

Координационная химия, 2021, T. 47, № 11, стр. 668-678
Влияние природы заместителей в оксиакридиновых лигандах на люминесцентные свойства и цитотоксичность цинковых комплексов
Т. В. Балашова 1, *, С. К. Полякова 1, 2, А. А. Кукинов 1, Р. В. Румянцев 1, Г. К. Фукин 1, В. А. Ильичев 1, Ю. А. Грачева 3, Т. А. Антоненко 3, Е. Р. Милаева 3, М. Н. Бочкарев 1, 2
1 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
2 Нижегородский государственный университет им. Н.И. Лобачевского
Нижний Новгород, Россия
3 Московский государственный университет им. М.В. Ломоносова
Москва, Россия
* E-mail: petrovsk@iomc.ras.ru
Поступила в редакцию 15.03.2021
После доработки 30.03.2021
Принята к публикации 01.04.2021
Аннотация
Синтезированы комплексы цинка с оксиакридиновыми лигандами Zn(Acr)2 и [Zn(Acr)Et]2 и методом РСА установлено их строение (CIF files CCDC № 2063229 и 2063230 соответственно). При возбуждении светом (λ = 405 нм) комплексы обнаружили эмиссию средней интенсивности с максимумами при 700 и 630 нм. Оба соединения проявили также слабые электролюминесцентные свойства. Из всего ряда полученных соединений цинка с оксиакридиновыми лигандами Zn(AcrR)2 и [Zn(AcrR)Et]2 (R = Me, Br, H) наиболее люминесцентно активными оказались комплексы с метильными заместителями в лигандах. Результаты проведенного МТТ-теста показали умеренную цитотоксическую активность полученных соединений.
Несмотря на многочисленные исследования в области органических светоизлучающих устройств (OLED), поиск новых эффективных люминофоров и изучение процессов трансформации энергии продолжаются во многих исследовательских центрах [1–4]. Комплексы металлов основной группы с лигандами оксихинолинового типа нашли широкое применение в качестве электролюминесцентных (ЭЛ) материалов: Zn [5], Cd [6], Cu [7], Al [8]. Среди них комплексы цинка привлекли особое внимание благодаря высокой фото- и электролюминесцентной активности, высокой термостойкости, легкой сублимации в вакууме и широкому спектру настраиваемых электронных свойств, легко изменяющихся при введении заместителей в лиганды. В ряду цинкорганических люминофоров оксихинолинолятные комплексы демонстрируют наиболее высокий квантовый выход фотолюминесценции [9]. 4-Гидроксиакридин можно представить, как производное 8-гидроксихинолина с расширенной π-системой [10, 11], что позволяет предположить наличие у него и комплексов на его основе люминесцентных свойств. Комплексы 4-гидроксиакридина с двухвалентными катионами, в том числе Zn2+, уже нашли применение в качестве эмиссионного компонента при разработке новых OLED [12, 13]. Кроме того, акридины относятся к группе природных соединений, обладающих выраженной антибактериальной и противоопухолевой активностью. Комплексные соединения акридинового ряда проявляют фармакологическую активность как противоопухолевые и противопаразитарные препараты. Ожидалось, что введение в структуру акридиновых лигандов алкильных заместителей должно привести к увеличению растворимости этих соединений в органических растворителях, что требуется для их применения в биомедицине и технологии OLED [14, 15].
В настоящей работе представлен синтез и фотофизические свойства комплексов цинка с пространственно затрудненным 1,3-ди-трет-бутилакридин-4-олом. Для учета изменения электронной структуры выполнены теоретические расчеты уровней энергии ВЗМО/НСМО. Обсуждается влияние заместителей и строения полученных соединений (cхема 1) на положение максимума люминесценции, а также ее интенсивность.

Схема 1 .
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтезы проводили в условиях, исключающих контакт с кислородом и влагой воздуха, с использованием стандартной техники Шленка. Ди-метоксиэтан (DME) сушили бензофенонкетилом натрия, пентан – над натрием по стандартным методикам и отбирали в вакууме непосредственно перед использованием. В работе применяли коммерческий реактив Et2Zn (Aldrich). Комплексы Zn(AcrMe)2 (I), [Zn(Et)(AcrMe)]2 (II), Zn(AcrBr)2 (III) и [Zn(Et)(AcrBr)]2 (IV) синтезировали, как описано ранее [16], лиганд 1,3-ди-трет-бутилакридин-4-ол [H(Acr)] – по методике [17].
Синтез Zn(Acr)2 (V). Раствор 26 мг (0.21 ммоль) диэтилцинка в 5 мл ДМЕ добавляли к раствору 129 мг (0.42 ммоль) H(Acr) в 10 мл ДМЕ. Реакционную смесь перемешивали в течение 30 мин при комнатной температуре. Удаляли все летучие продукты. Полученное вещество промывали холодным пентаном и высушивали в вакууме. Выход V в виде бордового порошка 113 мг (79%). Кристаллы, пригодные для РСА, получали при перекристаллизации комплекса V из смеси растворителей ДМЭ–диэтиловый эфир (1 : 1).
ИК-спектр (KBr; ν, cм–1): 1622 ср, 1589 сл, 1577 с, 1558 ср, 1514 с, 1438 с, 1408 ср, 1373 ср, 1344 с, 1306 ср, 1280 ср, 1253 ср, 1239 ср, 1219 ср, 1199 сл, 1146 ср, 1104 ср, 992 ср, 975 сл, 955 сл, 941 ср, 925 сл, 895 сл, 879 сл, 849 сл, 817 сл, 764 с, 744 с, 645 ср, 624 ср, 600 сл, 548 сл, 531 ср, 517 сл, 505 сл, 477 ср. ЯМР 1H (400 МГц; пиридин-d5; δ, м.д.): 10.02 (д., J = 8.9 Гц, 1H), 9.59 (с., 1H), 8.18 (д., J = 8.3 Гц, 1H), 8.06 (с., 1H), 7.89–7.83 (м., 1H), 7.56–7.51 (м., 1H), 2.00 (с., 9H), 1.71 (с., 9H).
Синтез [Zn(Et)(Acr)]2 (VI). Раствор 61 мг (0.49 ммоль) диэтилцинка в 5 мл ДМE добавляли к раствору 135 мг (0.41 ммоль) H(Acr) в 10 мл ДМЕ. Реакционную смесь перемешивали в течение 30 мин при комнатной температуре. При медленном удалении летучих продуктов и растворителя выпадают оранжевые кристаллы комплекса VI. Выход 160 мг (81%).
ИК-спектр (KBr; ν, cм–1): 1627 ср, 1593 сл, 1590 с, 1556 сл, 1512 с, 1410 ср, 1394 сл, 1348 с, 1303 сл, 1283 сл, 1275 сл, 1234 ср, 1218 сл, 1183 сл, 1167 сл, 1145 с, 1095 ср, 1026 сл, 1009 сл, 953 сл, 939 сл, 918 ср, 893 сл, 882 ср, 846 сл, 821 сл, 764 с, 745 с, 701 сл, 641 ср, 629 ср, 601 ср, 593 сл, 520 с, 476 сл. ЯМР 1H (400 МГц; пиридин-d5; δ, м.д.): 10.03 (д., J = 8.8 Гц, 1H), 9.60 (с., 1H), 8.19 (д., J = 8.2 Гц, 1H), 8.07 (с., 1H), 7.90–7.85 (м., 1H), 7.57–7.52 (м., 1H), 2.01 (с., 9H), 1.77 (т., J = 8.1 Гц, 3H), 1.72 (с., 9H), 1.32 (кв., J = 8.2 Гц, 2H).
ИК-спектры записывали на Фурье-спектрометре ФСМ-1201 в диапазоне 4000–400 см–1 (суспензии в вазелиновом масле). С,Н,N,S-элементный анализ выполняли на анализаторе Elementar Vario ELcube Analyzer. Спектры поглощения записывали в кварцевой кювете диаметром 1 см на спектрометре Per-kinElmer Lambda-25 от 200 до 700 нм. Спектры фотолюминесценции (ФЛ) регистрировали на спектрометре USB2000 при возбуждении диодным лазером 405 нм. Спектры ЯМР 1Н записывали в C5D5N при 298 К на спектрометре Bruker Avance III 400 с рабочей частотой 400 МГц. Химические сдвиги ядер 1Н приведены относительно ТМС.
Трехслойные OLED-устройства состава ITO/ TPD/complex/Bath/Yb (ITO = окись индия-олова (анод), TPD = N,N'-бис(3-метилфенил)-N,N'-дифенилбензидин (дырочно-транспортный слой), Bath = 4,7-дифенил-1,10-фенантролин (дырочно-блокирующий слой), Yb = катод) готовили напылением в вакуумной камере при 10–6 Торр по методике [18]. Толщину наносимых слоев контролировали с помощью кварцевого резонатора.
Спектры ЭЛ в видимом диапазоне и вольт-амперные (яркостные) характеристики получали на некапсулированных образцах с использованием флуориметра USB-2000 (Ocean Optics), источника питания PPE 3323 (GW Instek) и цифрового мультиметра GDM 8246 (GW Inste).
РСА комплексов V и VI проведен на автоматических дифрактометрах Bruker D8 Quest и Oxford Xcalibur Eos (графитовый монохроматор, МоКα-излучение, φ- и ω-сканирование, λ = 0.71073 Å) соответственно. Экспериментальные наборы интенсивностей интегрированы с помощью программ SAINT [19] для V и CrysAlisPro [20] для VI. Программы SADABS [21] (V) и алгоритм SCALE3 ABSPACK (VI) [20] использованы для введения поправок на поглощение. Структуры расшифрованы методом “dual-space” с использованием программы SHELXT [22]. Все неводородные атомы в комплексах V и VI уточнены полноматричным МНК по $F_{{hkl}}^{2}$ в анизотропном приближении с помощью программного пакета SHELXTL [23]. Атомы водорода помещены в геометрически рассчитанные положения и уточнены в модели наездника (Uизо(H) = 1.5Uэкв(C) для CH3 групп и Uизо(H) = 1.2Uэкв(C) для остальных групп). Основные кристаллографические характеристики и параметры рентгеноструктурного эксперимента для V и VI приведены в табл. 1.
Таблица 1.
Кристаллографические данные, параметры эксперимента и уточнения структур V и VI
| Параметр | Значение | |
|---|---|---|
| V | VI | |
| Брутто-формула | C42H48N2O2Zn | C46H58N2O2Zn2 |
| М | 678.19 | 801.68 |
| Температура, K | 100(2) | 100(2) |
| Сингония | Моноклинная | Моноклинная |
| Пр. гр. | C2/c | Pс |
| a, Å | 14.9598(6) | 10.1326(3) |
| b, Å | 17.4638(6) | 13.9696(3) |
| c, Å | 14.4507(5) | 15.3811(5) |
| α, град | 90 | 90 |
| β, град | 108.7204(12) | 109.095(4) |
| γ, град | 90 | 90 |
| V, Å3 | 3575.6(2) | 2057.38(11) |
| Z | 4 | 2 |
| ρ (выч.), мг/м3 | 1.260 | 1.294 |
| μ, мм–1 | 0.725 | 1.204 |
| Размер кристалла, мм | 0.19 × 0.17 × 0.04 | 0.51 × 0.22 × 0.10 |
| F(000) | 1440 | 848 |
| θ, град | 2.649–26.022 | 2.916–25.023 |
| Число отражений собранных/независимых | 24 674/3533 | 28 227/7246 |
| Rint | 0.0358 | 0.0490 |
| R1, wR2 (I > 2σ(I)) | 0.0353 0.0795 |
0.0356 0.0771 |
| R1, wR2 (по всем данным) | 0.0431 0.0832 |
0.0466 0.0822 |
| Абсолютный структурный параметр | –0.027(6) | |
| S | 1.052 | 1.033 |
| Остаточная электронная плотность (max/min), e/Å3 | 0.347/–0.352 | 0.452/–0.262 |
Структуры депонированы в Кембриджском банке структурных данных (№ 2063229 (V) и 2063230 (VI); ccdc.cam.ac.uk/structures/).
МТТ-тест. Жизнеспособность клеток исследовали с помощью MTT [3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолиум бромид] анализа по методу Никса с небольшими изменениями [24]. MCF-7, линии раковых клеток человека HCT-116 культивировали в среде DMEM (ПанЭко, Россия) с глутамином (ПанЭко, Россия) и антибиотиками (ПанЭко, Россия) в СО2 (5%) при 37°С. Соединения растворяли (20 мМ) в ДМСО и затем добавляли в среду для культивирования клеток в необходимой концентрации с максимальным содержанием ДМСО 0.5 об. %. Как показали контрольные эксперименты, в этих концентрациях ДМСО не влияет на жизнеспособность клеток. Ячейки были инкубированы в 96-луночных планшетах (7000 клеток/лунку) и обрабатывали различными концентрациями (от 0.01 до 100 мМ) тестируемых соединений, а также цисплатина при 37°C в течение 72 ч. Жизнеспособность клеток определяли с помощью теста МТТ, который количественно определяет активность дегидрогеназы. Затем клетки инкубировали при 37°C в течение 50 мин с раствором МТТ (10 мл, 5 мг/мл) (SigmaAldrich, Сент-Луис, США). Супернатант отбрасывали, а клетки растворяли в ДМСО (100 мл). Оптическую плотность раствора измеряли при 570 нм с использованием 96-луночного планшета (Anthos Zenyth 2000, Biochrom, Великобритания), а процент выживших клеток рассчитывали по поглощению необработанных клеток. Каждый эксперимент повторяли не менее 3 раз и каждую концентрацию тестировали трижды. Значения концентрации 50% ингибирования (IC50) со стандартным отклонением рассчитывали с помощью GraphPad Prism (версия 5.03).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Комплекс V получен из диэтилцинка и 1,3-ди-трет-бутилакридин-4-ола (схема 2 ). Реакция легко протекает при комнатной температуре в ДМЭ, образуются темно-бордовые кристаллы Zn(Acr)2. При проведении реакции в соотношении реагентов 1 : 1 или с избытком диэтилцинка образуется комплекс VI в виде ярко-оранжевых кристаллов. Полученные соединения хорошо растворимы в большинстве органических растворителей, устойчивы на воздухе как в твердом состоянии, так и в растворе.

Схема 2 .
По данным РСА, комплекс V имеет схожее строение с родственным комплексом, содержащим метильный заместитель в седьмом положении оксиакридинового лиганда [16]. Катион цинка связан с двумя бидентатно координированными оксиакридиновыми лигандами (рис. 1). Таким образом, координационное окружение Zn2+ в V представляет собой искаженный тетраэдр (τ4 = 0.73) [25], а координационное число равно четырем.
Рис. 1.
Молекулярная структура комплекса Zn(Acr)2 (V). Тепловые эллипсоиды приведены с 30%-ной вероятностью. Атомы водорода не показаны для наглядности. Основные расстояния и углы в комплексе V: Zn(1)–O(1) 1.9315(13), Zn(1)–N(1) 2.0253(14), O(1)–C(1) 1.327(2), N(1)–C(2) 1.355(2), C(1)–C(2) 1.445(3) Å и O(1)Zn(1)N(1) 84.07(6)°, O(1)Zn(1)O(1A) 114.46(8)°, O(1)Zn(1)N(1A) 128.51(6)°, N(1)Zn(1)O(1A) 128.51(6)°, N(1)Zn(1)N(1A) 122.67(8)°.

Основные геометрические характеристики в комплексе V находятся в хорошем согласии с ранее опубликованными родственными соединениями цинка (КЧ 4) [16, 26, 27]. Лиганды Acr в комплексе V практически плоские. Среднее отклонение атомов от плоскости составляет 0.03 Å. Интересно отметить, что в родственных соединениях [16, 26, 27] двугранный угол между плоскостями лигандов варьируется от 86.90° до 88.45°, тогда как в комплексе V значение этого параметра составляет 75.41°. Вероятно, такое взаимное расположение лигандов в V определяется наличием взаимодействий между лигандами соседних молекул. В кристалле формируются бесконечные цепочки (рис. 2). Плоскости лигандов параллельны друг другу, а расстояния между центрами ароматических систем A…B и C…D составляют 3.486 Å. Такие геометрические характеристики свидетельствуют о реализации межмолекулярного π…π-взаимодействия между оксиакридиновыми лигандами [28].
Рис. 2.
Фрагмент кристаллической упаковки комплекса V. Атомы водорода и метильные углероды трет-бутильных групп не приведены для наглядности.

Комплекс VI представляет собой несимметричный димер (рис. 3), молекулярное строение которого близко с ранее опубликованными родственными комплексами цинка [16]. Как и в комплексе V, каждый атом цинка в комплексе VI имеет искаженное тетраэдрическое окружение (τ4 = 0.77 и 0.75 для атомов Zn(1) и Zn(2) соответственно [25]). Геометрические характеристики оксиакридиновых лигандов в VI находятся в хорошем согласии с комплексом V. В свою очередь расстояния Zn–O и Zn–N в комплексе VI несколько длиннее, чем в комплексе V, что хорошо согласуется с ранее опубликованными данными для родственных димерных комплексов цинка [16, 29]. Металлоцикл ZnOZnO в комплексе VI практически плоский. Двугранный угол между плоскостями OZnO составляет 2.70°. Такое значение лучше совпадает с комплексом цинка, имеющим метильный заместитель, чем с комплексом, имеющим бромный заместитель в оксиакридиновом лиганде [16]. В отличие от комплекса V, в кристалле VI не реализуются межмолекулярные π…π-взаимодействия. Однако в молекуле плоскости лигандов располагаются под углом 29.79°, а расстояние между центрами пиридиновых фрагментов составляет 3.651 Å (рис. 3). Такие характеристики свидетельствуют о возможной реализации внутримолекулярного π…π-взаимодействия [28].
Рис. 3.
Молекулярная структура комплекса [Zn(Acr)(Et)]2 (VI). Тепловые эллипсоиды приведены с 30%-ной вероятностью. Атомы водорода не показаны для наглядности. Основные расстояния и углы в комплексе VI: Zn(1)–O(1) 2.054(3), Zn(1)–O(2) 2.078(3), Zn(2)–O(1) 2.090(3), Zn(2)–O(2) 2.046(4), Zn(1)–N(1) 2.083(4), Zn(2)–N(2) 2.084(4), Zn(1)–C(43) 1.980(5), Zn(2)–C(45) 1.964(6), Zn(1)…Zn(2) 2.9663(8), O(1)–C(1) 1.348(6), O(2)–C(22) 1.353(6), N(1)–C(2) 1.343(6), N(2)–C(23) 1.350(6), C(1)–C(2) 1.435(7), C(22)–C(23) 1.445(7) Å и O(1)Zn(1)O(2) 88.31(14)°, O(1)Zn(1)N(1) 79.18(14)°, O(2)Zn(1)N(1) 101.08(14)°, O(1)Zn(2)O(2) 88.19(14)°, O(2)Zn(2)N(2) 79.76(15)°, N(2)Zn(2)O(1) 100.52(15)°, C(43)Zn(1)O(1) 126.65(18)°, C(43)Zn(1)O(2) 123.86(19)°, C(43)Zn(1)N(1) 125.2(2)°, C(45)Zn(2)O(1) 123.2(2)°, C(45)Zn(2)O(2) 119.7(2)°, C(45)Zn(2)N(2) 130.6(3)°.

В спектрах поглощения растворов комплексов V и VI в ацетонитриле при комнатной температуре (рис. 4) наряду с интенсивной полосой при 300 нм и полосами между 320 и 380 нм, относящимися к π–π*-переходам в ароматических лигандах, наблюдается широкая полоса в области 510–535 нм, которую можно отнести к LMCT-переходу. Введение в ароматическое кольцо электронодонорного (–CH3) заместителя приводит к небольшому гипсохромному сдвигу, а электроноакцепторного (–Br) заместителя – к батохромному сдвигу в спектрах поглощения (рис. 4).
Рис. 4.
Спектры поглощения комплексов Zn: I, III, V (а) и II, IV, VI (б) в растворе CH3CN (2 × 10–5 М).

Модификация хелатирующего лиганда является эффективным способом регулировки оптических свойств комплексов металлов. Известно, что заместители в хинолиновом лиганде в зависимости от их природы и положения в кольце оказывают различное влияние на флуоресцентные свойства, изменяя длину волны излучения и его интенсивность [9, 30]. Существует хорошая корреляция между электронодонорными или акцепторными свойствами заместителей и положением максимумов полос в спектре люминесценции соединений [31]. На примере оксихинолинолятных комплексов цинка и алюминия было показано, что введение в пиридиновое кольцо гидроксихинолина электронодонорных групп или электроноакцепторных групп в бензольное кольцо приводит к синему сдвигу длины волны излучения [32, 33]. Как видно из рис. 5, введение метильного заместителя в положение С-7 оксиакридинового лиганда существенно увеличивает интенсивность ФЛ, как в случае комплексов Zn(AcrR)2, так и для комплексов [Zn(AcrR)(Et)]2 (R = H, CH3, Br) и одновременно смещает максимум ФЛ в коротковолновую область с 635 до 627 нм в случае гетеролигандных комплексов VI и II, но не оказывает влияния на спектры гомолигандных комплексов V и I. Введение бромного заместителя также влияет на положение максимума и интенсивность люминесценции. Для комплексов Zn(AcrR)2 максимум ФЛ смещается незначительно в красную область спектра с 690 до 705 нм, для комплексов [Zn(AcrR)(Et)]2 батохромное смещение максимума ФЛ составляет 50 нм (с 635 до 685 нм). В обоих случаях интенсивность ФЛ существенно уменьшается. Следует отметить, что интенсивность ФЛ для гетеролигандных комплексов II, IV и VI выше, чем для гомолигандных. Из всех полученных соединений самая интенсивная ФЛ наблюдается для гетеролигандного комплекса II с метильными заместителями в оксиакридиновых лигандах, что, по-видимому, связано с особенностями строения кристаллической решетки полученных соединений.
Рис. 5.
Спектры ФЛ твердых образцов комплексов I, III, V при λвозб 365 (а), 405 нм (в) и II, IV, VI при λвозб 365 (б), 405 нм (г) при комнатной температуре.

Для оценки влияния метильных и бромных заместителей на электронную структуру акридинолятных комплексов цинка были выполнены DFT-расчеты изолированных молекул соединений V и VI с использованием функционала B3LYP и полноэлектронного базиса 6-31G**. Следует отметить, что выбранный уровень расчетов адекватно воспроизводит геометрические параметры этих молекул в кристалле. Разница в длинах связей в координационной сфере атомов цинка не превышает 0.02 и 0.03 Å в комплексах V и VI соответственно. Квантово-химические расчеты для комплексов I–IV были выполнены ранее [16]. В табл. 2 приведены энергии молекулярных орбиталей соединений Zn(AcrR)2 и [Zn(AcrR)(Et)]2 (R = H, CH3, Br).
Таблица 2.
Энергии молекулярных орбиталей в комплексах I–VI
| Орбиталь | Комплекс | |||||
|---|---|---|---|---|---|---|
| I | II | III | IV | V | VI | |
| эВ | ||||||
| ВЗМО | –4.79 | –4.97 | –4.68 | –5.20 | –4.85 | –5.02 |
| НСМО | –2.13 | –2.45 | –2.16 | –2.59 | –2.22 | –2.33 |
| ВЗМО–НСМО | 2.66 | 2.72 | 2.52 | 2.61 | 2.63 | 2.68 |
Высшие занятые и низшие свободные молекулярные орбитали (ВЗМО и НСМО) в комплексах V и VI локализованы в основном на ароматических кольцах (рис. 6). НСМО в соединении VI представляет собой орбиталь чистого оксиакридинового лиганда без вклада атома металла. При переходе к ВЗМО вклад волновых функций каждого из атомов цинка увеличивается до 4%. В комплексе V небольшой вклад атома Zn (~1%) присутствует как в ВЗМО-, так и в НСМО-орбитали. Поэтому электронные переходы ВЗМО–НСМО могут рассматриваться как межлигандный перенос заряда с незначительным вкладом переноса заряда с металла на лиганд.
Рис. 6.
Изоповерхности B3LYP/6-31G** ВЗМО- и НСМО-орбиталей для комплексов V и VI (0.02 а. е.). Атомы водорода не показаны.

Как было показано на 8-оксихинолинолятных комплексах Al и Zn [2, 30] введение электронодонорных заместителей (–CH3) в пиридиновое кольцо лиганда приводит к незначительному гипсохромному сдвигу в спектрах поглощения и люминесценции. Аналогичная ситуация имеет место и в полученных нами комплексах. Введение в положение С-7 оксиакридинового лиганда атома брома приводит к уменьшению энергетической щели ВЗMO/НСMO. Вследствие этого максимумы ФЛ цинковых комплексов III и IV смещены в красную область по отношению к комплексам V и VI без метильных и бромных заместителей в лигандах (рис. 5).
ЭЛ-свойства комплексов V и VI изучали на трехслойных OLED-устройствах конфигурации ITO/TPD (25 нм)/комплекс (45 нм)/BATH (15 нм)/ LiF (0.5)/Al. Оба устройства обнаружили ЭЛ, хотя и с низкой интенсивностью (рис. 7).
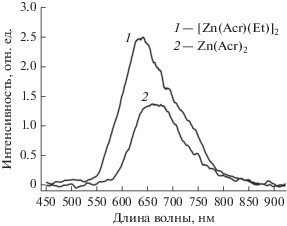
Увеличение энергетической щели ВЗМО/НСМО для комплекса VI приводит к гипсохромному сдвигу в спектре ЭЛ по сравнению с комплексом V. На рис. 8 представлены вольт-яркостные и вольт-амперные характеристики рассматриваемых устройств. Их рабочие характеристики приведены в табл. 3. Эффективность устройств на основе комплексов V и VI оказалась ниже изученных ранее комплексов I и II с метильными заместителями в лигандах: напряжение включения 9–10 В, максимальная яркость 15–16 кд/м2 при 30 В, выход по току 0.020–0.23 кд/А [16]. Устройства с комплексами III и IV, содержащими бромированные лиганды, не показали люминесценции даже при высоком напряжении [16]. Положение и форма полос в спектрах ЭЛ (рис. 7) комплексов V и VI такие же, как и в соответствующих спектрах ФЛ. Так же, как и в спектрах ФЛ, относительная интенсивность спектров ЭЛ комплекса VI в два раза больше, чем у комплекса V.
Рис. 8.
Вольт-яркостные (а) и вольт-амперные (б) характеристики OLED на основе комплексов V (2) и VI (1).

Таблица 3.
Рабочие характеристики диодов ITO/TPD/комплекс/BATH/LiF/Al*
| Комплекс | ηi, кд А–1 | ηр, лм Вт–1 | U, В** | Максимальная яркость, кд/м2 |
|---|---|---|---|---|
| Zn(Acr)2 | 0.83 (20 В) | 0.26 (20 В) | 11 | 1.5 (30 B) |
| [Zn(Acr)(Et)]2 | 3.15 (18 В) | 1.1 (18 В) | 6 | 2.5 (30 B) |
Как видно из табл. 2, введение этильных лигандов в цинковые комплексы существенно изменяет значения НСМО и ВЗМО. Можно предположить, что большее значение НСМО и ВЗМО приводит к удержанию дырок и электронов в органическом светоизлучающем слое, увеличивая эффективность электронно-дырочной рекомбинации. Благодаря своему строению в комплексах II и VI подвижность электронов увеличивается и уменьшается вероятность внутренних электронных переходов, поэтому соответствующие комплексы цинка обладают лучшими люминесцентными свойствами по сравнению с соединениями I и V.
Цинк входит в состав многих ферментов, присутствующих в живом организме. Известно, что комплексы цинка обладают широким спектром биологического действия, в частности антимикробной и противоопухолевой активностью [34–36]. В последние годы наблюдается рост интереса к металлокомплексам на основе гетероциклических ароматических лигандов, поскольку они представляют большой интерес для биохимии [37]. В этой связи 4-гидроксиакридин привлекает внимание благодаря своим фармакологическим свойствам [38]. Доказано, что он является отличным хелатирующим и интеркалирующим лигандом. Комплексы Ni(II) [11] и Pd(II) [39 ] на его основе оказались перспективными с точки зрения биологической, в частности противоопухолевой активности.
Комплексы Zn(II) I–V были протестированы на цитотоксичность в отношении клеточных линий рака человека. Цитотоксичность соединений in vitro оценивали на линиях HCT-116 (рак толстой кишки человека) и MCF-7 (аденокарцинома молочной железы человека). Активность соединений сравнивали с действием цисплатина. Результаты исследования показали, что наиболее активным является комплекс V, тогда как соединения I–IV обладают умеренной цитотоксичностью, что, вероятно, связано с влиянием природы заместителей в оксиакридиновых лигандах (табл. 4).
Таблица 4.
Значения IC50 (µM) для комплексов I–V и цисплатина по отношению к клеточным линиям HCT-116 и MCF-7 в МТТ-тесте
| Соединение | HCT-116 | MCF-7 |
|---|---|---|
| Zn(AcrMe)2 (I) | 147 ± 12 | 121 ± 11 |
| [Zn(Et)(AcrMe)]2 (II) | 43 ± 5 | 54 ± 10 |
| Zn(AcrBr)2 (III) | 85 ± 7 | 107 ± 11 |
| [Zn(Et)(AcrBr)]2 (IV) | 121 ± 11 | 116 ± 10 |
| Zn(Acr)2 (V) | 25 ± 5 | 31 ± 5 |
| Цисплатин | 7 ± 4 | 15 ± 3 |
Таким образом, синтезированы оксиакридинолятные комплексы цинка Zn(Acr)2 и [Zn(Acr)Et]2, методом РСА установлено их строение, а также исследованы люминесцентные свойства. При возбуждении светом (λ = 405 нм) соединения обнаружили эмиссию средней интенсивности с максимумами при 700 и 630 нм. Оба комплексы проявили также слабые электролюминесцентные свойства. Из всего ряда полученных соединений цинка I–VI наиболее люминесцентно активными оказались комплексы с метильными заместителями в оксиакридиновых лигандах (I и II). Для оценки влияния метильных и бромных заместителей на электронную структуру акридинолятов цинка были выполнены DFT-расчеты изолированных молекул. Введение электронодонорных заместителей (–CH3) в пиридиновое кольцо лиганда приводит к незначительному гипсохромному сдвигу в спектрах поглощения и люминесценции. Введение же атома брома приводит к уменьшению энергетической щели ВЗMO/НСMO и красному смещению максимума ФЛ цинковых комплексов. С целью исследования биологической активности был проведен МТТ-тест полученных соединений, результаты которого показали слабую цитотоксическую активность синтезированных цинковых комплексов. Наиболее активным оказалось соединение V, не имеющее ни метильных, ни бромных заместителей в лигандах.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Mitschke U., Bauerle P. // J. Mater. Chem. 2000. V. 10. P. 1471.
Chen C.H., Shi J. // Coord. Chem. Rev. 1998. V. 171. P. 161.
Bunzli J.-C.G., Piguet C. // Chem. Soc. Rev. 2005. V. 34. P. 1048.
De Sa G.F., Malta O.L., De Mello Donega C. // Coord. Chem. Rev. 2000. V. 196. P. 165.
Ghedini M., Deda M.L., Aiello I., Grisolia A. // Inorg. Chim. Acta. 2004. V. 357. P. 33.
Wang H., Wang W.S., Zhang H.S. // Talanta. 2001. V. 53. P. 1015.
Wei L.-J., Gao H.-L., Cui J.-Z. // Phosphorus, Sulfur Silicon Relat. Elem. 2012. V. 187. P. 1101.
Mishra A., Nayak P.K., Periasamy N. // Tetrahedron Lett. 2004. V. 45. P. 6265.
Jianbo H., Tingting Z., Yongjing C. et al. // J. Fluoresc. 2018. V. 28. № 5. P. 1121.
Mastropietro T.F., Szerb E.I., Deda M.L. et al. // Eur. J. Inorg. Chem. 2013. V. 2013. P. 2188.
Crispini A., Pucci D., Sessa S. et al. // New J. Chem. 2003, V. 27. P. 1497.
Hamada J., Sano K., Fujita M. et al. // Jpn. Kokai Tokkyo Koho. 1995. JP 07166159 A 19950627.
Cui Z., Li X., Li L. et al. // Bioorg. Med. Chem. 2016. V. 24. P. 261.
Kumar A., Kumar N., Roy P. et al. // Med. Chem. Res. 2015. V. 24. P. 3272.
Balashova T.V., Arsenyev M.V., Polyakova S.K., et al. // J. Mol. Struct. 2021. V. 1229. P. 129798.
Polyakova S.K., Balashova T.V., Rumyantcev R.V. et al. // Mendeleev Commun. 2021. V. 31. P. 262.
Katkova M.A., Pushkarev A.P., Balashova T.V. et al. // J. Mater. Chem. 2011. V. 21. P. 16611.
SAINT. Data Reduction and Correction Program. Madison (WI, USA): Bruker AXS., 2014.
Rigaku Oxford Diffraction. CrysAlis Pro software system. Version 1.171.40.84a. Wroclaw (Poland): Rigaku Corporation, 2020.
Krause L., Herbst-Irmer R., Sheldrick G.M., Stalke D. // J. Appl. Crystallogr. 2015. V. 48 № 1. P. 3.
Sheldrick G.M. // Acta Crystallogr. A. 2015. V. 71. № 1. P. 3.
Sheldrick G.M. // Acta Crystallogr. C. 2015. V. 71. P. 3.
Niks M., Otto M. // J. Immunol. Methods. 1990. V. 130. P. 149.
Yang L., Powell D.R., Houser R.P. // Dalton Trans. 2007. № 9. P. 955.
Wang D., Li S.-M., Zheng J.-Q. et al. // Inorg. Chem. 2017. V. 56. P. 984.
Czugler M., Neumann R., Weber E. // Inorg. Chim. Acta. 2001. V. 313. P. 100.
Janiak C. // Dalton Trans. 2000. № 21. P. 3885.
Bakewell C., Fateh-Iravani G., Beh D.W. et al. // Dalton Trans. 2015. V. 44. P. 12326.
Perez-Bolıvar C., Montes V.A., Anzenbacher P.Jr. // I-norg. Chem. 2006. V. 45. P. 9610.
Fazaeli Y., Amini M.M., Najafi E. et al. // J. Fluoresc. 2012. V. 22. P. 1263.
Sapochak L.S., Padmaperuma A., Washton N. et al. // J. Am. Chem. Soc. 2001. V. 123. P. 6300.
Kido J., Iizumi Y. // Chem. Lett. 1997. V. 10. P. 963.
Zhu T., Wang Y., Ding W. et al. // Chem Biol. Drug Des. 2015. V. 85. P. 385.
Li Y., Yang Z., Zhou M. et al. // J. Mol. Struct. 2017. V. 1130. P. 818.
Sankarganesh M., Dhaveethu Raja J., Adwin Jose P.R. et al. // J. Fluoresc. 2018. V. 28. № 4. P. 975.
Oehninger L., Rubbiania R., Ott I. // Dalton Trans. 2013. V. 42. P. 3269.
Matsumura K. // J. Am. Chem. Soc. 1927. V. 49. P. 810.
Pucci D., Albertini V., Bloise R. et al. // J. Inorg. Bi-ochem. 2006. V. 100. P. 1575.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия