

Координационная химия, 2020, T. 46, № 4, стр. 195-203
Новые бромсодержащие бис(арилимино)аценафтены и металлокомплексы на их основе
В. М. Макаров 1, Т. С. Копцева 1, В. Г. Соколов 1, В. А. Додонов 1, А. А. Скатова 1, *, Е. В. Баранов 1, И. Л. Федюшкин 1
1 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
* E-mail: skatova@iomc.ras.ru
Поступила в редакцию 03.10.2019
После доработки 23.10.2019
Принята к публикации 31.10.2019
Аннотация
По реакциям конденсации 5-бромаценафтенхинона с 2,6-диизопропиланилином и 4-бром-2,6-ди-изо-пропиланилином получены новые моно-и трибромзамещенные бис(арилимино)аценафтены: 1,2-бис[(2,6-диизопропилфенил)имино]-5-бромаценафтен (Dpp-Br-bian) (L1) и 1,2-бис[(4-бром-2,6-диизопропилфенил)имино]-5-бромаценафтен (п-Br-Dpp-Br-bian) (L2) соответственно. Соединения L1 и L2 выступают в качестве нейтральных лигандов в комплексах [(Dpp-Br-bian)ZnCl2] (I), [(п-Br-Dpp-Br-bian)AlCl3] (III) и [(п-Br-Dpp-Br-bian)GaCl3] (IV), полученных реакциями свободных дииминов с хлоридами соответствующих металлов. В свою очередь, реакция известного дибромпроизводного 1,2-бис[(4-бром-2,6-ди-изо-пропилфенил)иминоаценафтенa (п-Br-Dpp-bian) (L3) с хлоридом меди(I) также позволяет получить комплекс [(п-Br-Dpp-bian)CuCl] (II) с нейтральным дииминовым лигандом. Новые соединения L1, L2, I–IV охарактеризованы методами ЯМР-, ИК-спектроскопии и элементным анализом. Молекулярное строение I установлено методом РСА.
В течение последних десятилетий в химии переходных и непереходных металлов в качестве хелатирующих лигандов стали широко использоваться диимины, в том числе конформационно-жесткие бис(арилимино)аценафтены (Ar-bian). Несмотря на то что первые сообщения о них появились в 1990 гг. [1–4], в настоящее время изучение данных лигандов – одно из приоритетных направлений в химии координационных соединений, поскольку благодаря возможности варьирования органического заместителя при атоме азота можно направленно регулировать Льюисовскую кислотность металлокомплексов на их основе, а следовательно, их реакционную способность. К настоящему времени сообщалось о многочисленных комплексах переходных и непереходных металлов на основе Ar-bian [5], в которых лиганд может находиться в нейтральном, моно-, ди-, три- и тетраанионном [6] состояниях. Многие из этих соединений использовали в качестве эффективных катализаторов реакций органического синтеза [7–22], а также полимеризации олефинов [23–33] и циклических сложных эфиров [34–38].
Несмотря на огромный интерес к использованию лигандов Ar-bian в координационной химии, в литературе встречается лишь несколько представителей бромзамещенных производных данного ряда. Во всех случаях атомы брома находятся в пара-положениях арильного заместителя при атоме азота [24, 39, 40]. При этом производные никеля на основе п-Br-Dpp-bian являются эффективными катализаторами полимеризации алкенов [27, 28]. Учитывая этот факт в совокупности с данными об уникальных свойствах комплексов непереходных металлов с бис(арилимино)аценафтенами, такими как обратимое циклоприсоединение алкинов [16, 41], изотиоцианатов [42] и изоцианатов [43] к биядерным производным [(Dpp-b-ian)М–М(Dpp-bian)] (M = Ga, Al), способность комплекса галлия к двухэлектронному окислительному присоединению субстрата, подобно переходным металлам [44], а также способность комплекса магния к одноэлектронному восстановлению органических галогенидов и ароматических кетонов [45, 46], получение новых бромзамещенных лигандов ряда Ar-bian видится нам перспективным. Такие лиганды вызывают интерес не только с точки зрения металлокомплексного катализа, но и в качестве прекурсоров для синтеза новых аценафтен-1,2-дииминовых производных. Дальнейшая функционализация Ar-bian обеспечит их способность к формированию двумерных, трехмерных и полимерных каркасов за счет связывания функциональных групп с атомами металла как за счет редокс-активного дииминового фрагмента, так и за счет редокс-неактивных функциональных групп. В перспективе это позволит получать редокс-активные металлоорганические каркасы (MOFs) на основе редокс-активных органических лигандов, примеры которых относительно немногочисленны [47–49].
В настоящей статье мы сообщаем о синтезе моно-, ди- и трибромзамещенных бис(арилимино)аценафтенов, а также комплексов цинка, меди(I), алюминия и галлия на их основе. При этом аценафтен-1,2-диимины, содержащие атомы брома в нафталиновой части, получены впервые.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Соединения I–IV чувствительны к кислороду и влаге, поэтому все манипуляции по их синтезу, выделению и идентификации выполняли в вакууме с использованием техники Шленка или в атмосфере азота (Glovebox M. Braun). Толуол (Aldrich) сушили и хранили над натрийбензофеноном, пентан (Aldrich) – над натриевым зеркалом и отбирали конденсацией в вакууме непосредственно перед использованием. Другие растворители и реагенты, используемые для синтеза органических соединений, очищали по общепринятым методикам. Дейтеробензол (Aldrich) сушили над натрийбензофеноном и отбирали конденсацией в вакууме в ампулы для регистрации спектров ЯМР, содержащие образцы исследуемых соединений. 5-Бромаценафтен получали по методике [50].
ИК-спектры получали на спектрометре ФСМ-1201 в области 4000–450 cм–1 (образцы в вазелиновом масле). Спектры ЯМР 1H записывали на спектрометрах Bruker DPX-200 (200 МГц) и Bruker Avance III (400 МГц). Температуры плавления определяли в запаянных вакуумированных капиллярах. Элементный анализ выполняли сжиганием образцов в кислороде по методу Прегля.
Синтез 4-бром-2,6-ди-изо-пропиланилинa. К раствору 13.4 г (75.7 ммоль) 2,6-ди-изо-пропиланилина в 60 мл смеси этанол–уксусная кислота (2 : 1) в течение 10 мин добавляли 12.1 г брома (75.6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. По окончании реакции к смеси добавляли 20%-ный водный раствор NaOH до слабощелочной реакции. Органический слой экстрагировали диэтиловым эфиром (25 мл), дважды промывали водой и выдерживали в течение 15 ч над сульфатом магния. После удаления эфира образовалась красно-фиолетовая маслянистая жидкость – 4-бром-2,6-ди-изо-пропиланилин. Выход 18.9 г (97%).
Спектр ЯМР 1Н (200 МГц; CDCl3; 300 К; δ, м.д.; J, Гц): 7.17 (с., 2Н), 3.87 (c., 2Н), 2.92 (септ., 4Н, J = 6.8), 1.29 (д., 12Н, J = 6.8).
ИК-спектр (ν, cм–1): 3492 сл, 3405 сл, 2360 сл, 2337 сл, 1620 ср, 1575 сл, 1243 сл, 1209 сл, 866 ср, 838 сл, 670 сл. Спектры соответствуют литературным данным [51].
Синтез 5-бромаценафтенхинонa. К раствору 11.0 г (47.2 ммоль) 5-бромаценафтена и 0.5 г (1.3 ммоль) гептагидрата хлорида церия в уксусной кислоте (50 мл) при 40°С в течение часа небольшими порциями с постоянным перемешиванием добавляли дигидрат бихромата натрия (23.9 г, 80.2 ммоль). Реакционную смесь перемешивали 8 ч при 40°С, после чего добавляли 1 л холодной воды. Образовавшийся осадок отделяли фильтрованием (на воронке Бюхнера) и промывали водой. Образовавшееся твердое вещество вместе с 10%-ным раствором соды (200 мл) перемешивали в течение 30 мин при 80°С, затем отделяли фильтрованием (на воронке Бюхнера) и промывaли водой. Осадок экстрагировали 4%-ным водным раствором бисульфита натрия (500 мл) при 80°С в течение 30 мин. Полученный фильтрат при непрерывном перемешивании подкисляли концентрированной серной кислотой, достигая слабокислой реакции раствора. Выпавший осадок желтого цвета отделяли от раствора фильтрованием, последовательно промывали водой, этанолом и высушивали в вакууме. Выход 9.4 г (76%). Тпл > 238 (разл.).
Спектр ЯМР 1Н (200 МГц; DMSO-d6; 300 К; δ, м.д.; J, Гц): 8.37 (д., 1Н, J = 8.2), 8.24–8.09 (м., 2Н), 8.04 (д., 1Н, J = 7.6), 7.93 (д., 1Н, J = 7.2).
ИК-спектр (ν, cм–1): 2362 сл, 1730 с, 1684 сл, 1604 сл, 1576 сл, 1507 сл, 1304 сл, 1268 сл, 1209 сл, 1117 сл, 1083 сл, 1020 ср, 943 сл, 897 сл, 847 сл, 818 сл, 794 сл, 768 ср, 724 сл.
Синтез Dpp-Br-bian (L1). 5-Бромаценафтенхинон (3.0 г, 11.5 ммоль) в уксусной кислоте (50 мл) нагревали до 118°С. В полученный горячий раствор при постоянном перемешивании в течение часа по каплям прибавляли раствор 4.1 г (23.1 ммоль) 2,6-ди-изо-пропиланилина в уксусной кислоте (5 мл). По мере прибавления анилина происходило растворение осадка 5-бромаценафтенхинона и раствор приобретал темно-красный цвет. Реакционную смесь кипятили в течение 4 ч, затем выдерживали сутки при 5°С. Выпавший осадок красного цвета отделяли от раствора фильтрованием, последовательно промывали водой, этанолом, гексаном, диэтиловым эфиром и высушивали в вакууме. Выход 5.6 г (84%). Тпл > 265 (разл.).
Спектр ЯМР 1Н (400 МГц; CDCl3; 300 К; δ, м.д.; J, Гц): 8.10 (д., 1Н, J = 8.3), 7.62 (д., 1Н, J = 7.3), 7.48 (пс.т., 1Н), 7.31–7.27 (м., 5Н), 6.68 (д., 1Н, J = 7.3), 6.45 (д., 1Н, J = 7.8), 3.00 (септ, 4Н, J = 6.8), 1.24 (д., 12Н, J = 6.8), 0.98 (д., 6Н, J = 4.3), 0.97 (д., 6Н, J = 4.3).
ИК-спектр (ν, cм–1): 1743 сл, 1673 сл, 1654 ср, 1631 с, 1579 ср, 1523 ср, 1425 ср, 1361 сл, 1348 сл, 1324 ср, 1270 сл, 1237 сл, 1223 сл, 1188 ср, 1167 сл, 1108 сл, 1091 ср, 1043 ср, 1022 ср, 975 сл, 960 сл, 938 сл, 890 сл, 875 сл, 862 с, 836 ср, 816 сл, 803 сл, 781 с, 746 ср, 726 сл.
Синтез п-Br-Dpp-Br-bian (L2). 5-Бромаценафтенхинон (3.0 г, 11.5 ммоль) в уксусной кислоте (50 мл) нагревали до 118°С. В полученный горячий раствор при постоянном перемешивании в течение часа по каплям прибавляли раствор 6.0 г (23.4 ммоль) 4-бром-2,6-ди-изо-пропиланилина в уксусной кислоте (5 мл). По мере прибавления анилина происходило растворение осадка 5-бромаценафтенхинона и раствор приобретал темно-красный цвет. Реакционную смесь кипятили в течение 4 ч, затем выдерживали сутки при 5°С. Выпавший осадок красного цвета отделяли от раствора фильтрованием, последовательно промывали водой, этанолом, гексаном, диэтиловым эфиром и высушивали в вакууме. Выход 5.7 г (67%). Тпл > 215 (разл.).
Спектр ЯМР 1Н (400 МГц, CDCl3, 300 К, δ, м.д., J, Гц): 8.16 (д., 1Н, J = 8.5), 7.69 (д, 1Н, J = = 7.8), 7.55 (пс. т., 1Н, J = 7.6) 7.39 (с., 2Н), 7.38 (с., 2Н), 6.83 (д, 1Н, J = 7.0), 6.61 (д., 1Н, J = 7.8), 2.93 (септ, 4Н, J = 6.8), 1.21 (д., 12Н, J = 7.0), 0.97 (д., 6Н, J = 4.7), 0.95 (д., 6Н, J = 4.7).
ИК-спектр (ν, cм–1): 1711 с, 1676 ср, 1633 ср, 595 сл, 1582 сл, 1571 сл, 1327 ср, 1291 ср, 1271 сл, 1236 сл, 1209 сл, 1187 ср, 1171 сл, 1148 сл, 1093 сл, 1047 сл, 1029 сл, 970 сл, 942 сл, 924 сл, 865 ср, 846 сл, 824 сл, 781 ср, 750 сл, 728 ср, 627 сл, 478 сл.
Синтез п-Br-Dpp-bian (L3). Аценафтенхинон (5.0 г, 27.5 ммоль) в уксусной кислоте (50 мл) нагревали до 118°С. В полученный горячий раствор при постоянном перемешивании в течение часа по каплям прибавляли раствор 4-бром-2,6-ди-изо-пропиланилина (14.5 г, 56.6 ммоль) в уксусной кислоте (5 мл). По мере прибавления анилина происходило растворение осадка аценафтенхинона и раствор приобретал темно-красный цвет. Реакционную смесь кипятили в течение 4 ч, затем выдерживали сутки при 5°С. Выпавший осадок оранжевого цвета отделяли от раствора фильтрованием, последовательно промывали водой, этанолом, гексаном, диэтиловым эфиром и высушивали в вакууме. Выход 14.3 г (79%).
Спектр ЯМР 1Н (200 МГц; CDCl3; 300 К, δ, м.д.; J, Гц): 7.93 (д., 2Н, J = 8.3), 7.50–7.33 (м., 6Н), 6.78 (д., 2Н, J = 7.1), 2.96 (септ., 4Н, J = 6.8), 1.21 (д., 12Н, J = 6.8), 0.96 (д., 12Н, J = 6.8).
ИК-спектр (ν, cм–1): 1722 ср, 1678 сл, 1649 ср, 1597 сл, 1324 сл, 1277 ср, 1235 сл, 1189 сл, 1039 сл, 1015 сл, 944 сл, 924 сл, 862 ср, 835 ср, 822 сл, 785 ср, 722 сл.
Синтез [(Dpp-Br-bian)ZnCl2] (I). К раствору соединения L1 (0.29 г, 0.5 ммоль) в толуоле (20 мл) добавляли хлорид цинка (0.06 г, 0.5 ммоль). Смесь перемешивали 2 ч при 60°С. Полученный оранжевый раствор выдерживали в течение 24 ч при 10°С. Выпавшие оранжевые кристаллы отделяли от раствора декантацией, промывали холодным толуолом и высушивали в вакууме. Выход I 0.33 г (86%).
Спектр ЯМР 1Н (200 МГц; C6D6; 298 К; δ, м.д.; J, Гц): 7.62–7.50 (м., 1Н), 7.30–7.17 (м., 6Н), 7.15–6.97 (м., 5Н), 6.70 (д., 1Н, J = 7.8), 6.62–6.55 (м., 2Н), 6.28 (д., 1Н, J = 7.8), 3.77–3.55 (м., 4Н), 2.11 (с., 3Н), 1.47 (д., 12Н, J = 6.4), 0.82 (д., 6Н, J = 2.8), 0.79 (д., 6Н, J = 2.8).
ИК-спектр (ν, cм–1): 1661 с, 1625 с, 1593 ср, 1572 с, 1495 ср, 1417 ср, 1397 сл, 1365 сл, 1347 ср, 1326 ср, 1277 ср, 1253 ср, 1220 ср, 1208 ср, 1185 ср, 1170 сл, 1130 ср, 1118 ср, 1106 сл, 1096 сл, 1074 сл, 1043 сл, 1032 с, 974 с, 954 ср, 937 ср, 854 с, 823 ср, 800 с, 785 с, 759 с, 729 с, 695 с, 661 ср, 622 ср, 594 сл, 577 сл, 564 сл, 527 ср, 486 ср, 466 ср.
Синтез [(п-Br-Dpp-bian)CuCl] (II). К раствору соединения L3 (0.32 г, 0.5 ммоль) в толуоле (20 мл) добавляли 0.05 г хлорида меди (I) (0.5 ммоль). Смесь перемешивали 3 ч при 60°С. Полученный синий раствор выдерживали в течение 24 ч при 10°С. Выпавшие синие кристаллы отделяли от раствора декантацией, промывали холодным толуолом и высушивали в вакууме. Выход II 0.30 г (81%).
Спектр ЯМР 1Н (200 МГц; C6D6; 298 К; δ, м.д.; J, Гц): 7.58 (c., 4Н), 7.20–7.11 (м., 2Н), 6.83 (д., 2Н, J = 6.4), 6.64 (пс. т., 2Н), 3.42 (септ., 4Н, J = 6.4), 1.28 (д., 12Н, J = 6.4), 0.76 (д., 12Н, J = 6.4).
ИК-спектр (ν, cм–1): 1645 с, 1603 сл, 1573 ср, 1494 сл, 1365 сл, 1327 с, 1281 ср, 1232 сл, 1223 ср, 1187 с, 1149 сл, 1118 ср, 1086 сл, 1072 сл, 1043 ср, 1030 сл, 943 ср, 923 сл, 888 сл, 865 с, 845 сл, 829 ср, 819 с, 797 сл, 776 с, 729 с, 694 с, 617 ср, 533 ср, 499 сл, 465 с.
Синтез [(п-Br-Dpp-Br-bian)AlCl3] (III). Смесь диимина L2 (0.25 г, 0.34 ммоль) и AlCl3 (0.045 г, 0.34 ммоль) в толуоле (5 мл) перемешивали при 60°C в течение ночи. К полученному темно-оранжевому раствору добавляли пентан (20 мл). Из смеси толуол–пентан (9 : 1) выпадали оранжевые кристаллы. Выход III 0.21 г (62%).
Спектр ЯМР 1Н (400 МГц; C6D6; 298 К; δ, м.д.; J, Гц): 7.61 (д., 1H, J = 8.3), 7.52 (д., 4H, J = 8.5), 6.82 (д., 1H, J = 7.5), 6.77 (д., 1H), 6.65 (пс.т., 1H, J = 7.8), 6.46 (д., 1H, J = 7.8), 2.94 (уш.с., 4H), 1.15 (д., 6H, J = 6.3), 1.14 (д., 6H, J = 6.3), 0.8 (д., 6H, J = 6.8), 0.77 (д., 6H, J = 6.8).
ИК-спектр (ν, cм–1): 1674 ср, 1661 ср, 1634 ср, 1595 ср, 1574 с, 1423 ср, 1418 ср, 1366 ср, 1348 сл, 1319 с, 1265 ср, 1221 ср, 1207 сл, 1186 ср, 1152 ср, 1071 ср, 1030 ср, 972 с, 943 ср 889 сл, 876 ср, 864 ср, 845 сл, 822 с, 806 сл, 789 ср, 779 с, 741 ср, 638 сл, 611 сл, 604 сл, 529 сл, 496 о.с, 484 о.с.
Синтез [(п-Br-Dpp-Br-bian)GaCl3] (IV). Смесь диимина L2 (0.25 г, 0.34 ммоль) и GaCl3 (0.060 г, 0.34 ммоль) в толуоле (5 мл) перемешивали при 60°C в течение ночи. Все летучие продукты удаляли в вакууме и добавляли диэтиловый эфир (5 мл). Из эфирного раствора выпадали оранжевые кристаллы. Выход IV 0.24 г (65%).
Спектр ЯМР 1Н (400 МГц; C6D6; 298 К; δ, м.д.; J, Гц): 7.69 (уш. с., 1H), 7.64 (д., 4H, J = 5.3), 6.86 (уш. с., 1H), 6.77 (уш. с., 1H), 6.52 (уш. с., 1H), 6.15 (уш. с., 1H), 3.27 (уш. с., 2H), 2.94 (уш. с., 2H), 1.20 (уш. с, 12H), 0.81 (уш. с., 6H), 0.61 (уш. с., 6H).
ИК-спектр (ν, cм–1): 1661 ср, 1613 ср, 1587 сл, 1570 ср, 1418 ср, 1366 с, 1350 ср, 1321 ср, 1304 сл, 1277 сл, 1234 сл, 1221 сл, 1207 сл, 1182 ср, 1171 сл, 1152 сл, 1125 сл, 1092 сл, 1071 сл, 1061 сл, 1036 сл, 976 ср, 943 сл, 889 сл, 864 ср, 847 ср, 822 ср, 797 сл, 785 ср, 775 ср, 750 ср, 739 ср, 698 сл, 667 сл, 631 сл, 556 сл, 532 сл, 453 сл, 403 сл.
РСА I. Пригодные для РСА кристаллы соединения I получены кристаллизацией из толуола. Выбранный для анализа кристалл был покрыт минеральным маслом (Aldrich) и зафиксирован на стеклянном капилляре, затем помещен в холодный поток азота дифрактометра Agilent Xcalibur E (ω-сканирование, MoKα-излучение, λ = 0.71073 Å). Экспериментальные наборы интенсивностей интегрированы с помощью программы CrysAlisPro [52]. Структура решена прямым методом и уточнена полноматричным методом наименьших квадратов по $F_{{hkl}}^{2}$ с использованием программного пакета SHELXTL [53] в анизотропном приближении для неводородных атомов. Водородные атомы помещены в геометрически рассчитанные положения и уточнены изотропно. Учет поглощения произведен по программе SCALE3 ABSPACK [54]. Атомы брома и водорода разупорядочены по положениям 5 и 6 аценафтенового фрагмента. В кристалле I найдены разупорядоченные сольватные молекулы толуола и бензола в частном положении (на центре инверсии и на поворотной оси С3 соответственно). Основные кристаллографические характеристики и параметры рентгеноструктурного эксперимента для I приведены в табл. 1, некоторые длины связей и валентные углы – в табл. 2.
Таблица 1.
Кристаллографические данные, параметры эксперимента и уточнения структуры I
| Параметр | Значение |
|---|---|
| Брутто-формула | C41.50H45BrCl2N2Zn |
| М | 787.97 |
| Сингония | Тригональная |
| Пр. гр. | R$\bar {3}$ |
| Температура, К | 298(2) |
| Длина волны излучения, Å | 0.71073 |
| a, Å | 23.0682(4) |
| b, Å | 23.0682(4) |
| c, Å | 39.1825(8) |
| α, град | 90 |
| β, град | 90 |
| γ, град | 120 |
| V, Å3 | 18 057.2(7) |
| Z | 18 |
| ρ(выч.), г/см3 | 1.304 |
| μ, мм–1 | 1.770 |
| F(000) | 7326 |
| Размер кристалла, мм | 0.28 × 0.22 × 0.18 |
| Область измерений по θ, град | 2.89–26.37 |
| Индексы областей | –28 ≤ h ≤ 28, –28 ≤ k ≤ 28, –48 ≤ l ≤ 48 |
| Число измеренных отражений | 85 468 |
| Число независимых отражений (Rint) | 8148 (0.0483) |
| Число отражений с I > 2σ(I) | 5551 |
| Поправка на поглощение (макс/мин) | 0.779/0.696 |
| Данные/ограничения/параметры | 8148/92/490 |
| GOOF | 1.064 |
| R1, wR2 (I > 2σ(I)) | 0.0579, 0.1535 |
| R1, wR2 (по всем отражениям) | 0.0905, 0.1724 |
| Δρmax/Δρmin, e Å–3 | 0.920/–0.861 |
Таблица 2.
Некоторые длины связей (Å) и углы (град) в соединении I
| Связь | d, Å |
|---|---|
| Zn(1)–N(1) | 2.123(3) |
| Zn(1)–N(2) | 2.135(3) |
| Zn(1)–Cl(1) | 2.1905(14) |
| Zn(1)–Cl(2) | 2.1861(13) |
| N(1)–C(1) | 1.273(5) |
| N(2)–C(2) | 1.269(5) |
| C(1)–C(2) | 1.522(5) |
| Br(1)–C(8) | 1.917(4) |
| Угол | ω, град |
| N(1)ZnN(2) | 79.86(11) |
| Cl(2)Zn(1)Cl(1) | 122.39(6) |
Структура I зарегистрирована в Кембриджском банке структурных данных (CCDC № 1957209; ccdc.cam.ac.uk/getstructures).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Окислением 5-бромаценафтена бихроматом натрия в уксусной кислоте в присутствии хлорида церия (40°C, 9 ч) получен 5-бромаценафтенхинон (76%). Соединение охарактеризовано ЯМР- и ИК-спектроскопией. Последующая конденсация 5-бромаценафтенхинона с 2,6-ди-изо-пропиланилином в соотношении 1 : 2 в уксусной кислоте (118°C, 4 ч) (схема 1 ) привела к монобромзамещенному по нафталиновой части 1,2-бис[(2,6-ди-изо-пропилфенил)имино]-5-бромаценафтену (Dpp-Br-bian) (L1). Продукт реакции выделен в виде мелкокристаллического красного порошка с выходом 84%. По аналогичной реакция 5-бромаценафтенхинона с 4-бром-2,6-ди-изо-пропиланилином мы получили трибромзамещенное производное 1,2-бис[(4-бром-2,6-диизопропилфенил)ими-но]-5-бромаценафтен (L2) в виде мелкокристаллического вещества красного цвета с выходом 67% (схема 1 ). Отметим, что бромзамещенные по нафталиновой части бис(арилимино)аценафтены получены впервые. Конденсация 4-бром-2,6-ди-изо-пропиланилина с незамещенным аценафтенхиноном позволила получить известное соединение 1,2-бис[(4-бром-2,6-диизопропилфенил)имино]аценафтен (L3) [28]. Однако в данном случае в качестве растворителя использовали уксусную кислоту вместо толуола, что привело к сокращению времени реакции с 3 сут до 4 ч. Синтез соединений L1–L3 представлен на схеме 1 :
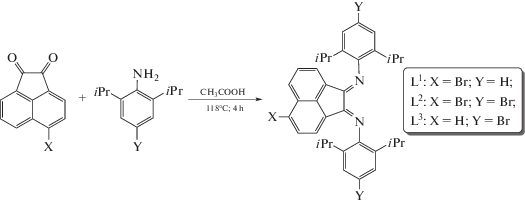
Схема 1 .
В ИК-спектрах новых соединений L1, L2 наблюдаются полосы поглощения при 1654, 1632 см–1 (L1) и 1711, 1676 см–1 (L2), отвечающие валентным колебаниям двойных связей C=N.
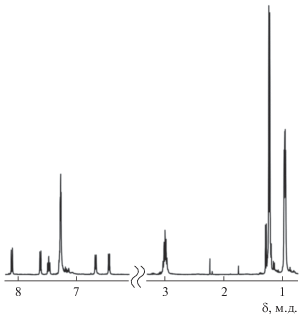
В спектре ЯМР 1H соединения L1 (рис. 1) присутствуют сигналы изо-пропильных групп 2,6-iPr2C6H3-заместителей: дублет удвоенной интенсивности (δ 1.24 (12Н) м.д.) и два перекрывающихся дублета (δ 0.98 (6Н) и 0.97 (6Н) м.д.) протонов восьми метильных групп, а также септет удвоенной интенсивности (δ 3.00 (4Н) м.д.) четырех метиновых протонов. Сигналы пяти протонов нафталиновой части расходятся и проявляются в виде четырех дублетов (δ 8.10 (1H), 7.62 (1H), 6.68 (1H) и 6.45 (1H) м.д.) и псевдотриплета δ 7.48 (1H) м.д. Ароматические протоны 2,6-iPr2C6H3-заместителей представлены мультиплетом в диапазоне 7.31–7.27 (5Н) м.д.
В спектре ЯМР 1H соединения L2 наблюдается тот же набор сигналов, что и для соединения L1: метиновые протоны изо-пропильных заместителей проявляются в виде септета при δ 2.93 (4Н) м.д., а протоны метильных групп изо-пропильных заместителей – в виде двух перекрывающихся дублетов (δ 0.97 (6Н), 0.95 (6Н) м.д.) и дублета удвоенной интенсивности при δ 1.21 м.д. (12H). В отличие от соединения L1, ароматические протоны 4-Br-2,6-iPr2-C6H2-заместителей в комплексе L2 представлены двумя синглетами при δ 7.39 (2Н) и 7.38 (2Н) м.д. Сигналы протонов нафталиновой части проявляются в виде четырех дублетов при δ 8.16 (1Н), 7.69 (1Н), 6.83 (1Н) и 6.61 (1Н) м.д. и псевдотриплета при δ 7.55 (1Н) м.д.
Реакция соединения L1 с хлоридом цинка (схема 2 ) протекает в толуоле при 60°С в течение 2 ч. Оранжевые кристаллы соединения [(Dpp-Br-bian)ZnCl2] (I) получены выдерживанием реакционной смеси в течение 24 ч при 10°С (выход 86%).
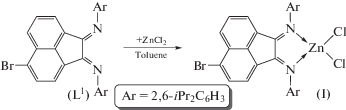
Схема 2 .
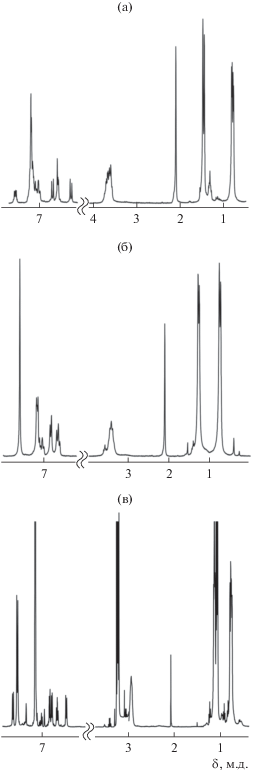
Спектр ЯМР 1H соединения I в дейтеробензоле (рис. 2а) содержит тот же набор сигналов, что и свободный бис(имино)аценафтен L1. Cигналы протонов метильных групп 2,6-iPr2C6H3-заместителей комплекса I: два перекрывающихся дублета при δ 0.82 (6Н) и 0.79 (6Н) м.д., дублет при δ 1.47 (12Н) м.д., а также мультиплет перекрывающихся сигналов метиновых протонов (δ 3.77–3.55 (4Н) м.д.). Дублеты при δ 6.28 (1Н) и 6.70 (1Н) м.д., а также сигналы в диапазонах δ 6.55–6.62 (2Н), 7.17–7.30 (6Н) и 7.50–7.62 (1Н) м.д. относятся к ароматическим протонам нафталинового фрагмента и фенильным заместителям при атомах азота. В кристаллической ячейке I присутствует молекула толуола, сигналы которого представлены мультиплетом при δ 7.15–6.97 (5Н) и синглетом при δ 2.11 (3Н) м.д.
Реакция соединения L3 с хлоридом меди(I) (cхема 3) протекает в толуоле при 60°С в течение 3 ч. Синие кристаллы комплекса [(п-Br-Dpp-b-ian)CuCl] (II) получены выдерживанием реакционной смеси в течение 24 ч при 10°С (выход 81%).
В ИК-спектрах комплексов I и II интенсивные полосы поглощения, характерные для колебаний связей C=N, проявляются при 1661 и 1625 см–1, а также при 1645 см–1 соответственно, что свидетельствует о нейтральной форме дииминового лиганда в этих соединениях.
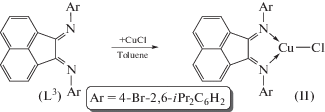
Схема 3 .
Поскольку молекула соединения II симметрична, спектр ЯМР 1H комплекса (рис. 2б) более простой по сравнению со спектром комплекса I с несимметричным лигандом. Так, в спектре ЯМР 1H соединения II наблюдаются сигналы 4-Br-2,6-iPr2C6H2-заместителей: два дублета (δ 0.76 (12Н) и 1.28 (12Н) м.д.) и септет δ 3.42 (4Н) м.д. изо-пропильных групп, а также синглет при δ 7.58 (4Н) м.д. ароматических протонов. Мультиплет δ 7.20–7.11 (2Н), дублет δ 6.83 (2Н) и псевдотриплет δ 6.64 (2Н) м.д. относятся к протонам нафталиновой части.
Реакция хлоридов алюминия и галлия с диимином L2 (cхема 4) в толуоле приводит к соединениям [(п-Br-Dpp-Br-bian)AlCl3] (III) и [(п-Br-Dpp-Br-bi-an)GaCl3] (IV), полученным в виде оранжевых кристаллов из пентана с выходами 62 и 65% соответственно.
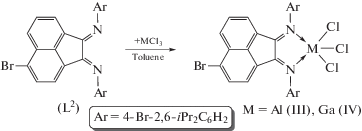
Схема 4 .
Из-за несимметричного строения соединений III и IV их спектры ЯМР 1Н довольно сложные. Так, спектр ЯМР 1Н комплекса IV (pис. 2в) содержит пять сигналов пяти неэквивалентных протонов нафталиновой части лиганда: дублеты при δ 7.61, 6.82, 6.77, 6.46 и псевдотриплет при 6.65 м.д. Дублет при δ 7.52 м.д. (4H) относится к протонам фенильных заместителей при атомах азота, уширенный синглет при 2.94 м.д. (4H) – к метиновым протонам изопропильных групп, четыре дублета при 1.15, 1.14, 0.8 и 0.77 м.д. (6H каждый) – к протонам групп CH3 изо-пропильных заместителей. Спектр ЯМР соединения IV содержит аналогичный набор сигналов.
ИК-спектры комплексов III и IV характеристичны для нейтральной формы Ar-bian лиганда: полосы поглощения при 1674, 1661 см–1 (III) и 1661 см–1 (IV) соответствуют валентным колебаниям двойных связей C=N.
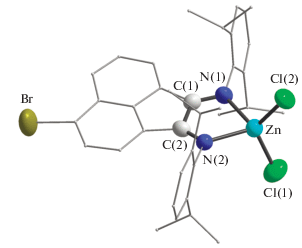
Согласно данным РСА комплекса I (рис. 3), координационное окружение атома цинка в нем – искаженный тетраэдр. Длины связей в ди-иминовом фрагменте (N(1)–C(1) 1.273(5), N(2)–C(2) 1.269(5), C(1)–C(2) 1.522(5) Å) подтверждают нейтральное состояние лиганда Dpp-Br-bian. Для сравнения в производном [(Cybu-bian)ZnCl2] (Cybu = циклобутил) c нейтральным диимином расстояния N(1)–C(1), N(2)–C(2) и C(1)–C(2) равны соответственно 1.282(3), 1.278(3) и 1.526(3) Å [55]. Тогда как, например, в соединениях [{(Dpp-bian)Zn–Ga(Dpp-bian)}{K(Thf)5}] [56] и [(Dpp-b-ian)Zn–Zn(Dpp-bian)] [57] с дианионным и анион-радикальным лигандами эти расстояния равны 1.377(3), 1.380(3), 1.362(3) и 1.3244(17), 1.3357(15), 1.4414(18) Å соответственно. Длины связей Zn–N(1) и Zn–N(2) в комплексе I (2.123(3) и 2.135(3) Å) заметно больше расстояний Zn–N в аналогичных бис(алкилимино)аценафтеновых нейтральных комплексах цинка [(Cybu-bian)ZnCl2] (2.081(2) и 2.085(2) Å) и [(Norb-bian)ZnCl2] (Norb = норборнил) (2.093(5) и 2.096(5) Å) [55], но хорошо согласуются с таковыми в соединении [(Dpp-bian)ZnCl2] (2.111(2) и 2.110(2) Å) [58]. Данные отличия объяснимы стерической загруженностью, обусловленной 2,6-iPr2C6H3-заместителями при атомах азота.
Рис. 3.
Молекулярная структура соединения I. Тепловые эллипсоиды приведены с 30%-ной вероятностью. Атомы водорода не показаны.
Таким образом, мы впервые получили бромзамещенные в нафталиновом кольце бис(арилимино)аценафтены и некоторые металлокомплексы на их основе. Такие аценафтен-1,2-диимины – удобные прекурсоры для дальнейшего получения функционализированных, например карбоксилзамещенных Ar-bian. Введение функциональных групп способно обеспечить формирование полимерных каркасов за счет связывания соседних молекул через атомы металла. Эта особенность сделает их удобными реагентами для получения металлоорганических координационных полимеров (MOF), содержащих редокс-активные органические дииминовые фрагменты. Такие соединения, возможно, будут проявлять свойства, которые в настоящее время для MOF неизвестны, например способность к обратимому переключению спектральных и магнитных характеристик, а также контролируемому регулированию реакционной способности благодаря настраиваемому изменению распределения заряда между металлом и редокс-активным органическим лигандом.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
van Asselt R., Elsevier C.J. // J. Mol. Catal. 1991. V. 65. № 3. P. L13.
van Asselt R., Gielens E.E.C.G., Rulke R.E. et al. // Chem. Commun. 1993. № 15. P. 1203.
van Asselt R., Gielens E.E.C.G., Rulke R.E. et al. // J. Am. Chem. Soc. 1994. V. 116. № 3. P. 977.
van Laren M.W., Elsevier C.J. // Angew. Chem. Int. Ed. 1999. V. 38. № 24. P. 3715.
Hill N.J., Vargas-Baca I., Cowley A.H. // Dalton Trans. 2009. № 2. P. 240.
Fedushkin I.L., Skatova A.A., Chudakova V.A. et al. // Angew. Chem. Int. Ed. 2003. V. 42. № 28. P. 3294.
Alonso J.C., Neves P., Pires da Silva M.J. et al. // Organometallics. 2007. V. 26. № 23. P. 5548.
Viganò M., Ragaini F., Buonomenna M.G. et al. // Chem. Cat. Chem. 2010. V. 2. № 9. P. 1150.
Gottumukkala A.L., Teichert J.F., Heijnen D. et al. // J. Org. Chem. 2011. V. 76. № 9. P. 3498.
de Fremont P., Clavier H., Rosa V. et al. // Organometallics. 2011. V. 30. № 8. P. 2241.
Li L., Lopes P.S., Rosa V. et al. // Dalton Trans. 2012. V. 41. № 17. P. 5144.
Rumble S.L., Page M.J., Field L.D. et al. // Eur. J. Inorg. Chem. 2012. V. 2012. № 13. P. 2226.
Moskalev M.V., Yakub A.M., Morozov A.G. et al. // Eur. J. Org. Chem. 2015. V. 2015. № 26. P. 5781.
Yakub A.M., Moskalev M.V., Bazyakina N.L. et al. // Russ. Chem. Bull. 2016. V. 65. № 12. P. 2887.
Yakub A.M., Moskalev M.V., Bazyakina N.L. et al. // Russ. Chem. Bull. 2018. V. 67. № 3. P. 473.
Fedushkin I.L., Moskalev M.V., Lukoyanov A.N. et al. // Chem. Eur. J. 2012. V. 18. № 36. P. 11264.
Fedushkin I.L., Nikipelov A.S., Morozov A.G. et al. // Chem. Eur. J. 2012. V. 18. № 1. P. 255.
Moskalev M.V., Skatova A.A., Chudakova V.A. et al. // Russ. Chem. Bull. 2015. V. 64. № 12. P. 2830.
Beltrani M., Carfagna C., Milani B. et al. // Adv. Synth. Catal. 2016. V. 358. № 20. P. 3244.
Marreiros J., Diaz-Couce M., Ferreira M. J. et al. // Inorg. Chim. Acta. 2019. V. 486. P. 274.
Yu X., Zhu F., Bu D. et al. // RSC Adv. 2017. V. 7. № 25. P. 15321.
Sandl S., Maier T. M., van Leest N. P. et al. // ACS Catal. 2019. V. 9. № 8. P. 7596.
Small B.L., Rios R., Fernandez E.R. et al. // Organometallics. 2010. V. 29. № 24. P. 6723.
Popeney C S., Guan Z. // Macromolecules. 2010. V. 43. № 9. P. 4091.
Miyamura Y., Kinbara K., Yamamoto Y. et al. // J. Am. Chem. Soc. 2010. V. 132. № 10. P. 3292.
Romain C., Rosa V., Fliedel C. et al. // Dalton Trans. 2012. V. 41. № 12. P. 3377.
Wang F., Tanaka R., Li Q. et al. // Organometallics. 2018. V. 37. № 9. P. 1358.
Liu J., Li Y., Li Y. et al. // J. Appl. Pol. Sci. 2008. V. 109. № 2. P. 700.
Soshnikov I.E., Bryliakov K.P., Antonov A.A. et al. // Dalton Trans. 2019. V. 48. № 23. P. 7974.
Liu Z.W.Q., Solan G.A., Sun W.-H. // Coord. Chem. Rev. 2017. V. 50. P. 8.
Guo L., Liu W., Chen C. // Mater. Chem. Front. 2017. № 2. P. 487.
Wang F., Chen C. // Polym. Chem. 2019. № 9. P. 354.
Soshnikov I.E., Bryliakov K.P., Antonov A.A. et al. // Dalton Trans. 2019. V. 8. № 3. P. 7974.
Brown L.A., Wekesa F.S., Unruh D.K. et al. // J. Pol. Sci. A. 2017. V. 55. № 17. P. 2824.
Kazarina O.V., Gourlaouen C., Karmazin L. et al. // Dalton Trans. 2018. V. 47. № 39. P. 13800.
Morozov A.G., Markelova E.S., Fedyushkin I.L. // Russ. J. Appl. Chem. 2018. V. 1. № 6. P. 1044.
Morozov A.G., Fedyushkin I.L., Aleinik D.Y. // J. Appl. Chem. 2016. V. 9. № 2. P. 2095.
Fedushkin I.L., Morozov A.G., Chudakova V.A. et al. // Eur. J. Inorg. Chem. 2009. № 33. P. 4995.
vanAsselt R., Elsevier C.J., Smeets W.J.J. et al. // Red. Trav. Chim. Pays-Bas. 1994. V. 113. P. 88.
Hasan K., Zysman-Colman E. // J. Phys. Org. Chem. 2013. V. 26. № 3. P. 274.
Fedushkin I.L., Nikipelov A.S., Lyssenko K.A. // J. Am. Chem. Soc. 2010. V. 132. № 23. P. 7874.
Zhang W., Dodonov V.A., Chen W. et al. // Chem. Eur. J. 2018. V. 24. № 56. P. 14994.
Dodonov V.A., Chen W., Zhao Y. et al. // Chem. Eur. J. 2019. V. 25. № 35. P. 8259.
Fedushkin I.L., SkatovaAA., Dodonov V.A. et al. // Inorg. Chem. 2014. V. 53. № 10. P. 5159.
Fedushkin I.L., Skatova A.A., Cherkasov V.K. et al. // Chem. Eur. J. 2003. V. 9. № 3. P. 778.
Fedushkin I.L., Skatova A A., Lukoyanov A.N. et al. // Russ. Chem. Bull. 2004. V. 3. № 2. P. 41.
Darago L.E., Aubrey M.L., Yu C.J. et al. // J. Am. Chem. Soc. 2015. V. 137. № 50. P. 15703.
Takaishi S., Hosoda M., Kajiwara T. et al. // Inorg. Chem. 2009. V. 48. № 19. P. 9048.
D’Alessandro D.M. // Chem. Commun. 2016. V. 52. № 58. P. 8957.
Дашевский М.М. Аценафтен. М.: Химия, 1966. 460 с.
Oskam J.H., Fox H.H., Yap K.B. et al. // J. Organomet. Chem. 1993. № 1–2. P. 185.
Data Collection. Reduction and Correction Program. CrysAlisPro 1.171.38.46 – Software Package, Rigaku OD, 2015.
Sheldrick G.M. SHELXTL. Version 6.14. Structure Determination Software Suite; Madison (WI, USA): Bruker AXS, 2003.
SCALE3 ABSPACK: Empirical Absorption Correction. CrysAlisPro 1.171.38.46 – Software Package, Rigaku OD, 2015.
Ragaini F., Gasperini M., Parma P. et al. // New J. Chem. 2006. V. 30. № 7. P. 1046.
Fedushkin I.L., Lukoyanov A.N., Tishkina A.N. et al. // Organometallics. 2011. № 3. P. 628.
Fedushkin I.L., Skatova A.A., Ketkov S.Y. et al. //Angew. Chem. Int. Ed. 2007. V. 46. № 23. P. 4302.
Villa M., Miesel D., Hildebrandt A. et al. // Chem. Cat. Chem. 2017. V. 9. № 16. P. 3203.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия