Simaan A.J., Riviere E. et al. // Dalton Trans. 2005. № 9. P. 1734.
Enachescu C., Hauser A., Girerd J.-J., Boillot M.-L. // Chem. Phys. Chem. 2006. V. 7. № 5. P. 1127.
Girerd J.-J., Boillot M.-L., Blain G., Rivière E. // Inorg. Chim. Acta. 2008. V. 361. № 14–15. P. 4012.
Harding D.J., Harding P., Phonsri W. // Coord. Chem. Rev. 2016. V. 313. P. 38.
Collet E., Boillot M.-L., Hebert J. et al. // Acta Crystallogr. 2009. V. 65. № 4. P. 474.
Kaszub W., Buron-Le Cointe M., Lorenc M. et al. // Eur. J. Inorg. Chem. 2013. № 5–6. P. 992.
Collet E., Moisan N., Baldé C. et al. // Phys. Chem. Chem. Phys. 2012. V. 14. № 8. P. 6192.
Tissot A., Shepherd H.J., Toupet L. et al. // Eur. J. Inorg. Chem. 2013. № 5–6. P. 1001.
Mayilmurugan R., Stoeckli-Evans H., Palaniandavar M. // Inorg. Chem. 2008. V. 47. № 15. P. 6645.
Yu F., Zhang Y.-M., Li A.-H., Li B. // Inorg. Chem. Commun. 2015. V. 51. P. 87.
Meer M.V.D., Rechkemmer Y., Breitgoff F.D. et al. // Dalton Trans. 2016. V. 45. № 20. P. 8394.
Shultz D.A., Bodnar S.H., Vostrikova K.E., Kampf J.W. // Inorg. Chem. 2000. V. 39. № 26. P. 6091.
Shultz D.A., Vostrikova K.E., Bodnar S.H. et al. // J. Am. Chem. Soc. 2003. V. 125. № 6. P. 1607.
Shultz D.A., Kumar R.K., Bin-Salamon S., Kirk M.L. // Polyhedron. 2005. V. 25. № 16–17. P. 2876.
Kirk M.L., Shultz D.A., Schmidt R.D. et al. // J. Am. Chem. Soc. 2009. V. 131. № 51. P. 18304.
Kirk M.L., Shultz D.A. // Coord. Chem. Rev. 2013. V. 257. № 1. P. 218.
Kirk M.L., Shultz D.A., Stasiw D.E. et al. // J. Am. Chem. Soc. 2013. V. 135. № 45. P. 17144.
Tichnell C.R., Shultz D.A., Popescu C.V. et al. // Inorg. Chem. 2015. V. 54. № 9. P. 4466.
Katayama K., Hirotsu M., Kinoshita I., Teki Y. // Dalton Trans. 2014. V. 43. № 35. P. 13384.
Katayama K., Hirotsu M., Ito A., Teki Y. // Dalton Trans. 2016. V. 45. № 25. P. 10165.
Frisch M.J., Trucks G.W., Schlegel H.B. et al. Gaussian-09. Revision E. 01. Wallingford: Gaussian, 2013.
Tao J.M., Perdew J.P., Staroverov V.N., Scuseria G.E. // Phys. Rev. Lett. 2003. V. 91. № 14. P. 146401.
Staroverov V.N., Scuseria G.E., Tao J., Perdew J.P. // J. Chem. Phys. 2003. V. 119. № 23. P. 12129.
Bannwarth A., Schmidt S.O., Peters G. et al. // Eur. J. Inorg. Chem. 2012. № 16. P. 2776.
Cirera J., Paesani F. // Inorg. Chem. 2012. V. 51. № 15. P. 8194.
Старикова А.А., Минкин В.И. // Коорд. химия. 2018. Т. 44. № 4. С. 229 (Starikova A.A., Minkin. V.I. // Russ. J. Coord. Chem. 2018. V. 44. № 8. Р. 483). https://doi.org/10.1134/S1070328418080079
Starikova A.A. // Chem. Papers. 2018. V. 72. № 4. P. 821.
Стариков А.Г., Старикова А.А., Чегерев М.Г., Минкин В.И. // Коорд. химия. 2019. Т. 45. № 2. С. 92 (Starikov A.G., Starikova A.A., Chegerev M.G., Minkin V.I. // Russ. J. Coord. Chem. 2019. V. 45. № 2. Р. 105). https://doi.org/10.1134/S1070328419020088
Cirera J., Via-Nadal M., Ruiz E. // Inorg. Chem. 2018. V. 57. № 22. P. 14097.
Стариков А.Г.,Старикова А.А., Минкин В.И. // Д-окл. PАН. 2016. Т. 467. № 3. С. 300 (Starikov A.G., Starikova A.A., Minkin V.I. // Dokl. Chem. 2016. V. 467. № 1. Р. 83).
Starikova A.A., Chegerev M.G., Starikov A.G., Minkin V.I. // Comp. Theor. Chem. 2018. V. 1124. P. 15.
Noodleman L. // J. Chem. Phys. 1981. V. 74. № 10. P. 5737.
Shoji M., Koizumi K., Kitagawa Y. et al. // Chem. Phys. Lett. 2006. V. 432. № 1–3. P. 343.
Minkin V.I., Starikov A.G., Starikova A.A. // Pure A-ppl. Chem. 2018. V. 90. №5. P. 811.
Bally T. // Nature Chem. 2010. V. 2. № 3. P. 165.
Старикова А.А., Метелица Е.А., Стариков А.Г. // Журн. структур. химии. 2019. Т. 60. № 8. С. 1272 (Starikova A.A., Metelitsa E.A., Starikov A.G. // J. Struct. Chem. 2019. V. 60. № 8. P. 1219).
Стариков А.Г., Чегерев М.Г., Старикова А.А., Минкин В.И. // Коорд. химия. 2019. Т. 45. № 10. С. 579 (Starikov A.G., Chegerev M.G., Starikova A.A., Minkin V.I. // Russ. J. Coord. Chem. 2019. V. 45. № 1. P. 675). https://doi.org/10.1134/S1070328419090082
Starikova A.A., Minkin V.I. // Comp. Theor. Chem. 2018. V. 1138. P. 163.
Rudebusch G.E., Zafra J.L., Jorner K. et al. // Nat. Chem. 2016. V. 8. № 8. P. 753.
Nakano M., Fukuda K., Champagne B. // J. Phys. Chem. C. 2016. V. 120. № 2. P. 1193.
Старикова А.А., Метелица Е.А., Минкин В.И. // Коорд. химия. 2019. Т. 45. № 6. С. 350 (Starikova A.A., Metelitsa E.A., Minkin V.I. // Russ. J. Coord. Chem. 2019. V. 45. № 6. P. 411). https://doi.org/10.1134/S1070328419060095
Minkin V.I., Starikov A.G., Starikova A.A. et al. // Dalton Trans. 2018. V. 47. № 44. P. 15948.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия