

Координационная химия, 2021, T. 47, № 10, стр. 640-650
Координационные полимеры палладия(I) с ненасыщенными дикарбоновыми кислотами со стабильными парамагнитными центрами
И. А. Ефименко 1, *, Н. Н. Ефимов 1, О. С. Ерофеева 1, Л. И. Демина 2, Н. П. Симоненко 1, А. В. Чураков 1, В. В. Минин 1
1 Институт общей и неорганической химии им. Н.С. Курнакова РАН
Москва, Россия
2 Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
Москва, Россия
* E-mail: ines@igic.ras.ru
Поступила в редакцию 30.10.2020
После доработки 17.03.2021
Принята к публикации 22.03.2021
Аннотация
Впервые осуществлен синтез координационных полимеров Pd(I) с ненасыщенными дикарбоновыми кислотами – малеиновой {[цис-Pd(C4H2O4)(H2O)] ∙ (H2O)}n (I), цитраконовой {[цис-Pd(C5H4O4)- (H2O)] ∙ 2H2O}n (II) и фумаровой {[транс-Pd(C4H2O4)(H2O)] ∙ (H2O)}n (III) со стабильными парамагнитными центрами в их полимерной матрице. На основании данных ИК- и ЭПР-спектроскопии (с подсчетом количества парамагнитных центров), а также результатов термогравиметрического анализа установлено, что в I–III строительными блоками являются, как минимум, четырехъядерные кластеры Pd(I), в которых атомы палладия связаны мостиковыми карбоксилатными группами. Каждая карбоксилатная группа координирована только одним атомом О, второй ее атом кислорода связан с ближайшим атомом палладия соседнего кластера, в результате образуется многомерный координационный полимер. Координационная сфера каждого атома Pd(I) в кластере дополняется молекулой воды и связью Pd–Pd. При нагревании I–III на первом этапе происходит потеря сольватных молекул воды, а при повышении температуры выше 100°С начавшаяся потеря координированных молекул воды сопровождается полным разложением комплексов. При взаимодействии I с CH3CN в метаноле протекает окислительно-восстановительная реакция, сопровождающаяся диспропорционированием I на комплекс Pd(II) и Pd(0) и образованием [Pd(HOOC–CH–CH(CH3O))- (CH3CN)2] (IV) (CIF file CCDC № 2039147).
Координационные полимеры – термин, введенный в координационную химию Д.С. Бейларом в 1964 г. [1]. В последнее десятилетие появилось большое количество публикаций, касающихся координационных полимеров, которые стали в настоящее время основой нового направления в материаловедении – создании металлоорганических каркасных материалов, привлекающих внимание исследователей широким потенциалом их практического применения. Активность изучения в последние десятилетия химии координационных полимеров обуслoвливается как их структурным разнообразием [2], так и уникальными функциональными характеристиками [3], такими как магнетизм [4–9], проводимость [10, 11] и различные области оптоэлектроники [12], а также использование в качестве основы каталитических и биометрических систем [13]. Такие материалы представляют собой комбинации различных металлических центров и органических связок. В качестве связок в настоящее время широко используются алифатические и ароматические карбоксилаты. Ненасыщенные дикарбоксилаты в этом качестве изучены совсем мало. Вместе с тем в ненасыщенных макромолекулярных карбоксилатах органические связки, в дополнение к связи металл–карбоксилатная группа, содержат потенциально способные к раскрытию кратные связи, обеспечивающие возможность формирования новых структур с интересными физическими свойствами [14, 15]. Основные результаты использования в качестве связок непредельных кислот на примере комплексов щелочных, щелочноземельных и ряда переходных металлов изложены в монографиях и обзорах [16–20].
Малеиновая, цитраконовая и фумаровая кислоты с сопряженной двойной связью, будучи полидентатными лигандами, могут рассматриваться в качестве органических связок, формирующих при координации с металлом каркасные системы различной топологии, как следствие различного расположения функциональных групп в них. Строение дикарбоновых кислот представлено на схеме 1 .
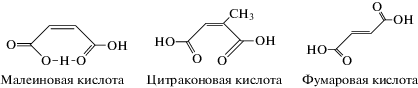
Так, 2D- и 3D-полимерные малеинаты и фумараты Fe, Co, Ni, Cu, Mn, Ag показали широкое разнообразие образующихся каркасов [2, 7, 13, 21–26]. Исследование дикарбоксилатов Cu(II), Mo(II), Ru(II, III), Rh(II) показали возможность создания пористых материалов, в которых величина пор и их окклюдирующая способность определяется, как природой металла, так и дикарбоновой кислоты [22, 23].
Что касается карбоксилатов палладия, то ранее в наших исследованиях показано, что при взаимодействии ацетата Pd(II) с ненасыщенными монокарбоновыми кислотами в ацетоне образуются трехядерные кластеры Pd(II) с мостиковыми карбоксилатными группами [PdL2]3 [27]. В реакциях с ненасыщенными дикарбоновыми кислотами (малеиновой и цитраконовой) в ацетоне при избытке кислоты образуются полимерные комплексы Pd(II) [PdL2Н2О]n, в которых одна молекула кислоты связана с Pd(II) обеими мостиковыми кабоксилатными группами, а вторая – только одной. Вторая депротонированная группа этой кислоты образует водородную связь с координированной молекулой Н2О ближайшего атома Pd [28].
Информации о комплексах Pd(I) с дикарбоновыми кислотами в литературе найдено не было. Первые координационные полимеры Pd(I) были получены нами при взаимодействии ацетата Pd(II) с монокарбоновыми кислотами – сорбиновой и 4‑пентеновой – в метаноле, в полимерной матрице которых методом ЭПР было зафиксировано существование стабильных парамагнитных центров [29]. Авторами [30, 31] показана возможность появления неспаренных электронов только при облучении обменных цеолитов (NaX, CaX), допированных монокристаллами K2PdCl4 при температуре не выше 77 К. В [32] была показана высокая каталитическая активность NaPdX- и CaPdX-цеолитов при димеризации этилена в н‑бутен. На первом этапе в процессе димеризации в системе появляются полученные in situ парамагнитные частицы Pd+C2H4 и Pd+(C2H4)2, которые по окончании процесса превращаются в парамагнитные частицы Pd+C4H8 с последующим восстановлением палладия до Pd(0).
Настоящая работа – продолжение начатых нами исследований полимерных ненасыщенных карбоксилатных комплексов Pd(I) со стабильными парамагнитными центрами Pd+ в их матрице, которое включало в себя разработку методов синтеза полимерных комплексов Pd(I) с дикарбоновыми кислотами (малеиновой, цитраконовой, фумаровой), изучение условий образования и стабильность парамагнитных центров и превращений в полимерной матрице Pd(I).
ЭКСПЕРИМЕТАЛЬНАЯ ЧАСТЬ
Для синтеза комплексов палладия с цис-малеиновой (С4Н4О4), цис-цитраконовой (С5Н6О4) и транс-фумаровой (С4Н4О4) кислотами в качестве исходного соединения использовали ацетат палладия Pd3(μ-MeCO2)6, полученный по разработанной нами методике [27]. В работе использовали реактивы без дополнительной очистки: малеиновая, цитраконовая, фумаровая кислоты (Sigma-Aldrich), метиловый спирт CH3OH, ацетон, ацетонитрил CH3CN (Химмед).
Синтез {[цис-Pd(C4H2O4)(H2O)] ∙ (H2O)}n (I). В круглодонную колбу помещали раствор Pd3(CH3CO2)6 0.224 г (1 ммоль) в CH3OH и 0.116 г (1 ммоль) малеиновой кислоты. Раствор перемешивали при комнатной температуре в течение ~2.5 ч до образования светло-коричневого осадка, который отфильтровали через фильтр Шотта № 16, промывали метанолом (3 × 10 мл) и высушивали в вакууме. Выход 56%.
Синтез {[цис-Pd(C5H4O4)(H2O)] ∙ 2H2O}n (II) выполняли по методике, аналогичной для I, с эквимолярным соотношением Pd3(CH3CO2)6 0.224 г (1 ммоль) и 0.130 мл (1 ммоль) цитраконовой кислоты в CH3OH. Синтез вели не более ~1.5 ч до образования осадка светло-коричневого цвета. При увеличении времени проведения синтеза начинается восстановление реакционного раствора до Pdмет. Выход 68%.
Синтез {[транс-Pd(C4H2O4)(H2O)] ∙ (H2O)}n (III). Синтез выполняли по методике, аналогичной для I, с эквимолярным соотношением Pd3(CH3CO2)6 0.224 г (1 ммоль) и 0.116 г (1 ммоль) фумаровой кислоты в CH3OH до образования осадка светло-коричневого цвета. Выход 45%.
Синтез [Pd(HOOC–CH–CH(CH3O))(CH3CN)2] (IV). В маточный раствор синтеза I после отделения осадка добавляли 0.116 г (1 ммоль) малеиновой кислоты и 5 мл CH3CN. При этом цвет реакционной смеси из светло-коричневого становился светло-зеленым. Из полученного раствора через 5 сут сформировывался кристаллический осадок светло-зеленого цвета (c микровкраплениями металлического Pd), из которого был отобран монокристалл.
Элементный анализ проводили на анализаторе фирмы Carlo Erba Instruments CHNSOEA 1108.
ИК-спектры кристаллических образцов регистрировали в интервале 4000–550 см–1 методом НПВО на ИК-Фурье спектрометре NEXUS фирмы NICOLET (однолучевой, сканирующий, светоделитель CsI, детектор TGS-CsI, фотометрическая точность 0.1%, разрешение 2 см–1) с использованием приставки MIRacle фирмы PIKETechnologies с алмазным кристаллом. Образцы наносили непосредственно на алмазный кристалл без дополнительной пробоподготовки.
КР-спектры регистрировали на спектрометре комбинационного рассеяния in Via “Renishaw” с длиной волны лазерного излучения λ = 633нм и мощностью менее 1 мВт.
Cпектры ЭПР комплексов регистрировали c использованием спектрометра ЭПР Elexsys E680X фирмы BRUKER в X-диапазоне (рабочая частота ~9.8 ГГц) при комнатной температуре. Исследуемые вещества после синтеза помещали в кварцевые ампулы и хранили в ампулах в течение всего срока эксперимента. Нагревание до 40°С проводили непосредственно в ампулах. Спектры исследуемых соединений корректировали с учетом спектра пустого резонатора. Концентрацию неспаренных электронов определяли двойным интегрированием спектров ЭПР и последующим анализом результатов с помощью функции SpinCount ЭПР-спектрометра, калиброванного эталоном Alanine Spin Concentration Sample фирмы BRUKER с концентрацией неспаренных электронов 1.75 × 1017(±10%).
Дифференциально-термический анализ проводили на приборе синхронный термоанализатор SDT Q600 (TA Instruments) в изотермических условиях при 40°С и постоянно повышающейся температуре в интервале 25–250°С со скоростью нагревания 5°C/мин.
РСА IV выполнен на автоматическом дифрактометре Bruker SMART APEX II при 240 K (MoKα-излучение, λ = 0.71073 Å, графитовый монохроматор). Адсорбционная коррекция введена на основании измерений интенсивностей эквивалентных отражений [33, 34]. Структура расшифрована прямым методом; все неводородные атомы уточнены полноматричным анизотропным МНК по F 2 (SHELXTL) [33]. Все атомы водорода помещены в рассчитанные позиции и уточнены с использованием схемы “наездник”. Исследованный кристалл представлял собой рацемический двойник с соотношением компонент 0.56(5) : 0.44(5).
Параметры кристаллической структуры IV депонированы в Кембриджском банке структурных данных (КБСД) (№ 2039147; deposit@ccdc.cam.ac.uk или http://www.ccdc.cam.ac.uk/data_request/cif).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Синтезированные в работе полимерные комплексы Pd(I) {[цис-Pd(C4H2O4)(H2O)] ∙ (H2O)}n (I), {[цис-Pd(C5H4O4)(H2O)] ∙ 2H2O}n (II) {[транс-Pd(C4H2O4)(H2O)] ∙ (H2O)}n (III) – результат реакции ацетата Pd(II) в метаноле с малеиновой, цитраконовой и фумаровой кислотами при соотношении реагентов 1 : 1, сопровождающийся восстановлением Pd(II) и образованием координационных полимеров Pd(I). В соответствии с аналитическими данными полученные соединения, характеризующиеся соотношением Pd : дикарбоксилат-ион : H2O, равным 1 : 1 : (H2O)n, где n = 2 (I, III) и 3 (II), практически нерастворимы в стандартных органических растворителях.
Строение I–III определяется прежде всего строением исходных кислот (схема 1
), а именно расположением карбоксилатных групп относительно двойной связи: цис-расположением в малеиновой и цитраконовой кислотах и транс-расположением в фумаровой кислоте. Анализ данных о строении малеиновой и фумаровой
кислот методом квантовой химии позволил авторам [35] выявить наиболее стабильные конформеры малеиновой и фумаровой кислот. Наиболее стабильный
почти плоский конформер малеиновой кислоты содержит семичленное кольцо 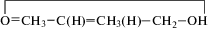 , образуемое формирующейся внутримолекулярной водородной связью –ОН…О=С– между карбоксилатными
группами одной молекулы малеиновой кислоты [35]. Полученные данные подтверждены расчетными и экспериментальными данными ИК-спектроскопии,
а именно положением как несопряженных, так и сопряженных ОН- и СО-групп. Рассматривая
результаты исследования фумаровой кислоты, авторы [35] выделили три наиболее стабильных конформера с мало отличающимися ИК-спектроскопическими
данными, представляющими собой широкие полосы в области 1760–1765 см–1, что является следствием сильного сопряжения в фумаровой кислоте транс-расположенных карбоксилатных групп по отношению к двойной связи.
, образуемое формирующейся внутримолекулярной водородной связью –ОН…О=С– между карбоксилатными
группами одной молекулы малеиновой кислоты [35]. Полученные данные подтверждены расчетными и экспериментальными данными ИК-спектроскопии,
а именно положением как несопряженных, так и сопряженных ОН- и СО-групп. Рассматривая
результаты исследования фумаровой кислоты, авторы [35] выделили три наиболее стабильных конформера с мало отличающимися ИК-спектроскопическими
данными, представляющими собой широкие полосы в области 1760–1765 см–1, что является следствием сильного сопряжения в фумаровой кислоте транс-расположенных карбоксилатных групп по отношению к двойной связи.
Следует отметить, что представленные в [35] исследования методом ИК-спектроскопии получены на образцах мономеров кислот высокой чистоты, приготовленных по специальной методике и выполнены при температуре жидкого азота. Используемые в нашей работе кислоты не подвергались специальной очистке. Некоторое расхождение в положении полос в ИК-спектрах используемых кислот с их положением в [35] обусловлены различием методов регистрации спектров и их пробоподготовки. Тем не менее ИК-спектроскопические характеристики используемых нами кислот принципиально не отличались от данных [35]. Так, подтверждением существования семичленного цикла в используемой в настоящей работе малеиновой кислоте являются полосы внешних и входящих в семичленный цикл групп С=О при 1812 и 1703 см–1, аналогичные полосам этих групп при 1808 и 1722 см–1 в [35], а также появление полос при 2164, 1887 см–1, связанных с образованием водородной связи, формирующей семичленный цикл в малеиновой кислоте. Сложная система сопряженных связей С=С и С=О линейной молекулы фумаровой кислоты описывается в ИК-спектре широкой полосой при 1691 см–1, в которой невозможно выделить отдельные колебания С=С и С=О. В спектре цитраконовой кислоты (СН3-группы у двойной связи вместо протона) наблюдаются полосы при 1691 см–1 ν(С=О), так и при 1640 см–1 ν(C=C). Спектры малеиновой, цитраконовой и фумаровой кислот содержат широкую область поглощения с рядом полос в интервале 3100–2100 см–1, характеризующих образование внутри- и межмолекулярных водородных связей. На фоне широкой полосы обозначаются пики полос при 3057 (I), 3060 (II) и 3082 (III), отвечающих валентным колебаниям ν(CH) и 2994, 2961 см–1 (II), соответствующих колебаниям ν(CH3). Типичные для карбоновых кислот δ(СОН) и ν(COH) проявляются в характерных для данного класса соединений диапазонax 1450–1420 и 1300–1200 см–1 соответственно. Такой подробный анализ спектров, используемых нами кислот, с учетом данных [35], был необходим для определения изменений в ИК-спектрах, происходящих в карбоксилатных группах при их координации палладием.
В спектрах I–III исчезают полосы сопряженных и несопряженных ОН- и С=О-групп кислот и появляются полосы при 1555 (I), 1539 (II) и 1516 (III) см–1, характеризующие νas(COO), и при 1382 (I), 1374 (II) и 1383 см–1(III), обусловленные νs(COO). Этот набор полос свидетельствует о мостиковой координации карбоксилатных групп обеих кислот. При этом в I–III при сохранении соотношения Pd : дикарбоксилат-ион = 1 : 1 каждый атом палладия координирует только один атом кислорода одной карбоксилатной группы дикарбоновой кислоты, а второй атом кислорода каждой мостиковой карбоксилатной группы координируется ближайшим атомом палладия соседнего кластера. Таким образом, одна молекула дикарбоновой кислоты, связывая четыре атома палладия, формирует полимерную сетку с объемными полостями (порами), что проявляется в уширении полос, соответствующих колебаниям νas, и наблюдается только в ИК-спектрах I–III.
Присутствие молекул воды в I–III проявляется в их ИК-спектрах в виде широкой полосы, отвечающей колебаниям ν(OH), в области 3300–3200 см–1 [21]. Полосы δ(НОН) в I–III перекрываются полосами, отвечающими колебаниям νas(COO) этих комплексов. Координационная сфера каждого атома Pd(I) дополняется одной координированной молекулой воды и связью Pd–Pd, наличие которой подтверждается полосой при 306 см–1 ν(Pd–Pd) в КР-спектре. С учетом изложенного выше и рентгеноструктурных данных полимерных матриц малеината и фумарата Ag(I), основой которых является биядерный малеинат Ag(I) и четырехъядерный фумарат Ag(I) [23], было сделано заключение о четырехъядерном кластере, как строительном блоке образующихся координационных полимеров I–III. Каждая молекула дикарбоновой кислоты связывает четыре атома палладия, что и обуслoвливает формирование полимерной сетки I–III. Как видно из схемы 2 , каркасы кластеров I и III имеют принципиально схожее строение, но за счет разного направления карбоксилатных групп в малеиновой (цис-расположения в I) и фумаровой кислотах (транс-расположения в III) координационные полимеры должны формироваться кластерными единицами с разной величиной пор в них.
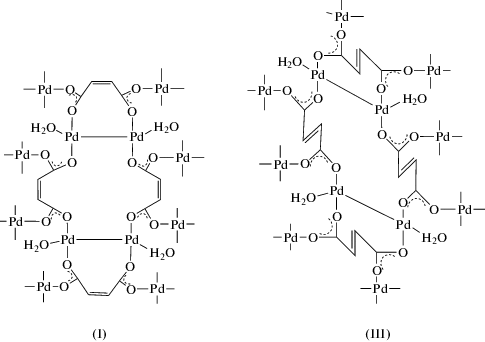
Схема 2 .
Известно, что спектроскопия ЭПР является чрезвычайно чувствительным методом определения наличия неспаренных электронов в веществах и материалах различной природы, в частности в координационных соединениях [36]. На рис. 1 представлены спектры ЭПР соединений I–III при комнатной температуре, аналогичные спектрам комплексов Pd(I) с сорбиновой и 4-пентеновой кислотами [29]. Наличие неспаренных электронов в соединениях I–III, по данным ЭПР, свидетельствует о том, что на момент выделения комплекса процесс формирования связи Pd–Pd в их полимерной матрице полностью не завершается, но замедляется в твердофазной матрице при выделении комплексов I–III.
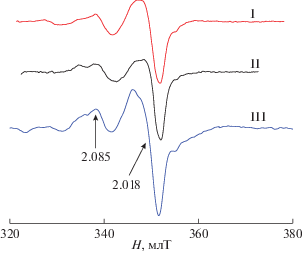
Измерение концентрации неспаренных электронов позволяет выявить влияние природы карбоксилатного лиганда на процессы формирования парамагнитных центров в их полимерной матрице. Из табл. 1 видно, что концентрация практически одинаковая в I и II с цис-расположенными карбоксилатными группами и почти на порядок выше в соединении III с их транс-расположением.
Таблица 1.
Концентрация неспаренных электронов в полимерной матрице I–III по данным ЭПР
| Соединение | Концентрация неспаренных электронов, спин/г | ||
|---|---|---|---|
| после синтеза | после нагревания при 40°С, 2 ч | после хранения 4 мес. | |
| I | 2.52 × 1017 | 1.57 × 1017 | 1.08 × 1017 |
| II | 2.46 × 1017 | 1.18 × 1017 | 5.02 × 1016 |
| III | 1.75 × 1018 | 1.16 × 1018 | 8.74 × 1017 |
Следует заметить, что на количество парамагнитных центров в полимерных матрицах I–III может влиять изменение условий их синтеза. Так, замена метилового спирта на ацетон в синтезе цитраконата Pd(I) II приводит к увеличению количества неспаренных электронов до 2.45 × 1018 спин/г против 2.46 × 1017 спин/г при синтезе II в метаноле. Уменьшение температуры синтеза I сопровождается увеличением количества неспаренных электронов в образце до 1.05 × 1019 спин/г по сравнению с их содержанием в I, полученном при комнатной температуре (2.52 × 1017 спин/г). Изложенные выше результаты об увеличении числа неспаренных электронов при замене метанола ацетоном или при снижении температуры реакции свидетельствуют о замедлении процессов формирования связей Pd–Pd в полимерной матрице I и в описанных выше условиях синтеза II при сохранении брутто-формулы. В связи с изложенным выше представляет несомненный интерес изучение влияния повышения температуры на процессы, происходящие в полимерных матрицах соединений I–III.
Данные о процессах, идущих в I–III при их нагревании, были получены методом ТГ и ДТА. На первом этапе было проведено исследование весовых изменений в соединениях I–III в изотермических условиях при 40°С (2 ч) и соответствующих изменений концентрации неспаренных электронов в них при тех же условиях. Комплексы I и III теряют в массе до 6%, что несколько ниже значения потери одной сольватной молекулы воды в расчете на один атом Pd (7%) как следствия прерывания процесса при 40°С через 2 ч. Термогравиграмма II, записанная в тех же условиях, демонстрирует убыль массы в 12%, что соответствует процессу потери двух сольватных молекулы воды.
По данным спектроскопии ЭПР, после изотермической выдержки I–III (40°С, 2 ч) и последующем хранении их при комнатной температуре в течение 4 мес. наблюдается некоторое уменьшение концентрации неспаренных электронов в их полимерных матрицах (табл. 1), что, хотя и свидетельствует о продолжающемся медленном процессе формирования связей Pd–Pd, позволяет оценивать полученные дикарбоксилатные полимеры Pd(I) как координационные полимеры со стабильными парамагнитными центрами.
Термогравиграммы I–III, полученные при их нагревании в интервале 30–250°С со скоростью 5°C/мин, демонстрируют плавный ход дифференциальных кривых в интервале 35–50°С, который отвечает весовой потере по одной сольватной молекуле воды. При 90°С наблюдается эндотермический процесс, сопровождающийся увеличением потери массы по сравнению с потерей только сольватных молекул (7%) и отвечающий началу процесса потери координированной молекулы воды. Однако выше 100°С эндотермический процесс непрерывно переходит в экзотермический процесс полного распада комплекса, завершающийся при 135–150°С. Масса твердого остатка соответствует содержанию Pd в исходных комплексах I и III (41%). На термогравиграмме II наблюдается потеря веса ~12%, отвечающая потере двух сольватных молекул воды. Далее, в интервале 150–175°С происходит резкая общая потеря веса ~63%, свидетельствующая о полном разложении комплекса.
Как было показано в [29], потеря координационной сферой мокарбоксилата Pd(I) – сорбата Pd(I) – координированной молекулы воды сопровождается его превращением в диамагнитный комплекс. Однако, как показало исследование дикарбоксилатов Pd(I), удаление координированной молекулы воды в I–III сопровождается полным разложением комплексов, что не позволяет получить их безводные диамагнитные аналоги, подобно сорбату Pd(I) [29].
Влияние природы образующих I–III карбоновых кислот на их реакционную способность показано при изучении взаимодействия I–III с CH3CN. Комплекс III при взаимодействии с CH3CN быстро восстанавливается до Pd(0) как в метаноле, так и в ацетоне. Комплекс II при реакции с CH3CN восстанавливается в метаноле. Комплекс I в реакции с CH3CN в ацетоне восстанавливается до Pd(0), а в метаноле образуется моноядерный комплекс Pd(II) – [Pd(HOOC–CH–CH(CH3O))(CH3CN)2] (IV), строение которого подтверждено данными РСА (рис. 2). Образование комплекса IV – результат проходящих в реакции двух процессов. В первом – в результате окислительно-восстановительной реакции Pd(I) в исходном комплексе диспропорционирует на образующийся комплекс Pd(II) и Pd(0), микровкрапления которого наблюдаются в кристаллической массе IV. Вторая реакция – восстановление двойной связи до одинарной – сопровождается образованием пятичленного цикла, координируемого Pd(I) связью β-углерода восстановленной двойной связи и кислородом одной карбоксилатной группы малеиновой кислоты. Вторая протонированная карбоксилатная группа остается свободной. Координационная сфера Pd(II) дополняется двумя молекулами ацетонитрила. В табл. 2 и 3 представлены кристаллографические данные IV.
Рис. 2.
Обе кристаллографически независимые молекулы в структуре IV. Тепловые эллипсоиды показаны с 50%-ной вероятностью.
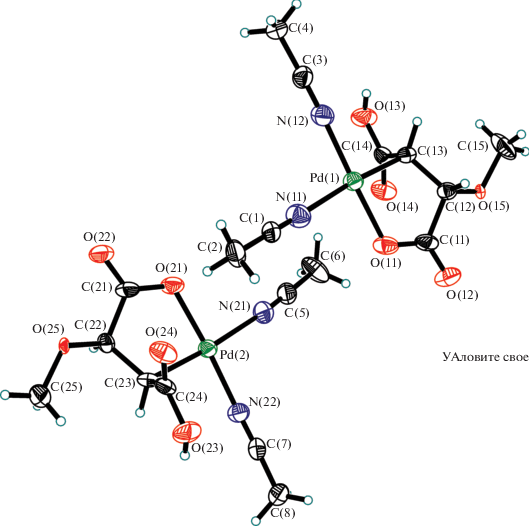
Таблица 2.
Кристаллографические данные и детали уточнения структуры IV
| Параметр | Значение |
|---|---|
| Брутто-формула | C9H12N2O5Pd |
| М | 334.61 |
| Размер кристалла, мм | 0.40 × 0.20 × 0.20 |
| Сингония | Ромбическая |
| Пр. гр. | Pna21 |
| a, Å | 13.3046(4) |
| b, Å | 14.3496(4) |
| c, Å | 12.7197(3) |
| V, Å3 | 2428.39(11) |
| Z | 8 |
| ρ(выч.), г/см3 | 1.830 |
| μ(MoKα), мм–1 | 1.539 |
| F(000) | 1328 |
| Область θ, град | 2.09–30.00 |
| Интервалы индексов | –18 ≤ h ≤ 18, –19 ≤ k ≤ 20, –17 ≤ l ≤ 16 |
| Всего отражений | 27 856 |
| Независимых отражений(Rint) | 6400(0.0321) |
| Отражений с I > 2σ(I) | 5670 |
| Число уточняемых параметров | 314 |
| R1 по I > 2σ(I) | 0.0588 |
| wR2 (все данные) | 0.1452 |
| GOОF по F 2 | 1.125 |
| Δρmin/Δρmax, e/Å3 | –0.584/3.394 |
Таблица 3.
Длины связей (d) в структуре IV
| Связь | Длина связи, Å |
|---|---|
| Pd(1)–N(12) | 1.974(5) |
| Pd(2)–N(22) | 1.981(4) |
| Pd(1)–O(11) | 1.995(4) |
| Pd(2)–O(21) | 2.000(4) |
| Pd(1)–C(13) | 2.043(5) |
| Pd(2)–C(23) | 2.030(5) |
| Pd(1)–N(11) | 2.115(5) |
| Pd(2)–N(21) | 2.122(5) |
В псевдосимметричной структуре IV присутствуют две кристаллографически независимые молекулы комплекса с очень близкими геометрическими параметрами, о чем свидетельствует результат взаимного наложения молекул с использованием метода наименьших квадратов (рис. 3).
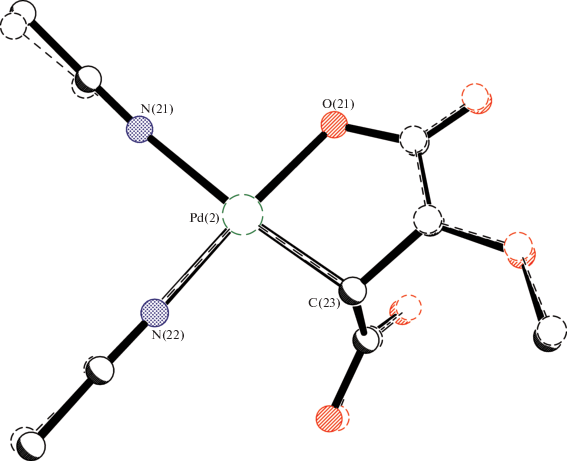
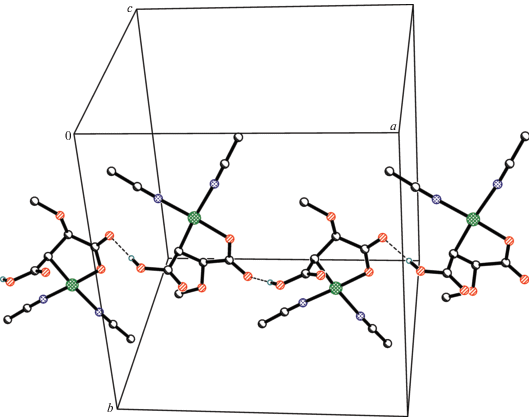
Центральные атомы палладия находятся в плоско-квадратном окружении с цис-углами между лигандами, лежащими в пределах 83.44(17)°–93.6(2)°. Как и исследовало ожидать, наименьшие значения этих углов относятся к эндоциклическим OPdC. Отклонения атомов Pd от плоскостей четырех базовых атомов не превышают 0.02 Å. Координационное окружение атомов металла состоит из хелатирующего О,С-лиганда и двух молекул ацетонитрила в цис-положениях относительно друг друга. Интересно отметить, что расстояния Pd–N в обеих молекулах различаются более чем на 0.1 Å, что, очевидно, вызвано сильно различным транс-влиянием противолежащих лигандов. Длины связей Pd–O близки к средней величине, найденной для немостиковых карбоксилатных комплексов палладия по КБСД [37] – 2.043 Å (876 структур, вер. 5.40, август 2019). То же самое наблюдается и для связи Pd–CAlk, средняя величина которой для плоско-квадратных комплексов палладия составляет 2.057 Å исходя из данных КБСД (по 1487 структурам). В обеих молекулах пятичленный металлоцикл принимает конформацию конверта, где в качестве клапана выступают атомы углерода С(13) и С(23), связанные с металлом. В кристалле соседние молекулы связаны в цепочки вдоль оси а за счет сильных водородных связей (O…O 2.649(7), 2.652(7) Å) между карбоксильными группами (рис. 4).
Таким образом, впервые осуществлен синтез координационных полимеров Pd(I) с ненасыщенными дикарбоновыми кислотами – малеиновой, цитраконовой и фумаровой – со стабильными парамагнитными центрами в их полимерной матрице. Методами ИК-, ЭПР-спектроскопии и термогравиметрического анализа установлено, что строительными блоками комплексов I–III могут являться кластеры, каркас которых состоит из четырех атомов палладия, координирующих четыре молекулы кислоты, координационная сфера, каждого атома Pd(I) в которых дополняется одной молекулой воды и связью Pd–Pd. При нагревании I–III при 40°С происходит потеря только сольватных молекул воды. В условиях записи термогравиграмм I–III при 30–250°С после потери сольватных молекул воды начинается процесс потери координационных молекул воды, который сопровождается полным их до Pdмет.
При взаимодействии I с CH3CN в метаноле протекающая окислительно-восстановительная реакция сопровождается диспропорционированием I на комплекс Pd(II) и Pd(0). В образовавшемся комплексе [Pd(HOOC–CH–CH(CH3O))(CH3CN)2] (IV), координационная сфера Pd(II) формируется за счет координации двух молекул CH3CN и пятичленного цикла, образовавшегося после восстановления двойной связи и связанного с Pd(II) связью Pd–β-C и атомом кислорода одной карбоксилатной группы малеиновой кислоты.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Bailar J.C. // J.R.J. Inorg. React. 1964. V. 1. P. 1.
Robin A.Y., Fromm K.M. // Coord. Chem. Rev. 2006. V. 250. P. 2127.
Noro S.-I., Miyasaka H., Kitagawa S. et al. // Inorg. Chem. 2005. V. 44. P. 133.
Zaworotko M.J. // Chem. Soc. Rev. 1994. P. 283.
Robson R.J. // Dalton Trans. 2000. P. 3735.
Moulton B., Zaworotko M.J. // Chem. Rev. 2001. V. 101. P. 1629.
Kim K. // Chem. Soc. Rev. 2002. V. 31. P. 96.
Miyasaka H., Matsumoto N., Okawa H. et al. // J. Am. Che. Soc. 1996. V. 118. P. 981.
Coronado E., Gaslan-Mascaros J.R., Gomez-Garcia C.J. et al. // Nature. 2000. V. 408. P. 447.
Hunig S., Kemmer M., Meixner H. et al. // Eur. J. Inorg. Chem. 1999. P. 899.
Kitagawa H., Onodera N., Sonoyama T. et al. // J. Am. Chem. Soc. 1999. V. 121. P. 10068.
Hou H., Meng X., Song Y. et al. // Inorg. Chem. 2002. V. 41. P. 4068.
Barthelet K., Marrot J., Riou D. et al. // Angew. Chem. Int. Ed. 2002. V. 41. P. 281.
Kim J., Chen B., Reinkek T. M. et al. // J. Am. Chem. Soc. 2001. T. 123. 8239.
Джардималиева Г.И., Помогайло А.Д. // Успехи химии. 2008. T. 77. № 3. С. 270.
Kickelbick G. // Prog. Polym. Sci. 2003. V. 28. № 1. P. 83.
Pomogailo A.D., Savostyanov V.S. Synthesis and Polymerization of Metal-Containing Monomers. BocaRaton (FL): CRCPress, 1994.
Порай-Кошиц М.А. // Итоги науки и техники. Кристаллохимия. Т. 1 / Под ред. Гилинской Э.Ф. Москва: Изд-во ВИНИТИ, 1981. С. 3.
Писаревский А.П., Мартыненко Л.И. // Коорд. химия. 1994. Т. 20. С. 324.
Кискин М.А., Еременко И.Л. // Успехи химии. 2006. T. 75. С. 627.
Porollo N.P., Aliev Z.G., Dzhardimalieva G.I. et al. // Russ. Chem. Bull. 1997. V. 46. № 2. P. 362.
Mori W., Takamizawa S. // J. Solid. State Chem. 2000. V. 152. P. 120.
Mori W., Sato T., Ohmura T. et al. // J. Solid. State Chem. 2005. V. 178. P. 2552
Bar A.K., Chakrabarty R., Mukherjee P.T. // Organometallics. 2008. V. 27. P. 3806.
Neo Y.C., Yeo J.S.L., Low P.M.N. et al. // J. Organomet. Chem. 2002. V. 658. P. 159.
Fernandes R.L., Takashi P.M., Frem R.C.G. et al. // J. Term. Analit. Calorim. 2009. V. 97. P. 153.
Ефименко И.А., Анкудинова П.В., Кузьмина Л.Г. и др. // Журн. неорган. химии. 2015. Т. 60. № 7. С. 935 (Efimenko I.A., Ankudinova P.V., Kuz’mina L.G. et al. // Russ. J. Inorg. Chem. 2015. V. 6. № 7. P. 848). https://doi.org/10.1134/S0036023615070050
Ефименко И.А., Демина Л.И., Анкудинова П.В. и др. // Журн. неорган. химии. 2016. Т. 61. № 10. С. 1309 (Efimenko I.A., Demina L.I., Ankudinova P.V. et al. // Russ. J. Inorg. Chem. 2016. V. 61. № 10. P. 1252). https://doi.org/10.1134/S0036023616100090
Efimenko I.A., Erofeeva O.S., Ugolkova E.A. et al. // Mend. Commun. 2018. V. 28. P. 632.
Prakash A.M., Wasowicz T., Kevan L. // J. Phys. Chem. 1997. V. 101. P. 1985.
Stokes L.S., Murphy D.M., Farley R.D. et al. // Phys. Chem. Chem. Phys. 1999. V. 1. P. 621.
Ghosh A.K., Kevan L. // J. Am. Chem. Soc. 1988. V. 110. P. 8044.
Sheldrick G.M. SADABS. Program for Scaling and Correction of Area Detector Data. Göttingen (Germany): Univ. of Göttingen, 1997.
Sheldrick G.M. // Acta Crystallogr. A. 2008. V. 64. № 1. P. 112.
Macoas E.M.S., Fausto R., Lundell J. et al. // J. Phys. Chem. A. 2001. V. 105. P. 3922.
Ракитин Ю.В., Ларин Г.М., Минин В.В. Интерпретация спектров ЭПР координационных соединений. М.: Наука, 1993.
Groom C.R., Bruno I.J., Lightfoot M.P. et al. // Acta Crystallogr. B. 2016. V. 72. P. 171.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия