

Координационная химия, 2021, T. 47, № 4, стр. 199-207
Применение модели молекулярного инвариома для исследования взаимодействий с участием атомов фтора в комплексе {${\text{Yb}}_{{\text{2}}}^{{{\text{II}}}}$(μ2-OCH(CF3)2)3(μ3-OCH(CF3)2)2YbIII(OCH(CF3)2)2(THF)(Et2O)}
Р. В. Румянцев 1, Г. К. Фукин 1, *, Е. В. Баранов 1, А. В. Черкасов 1, Е. А. Козлова 1
1 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
* E-mail: gera@iomc.ras.ru
Поступила в редакцию 26.04.2020
После доработки 19.06.2020
Принята к публикации 25.06.2020
Аннотация
Проведено сравнение распределения электронной плотности, полученной с помощью квантово-химических (DFT) расчетов и модели молекулярного инвариома в тримерном комплексе иттербия с гексафторизопропоксидными лигандами {${\text{Yb}}_{{\text{2}}}^{{{\text{II}}}}$(μ2-OR)3(μ3-OR)2YbIII(OR)2(THF)(Et2O)} (I), где R – CH(CF3)2, THF – тетрагидрофуран. Основные топологические характеристики электронной плотности в критических точках (3, –1), соответствующих взаимодействиям в координационной сфере атомов иттербия, полученные двумя исследованными подходами находятся в отличном согласии. Наибольшее расхождение между DFT-расчетами и моделью молекулярного инвариома наблюдается при исследовании внутримолекулярных взаимодействий с участием атомов фтора (F···F, F···H и F···O) в структуре I. Оптимизация геометрии приводит к большему количеству таких взаимодействий в комплексе. При этом также возрастает энергия, соответствующая этим взаимодействиям. Однако значения основных топологических характеристик для взаимодействий F···X (X = F, H, O), которые удается локализовать в рамках обоих методов, остаются в пределах индекса переносимости. Анализ деформационной электронной плотности показал, что независимо от способа получения распределения электронной плотности реализация взаимодействий Fδ–···Fδ– обусловлена соответствием области концентрации электронной плотности на одном из атомов фтора области разрежения электронной плотности на втором атоме фтора.
Исследование распределения электронной плотности (ЭП) является важным инструментом в решении теоретических и практических задач современной химической науки [1–3]. Прецизионные рентгеноструктурные исследования позволяют получить экспериментальное распределение ЭП, рассчитать электростатический потенциал, энергию внутри- и межатомных взаимодействий в кристалле, выявить структурообразующие факторы, определяющие строение, установить фрагменты, которые слабо меняются в родственных соединениях [4]. Такое исследование особенно важно для соотнесения свойств соединений с его строением. Однако к образцу для проведения высокоточного рентгеновского эксперимента предъявляется ряд требований, и получение подходящего монокристалла является весьма трудоемкой задачей. Этот факт существенно ограничивает экспериментальное исследование электронной плотности в практически важных соединениях. Компромиссным решением является получение распределения ЭП в соединениях на основе теоретических расчетов методом функционала плотности (DFT) [5–7].
Для изучения распределения ЭП в органических и элементоорганических соединениях с относительно легкими атомами (периодов 1–3) была разработана методика на основе инвариома (инвариантного атома) [8, 9]. Это экспериментально-теоретический подход, который позволяет получать распределение ЭП на основании данных рутинных рентгенодифракционных исследований и теоретических расчетов. Для металлоорганических соединений применяется усовершенствованный метод, который основан на определении молекулярного инвариома [10–12]. Применение этого метода позволяет получать информацию о топологии ЭП в молекулах с хорошей точностью [13, 14]. Однако большинство проведенных в этой области исследований посвящены переносимости топологических характеристик в координационной сфере атома металла [15–17].
Среди структурных особенностей многих биологически активных соединений является наличие внутримолекулярных слабых невалентных взаимодействий. Их реализация играет значительную роль для определения конформации молекулы и проявляемых физико-химических свойств [18]. Не менее важна реализация внутримолекулярных невалентных взаимодействий в металлоорганических комплексах. Так, в комплексах лантанидов с фторированными лигандами возможна реализация взаимодействий C–F → Ln. За счет этого, в частности, проявляется каталитическая активность данных комплексов [19, 20]. Не менее важно, что реализация таких взаимодействий определяет термическую стабильность данных комплексов [21, 22].
Некоторые комплексы лантанидов с гексафторизопропоксидными лигандами проявляют перспективные люминесцентные свойства [23, 24]. Проведенное нами недавно исследование топологии ЭП в тримерном комплексе европия продемонстрировало наличие многочисленных внутримолекулярных взаимодействий с участием атомов фтора F···X (X = Eu, F, H, O) [25].
Настоящая работа посвящена исследованию принципиальной возможности получения информации о топологических характеристиках ЭП в критических точках, соответствующих взаимодействиям с участием атомов фтора в комплексе {${\text{Yb}}_{{\text{2}}}^{{{\text{II}}}}$(μ2-OCH(CF3)2)3(μ3-OCH(CF3)2)2YbIII(OCH(CF3)2)2- (THF)(Et2O)} (I) с помощью модели молекулярного инвариома.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Полную оптимизацию геометрии комплекса I проводили методом DFT, реализованным в программном пакете Gaussian 09 [26], с использованием гибридного функционала B3LYP. Для органической части использовали базис 6-31 + G*, [27–31] для атомов лантанида – псевдопотенциал ECP28MWB [32]. Функцию электронной плотности (EDF) для ECP рассчитывали с использованием программы Molden2aim.
Топологический анализ полученной в результате DFT расчета теоретической функции распределения электронной плотности проводили с помощью программы AIMALL [33].
Для проведения экспериментально-теоретического уточнения использовали рутинный массив экспериментальных рентгенодифракционных данных. Одноточечный расчет изолированной молекулы комплекса I проводили методом функционала плотности (B3LYP/DZP) [34] в программе Gaussian 09 [26]. Затем молекулу комплекса помещали в псевдокубическую ячейку (а = 30 Å). С помощью программы Tonto [35] рассчитывали структурные амплитуды (sinθ/λ = 1.155 Å–1). На основе рассчитанных структурных амплитуд для кубической ячейки с помощью программы MoPro [36] получали значения заселенностей сферически-симметричной валентной оболочки (Pval) и описывающих ее деформацию мультипольных параметров (Plm) вместе с соответствующими коэффициентами расширения–сжатия (k, k') для каждого из атомов в комплексе I. Полученные значения Pval, Plm, k и k' использовали (но сами не уточнялись) для уточнения координат атомов и их тепловых параметров по экспериментальным отражениям рутинного набора данных в реальной симметрии комплекса I. Топологический анализ экспериментально-теоретической функции распределения электронной плотности проводили с помощью программы WinXPRO [37].
Анализ распределения деформационной электронной плотности (ДЭП) в области невалентных взаимодействий между атомами фтора в комплексе I проводили с использованием программ MoPro [36] и Multiwfn v. 3.3.8 [38].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
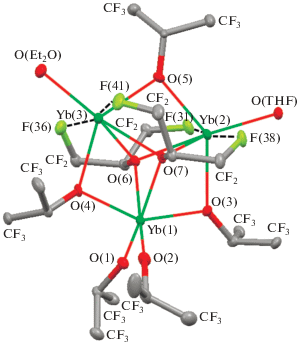
Кристаллическое строение комплекса I установлено методом рентгеноструктурного анализа (РСА) и подробно описано нами ранее [23]. Молекулярное строение комплекса представлено на рис. 1. Анализ геометрических характеристик позволяет по аналогии с родственным комплексом европия [25] предположить наличие большого числа внутримолекулярных невалентных взаимодействий с участием атомов фтора в структуре.
Рис. 1.
Молекулярное строение комплекса I. Тепловые эллипсоиды приведены с 30%-ной вероятностью. Атомы водорода не приведены для ясности.
Для проверки этой гипотезы мы провели квантово-химическое (DFT) исследование комплекса I методом функционала плотности (B3LYP/6-31 + G*, ECP28MWB) с оптимизацией геометрии. Далее для удобства модель комплекса I, полученная с использованием DFT-расчета, будет именоваться Iisol. Одновременно с этим по известной методологии был рассчитан молекулярный инвариом комплекса I и проведено уточнение по рутинному набору экспериментальных данных (Iinv). Таким образом, было получено экспериментально-теоретическое распределение ЭП в комплексе Iinv. Молекулярные графы для Iisol и Iinv моделей, демонстрирующие все взаимодействия в координационной сфере атомов иттербия, представлены на рис. 2.
Рис. 2.
Теоретический (Iisol) (a) и экспериментально-теоретический (Iinv) (б) молекулярный граф комплекса I. Для наглядности приведены только избранные КТ(3, –1), отвечающие валентным взаимодействиям и невалентным контактам C–F → Ln в координационной сфере атомов иттербия. Атомы водорода не приведены.
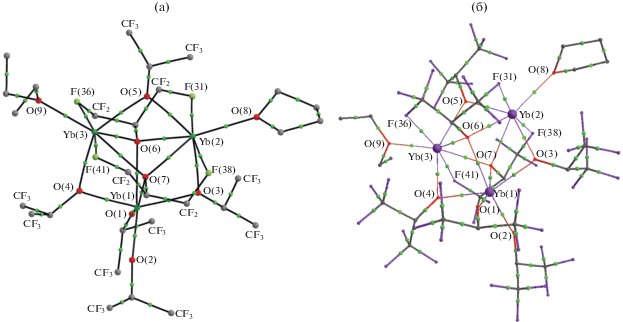
Следует отметить, что модель молекулярного инвариома более точно воспроизводит основные геометрические характеристики в комплексе I, полученные при проведении рутинного РСА, чем теоретическое квантово-химическое DFT-исследование (табл. 1). Среднее отклонение связей Yb–O и Yb···F для Iinv составляет 0.003 Å, тогда как для Iisol это значение равно 0.024 Å. При этом наибольшие изменения не превышают 0.009 и 0.094 Å соответственно. Интересно отметить, что как в случае Iisol, так и для Iinv наибольшие отклонения от данных РСА наблюдаются для координационных связей с нейтральными растворителями Yb(2)–O(8)THF и Yb(3)–O(9)EtOEt.
Таблица 1.
Расстояния связей Yb–O и контактов Yb···F в I по данным рутинного РСА, теоретических DFT расчетов (Iisol) и экспериментально-теоретических (с молекулярным инвариомом) (Iinv) исследований
| Связь | I, Å | Iisol, Å | Iinv, Å | Связь | I, Å | Iisol, Å | Iinv, Å |
|---|---|---|---|---|---|---|---|
| Yb(1)–O(1) | 2.075(3) | 2.0722 | 2.081(3) | Yb(2)–O(8) | 2.402(2) | 2.4586 | 2.411(3) |
| Yb(1)–O(2) | 2.075(3) | 2.0646 | 2.083(3) | Yb(3)–O(4) | 2.392(3) | 2.3762 | 2.391(3) |
| Yb(1)–O(3) | 2.239(2) | 2.2617 | 2.241(3) | Yb(3)–O(5) | 2.355(3) | 2.3864 | 2.359(3) |
| Yb(1)–O(4) | 2.248(2) | 2.2644 | 2.248(3) | Yb(3)–O(6) | 2.391(2) | 2.3970 | 2.395(3) |
| Yb(1)–O(6) | 2.371(2) | 2.4033 | 2.370(3) | Yb(3)–O(7) | 2.513(2) | 2.5191 | 2.516(3) |
| Yb(1)–O(7) | 2.359(2) | 2.3853 | 2.360(3) | Yb(3)–O(9) | 2.392(3) | 2.4861 | 2.400(3) |
| Yb(2)–O(3) | 2.394(3) | 2.3948 | 2.395(3) | Yb(2)–F(31) | 2.568(2) | 2.5871 | 2.570(3) |
| Yb(2)–O(5) | 2.366(3) | 2.3975 | 2.367(3) | Yb(2)–F(38) | 2.644(2) | 2.6523 | 2.649(3) |
| Yb(2)–O(6) | 2.513(2) | 2.5110 | 2.518(3) | Yb(3)–F(36) | 2.652(2) | 2.6120 | 2.653(3) |
| Yb(2)–O(7) | 2.415(2) | 2.4222 | 2.415(3) | Yb(3)–F(41) | 2.522(3) | 2.5692 | 2.526(3) |
Атомные заряды на органическом фрагменте комплекса I, полученные на основании теоретического (Iisol) и экспериментально-теоретического (Iinv) распределения ЭП, находятся в хорошем согласии между собой (табл. 2). Все атомы фтора и кислорода имеют отрицательный заряд. Наибольшая разница в зарядах наблюдается на атомах нейтрально координированных растворителей THF и Et2O. Атомы иттербия Yb(2) и Yb(3) в I двухвалентны, тогда как Yb(1) имеет формальный заряд +3. Такое представление хорошо согласуется с теоретическими атомными зарядами в Iisol. Заряды на атомах иттербия в Iinv совпадают с данными DFT на полуколичественном уровне. При этом сохраняется пропорциональная разница между зарядом на формально трехвалентном атоме Yb(1) и двухвалентных атомах Yb(2, 3). Анализ литературных данных показал, что теоретически рассчитанные значения зарядов на атомах металла в изолированных молекулах более положительны по сравнению со значениями, полученными на основании прецизионных рентгенодифракционных исследований [16, 39]. Таким образом, менее положительные значения зарядов атомов иттербия в Iinv вполне ожидаемы.
Таблица 2.
Атомные заряды в комплексе I, полученные на основании Iisol и Iinv распределения ЭП
| Атом | Iisol, e | Iinv, e |
|---|---|---|
| Yb(1) | 2.24 | 1.02 |
| Yb(2) | 1.67 | 0.78 |
| Yb(3) | 1.66 | 0.79 |
| q(F) | –0.65…–0.63 | –0.62…–0.52 |
| q(C) | –0.02…+1.75 | –0.67…+1.66 |
| q(O) | –1.26…–1.08 | –1.05…–0.84 |
| q(H) | 0.01–0.12 | 0.23–0.32 |
Исследование топологии теоретического и экспериментально-теоретического распределения ЭП в комплексе I проведено в рамках теории Р. Бейдера “Атомы в молекулах” [40]. Сравнение основных топологических характеристик Iisol и Iinv приведено в табл. 3. Основные топологические характеристики, соответствующие взаимодействиям в координационной сфере атомов иттербия, находятся в хорошем согласии между собой. Все взаимодействия Yb(II)–O, Yb(II)···F, а также Yb(III) с μ3-мостиковыми гексафторизопропоксидными лигандами соответствуют типу закрытых оболочек (∇2ρ(r)) > 0, he(r) > 0) как в Iisol, так и в Iinv. В свою очередь, все связи трехвалентного Yb(1) с терминальными и μ2-мостиковыми гексафторизопропоксидными лигандами относятся к типу промежуточных взаимодействий (∇2ρ(r)) > 0, he(r) < 0) (табл. 3). Важно отметить, что обе модели – теоретическая и экспериментально-теоретическая – позволяют обнаружить по четыре внутримолекулярных C–F → Yb взаимодействия (рис. 2). Согласно корреляции Эспинозы – Моллинса –Лекомта [41], энергия этих взаимодействий лежит в интервалах 6.26–7.90 и 5.71–8.44 ккал/моль для Iisol и Iinv соответственно. Представленные величины лишь незначительно меньше соответствующих значений для координационных связей Yb(2)–O(8)THF и Yb(3)–O(9)EtOEt и взаимодействий с μ3-мостиковыми гексафторизопропоксидными лигандами. Энергия Yb–O(изо-PrF) систематически увеличивается от μ3-мостиковых к терминальным лигандам как в Iisol, так и в Iinv. Такое распределение ЭП в координационной сфере атомов лантанидов в I находится также в хорошем согласии с родственным комплексом европия {${\text{Eu}}_{{\text{2}}}^{{{\text{II}}}}$(μ2-OCH(CF3)2)3(μ3-OCH(CF3)2)2EuIII(OCH(CF3)2)2-(DME)2} [25].
Таблица 3.
Основные топологические характеристики связей Yb–O и контактов Yb···F в Iisol и Iinv
| Связь | ρ(r), a.e. | ∇2ρ(r), a.e. | he(r), a.e. | |EEML|*, ккал/моль | ||||
|---|---|---|---|---|---|---|---|---|
| Iisol | Ιinv | Iisol | Ιinv | Iisol | Ιinv | Iisol | Ιinv | |
| Yb(1)–O(1) | 0.088 | 0.088 | 0.498 | 0.477 | –0.008 | –0.011 | 43.69 | 44.13 |
| Yb(1)–O(2) | 0.089 | 0.087 | 0.501 | 0.472 | –0.009 | –0.010 | 44.70 | 43.30 |
| Yb(1)–O(3) | 0.060 | 0.063 | 0.284 | 0.318 | –0.001 | –0.002 | 23.14 | 26.52 |
| Yb(1)–O(4) | 0.060 | 0.063 | 0.281 | 0.314 | –0.001 | –0.002 | 22.87 | 25.97 |
| Yb(1)–O(6) | 0.043 | 0.049 | 0.193 | 0.227 | 0.001 | 0.000 | 14.58 | 17.57 |
| Yb(1)–O(7) | 0.045 | 0.050 | 0.203 | 0.234 | 0.001 | 0.000 | 15.38 | 18.22 |
| Yb(2)–O(3) | 0.042 | 0.045 | 0.204 | 0.209 | 0.002 | 0.001 | 15.02 | 15.68 |
| Yb(2)–O(5) | 0.042 | 0.047 | 0.203 | 0.225 | 0.002 | 0.001 | 14.90 | 17.07 |
| Yb(2)–O(6) | 0.033 | 0.036 | 0.150 | 0.152 | 0.002 | 0.001 | 10.69 | 10.99 |
| Yb(2)–O(7) | 0.040 | 0.044 | 0.191 | 0.201 | 0.002 | 0.001 | 13.95 | 15.19 |
| Yb(2)–O(8) | 0.036 | 0.042 | 0.174 | 0.201 | 0.002 | 0.002 | 12.13 | 14.26 |
| Yb(3)–O(4) | 0.044 | 0.045 | 0.215 | 0.210 | 0.002 | 0.001 | 15.84 | 15.75 |
| Yb(3)–O(5) | 0.044 | 0.049 | 0.210 | 0.232 | 0.001 | 0.001 | 15.52 | 17.82 |
| Yb(3)–O(6) | 0.043 | 0.046 | 0.206 | 0.212 | 0.001 | 0.001 | 15.22 | 16.21 |
| Yb(3)–O(7) | 0.032 | 0.036 | 0.146 | 0.152 | 0.002 | 0.001 | 10.41 | 11.06 |
| Yb(3)–O(9) | 0.034 | 0.043 | 0.162 | 0.207 | 0.002 | 0.002 | 11.23 | 14.83 |
| Yb(2)···F(31) | 0.022 | 0.026 | 0.112 | 0.119 | 0.002 | 0.003 | 7.50 | 7.31 |
| Yb(2)···F(38) | 0.019 | 0.023 | 0.095 | 0.094 | 0.002 | 0.003 | 6.26 | 5.71 |
| Yb(3)···F(36) | 0.021 | 0.022 | 0.106 | 0.093 | 0.002 | 0.003 | 7.07 | 5.64 |
| Yb(3)···F(41) | 0.023 | 0.029 | 0.118 | 0.135 | 0.002 | 0.003 | 7.90 | 8.44 |
* Энергия взаимодействий рассчитана согласно корреляции Эспинозы – Моллинса – Лекомта [41].
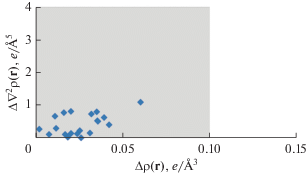
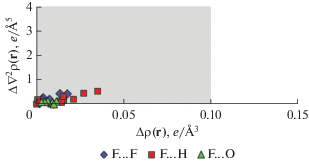
Разница в значениях ρ(r) и ∇2ρ(r) в критических точках (3, –1), соответствующих взаимодействиям Yb–O и Yb···F между моделями Iisol и Iinv, не превышает 0.009 и 0.046 а.е. соответственно. Таким образом, значения основных топологических характеристик для взаимодействий в координационной сфере атомов иттербия лежат в пределах индексов переносимости этих величин (ρ(r) = = 0.1 e Å–3 (0.015 а.е.), ∇2ρ(r) = 4 e Å–5 (0.17 а.е.)) [42] (рис. 3).
Рис. 3.
Графическое представление модулей отклонения в значениях ρ(r) и ∇2ρ(r), в критических точках (3, –1), соответствующих взаимодействиям в координационной сфере атомов иттербия, между Iisol и Iinv. Серая область демонстрирует границы индекса переносимости.
Оба метода – теоретический и экспериментально-теоретический – ожидаемо позволяют обнаружить в структуре комплекса I многочисленные внутримолекулярные невалентные взаимодействия F···F, F···H и F···O (рис. 4). Всего исследование топологии теоретической ЭП позволило обнаружить следующие взаимодействия: 24 F···F, 24 F···H и 14 F···O. Следует отметить, что исследование топологии экспериментально-теоретической ЭП позволяет обнаружить гораздо меньше взаимодействий с участием атомов фтора: 11 F···F, 13 F···H и 6 F···O. При этом абсолютные значения энергии этих взаимодействий, а также суммарные значения энергии всех взаимодействий с участием атомов фтора в Iinv ниже, чем в модели Iisol (табл. 4). Расстояния, соответствующие взаимодействиям F···F, F···H и F···O в комплексе I, варьируются в достаточно широких пределах независимо от метода получения распределения ЭП. Однако в Iisol интервал определения взаимодействий с участием атомов фтора несколько шире, а максимальные расстояния, на которых находятся взаимодействующие атомы, длиннее (табл. 4). Значительным оказывается также разброс энергии для соответствующих взаимодействий (0.65–4.11, 0.23–2.25 и 0.48–2.23 ккал/моль в Iisol и 0.53–1.67, 0.33–1.54 и 0.86–1.66 ккал/моль в Iinv).
Рис. 4.
Теоретические (Iisol) (а, в, д) и экспериментально-теоретические (Iinv) (б, г, е) молекулярные графы комплекса I. Для наглядности по отдельности приведены только избранные КТ(3, –1), отвечающие взаимодействиям F···F (а, б), F···H (в, г) и F···O (д, е). Атомы водорода не приведены.
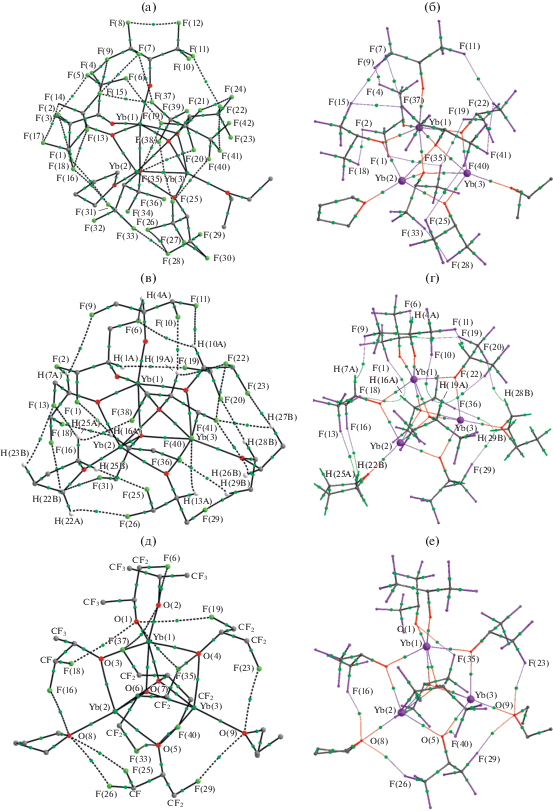
Таблица 4.
Внутримолекулярные взаимодействия с участием атомов фтора в Iisol и Iinv
| Контакт | Количество контактов, шт | Диапазон изменения расстояний, Å | Диапазон изменения энергии контактов, ккал/моль | Общая энергия контактов, ккал/моль | ||||
|---|---|---|---|---|---|---|---|---|
| Iisol | Iinv | Iisol | Iinv | Iisol | Iinv | Iisol | Iinv | |
| F···F | 24 | 11 | 2.651–3.183 | 2.768–3.164 | 0.65–4.11 | 0.53–1.67 | 55.22 | 10.93 |
| F···H | 24 | 13 | 2.341–3.146 | 2.400–2.791 | 0.23–2.25 | 0.33–1.54 | 26.14 | 11.86 |
| F···O | 14 | 6 | 2.925–3.467 | 2.901–3.167 | 0.48–2.23 | 0.86–1.66 | 16.93 | 7.26 |
Такая разница в количестве и энергии внутримолекулярных невалентных взаимодействий с участием атомов фтора в комплексе I определяется проведением оптимизации геометрии при теоретическом исследовании распределения ЭП в молекуле. Анализ геометрии взаимодействий F···X (X = F, H, O) показал, что модель Iinv значительно точнее воспроизводит геометрические характеристики, определенные в рутинном РСА, чем модель Iisol. Средние отклонения в расстояниях F···F, F···H и F···O составляют 0.112, 0.184 и 0.062 Å в Iisol и 0.005, 0.047 и 0.009 Å в Iinv. Оптимизация молекулярного строения при квантово-химическом расчете приводит к наиболее энергетически выгодной конформации и не учитывает межмолекулярного взаимодействия в кристалле. Интересно отметить, что в процессе оптимизации геометрии наибольшие изменения характерны для взаимодействий Fδ–···Hδ+. Расстояния между этими атомами в результате оптимизации значительно уменьшаются. Следовательно, стоит ожидать, что модель Iisol несколько переоценивает энергию этих взаимодействий. Суммарный вклад всех взаимодействий Fδ–···Fδ– играет важную роль в стабилизации молекулярного строения комплекса I независимо от подхода к исследованию распределения ЭП. Этот вывод хорошо согласуется с ранее проведенным квантово-химическим исследованием родственного комплекса европия [25].
Несмотря на такое значительное различие в энергиях взаимодействий, основные топологические характеристики для взаимодействий с участием атомов фтора, которые удается локализовать в рамках обоих подходов к исследованию распределения ЭП в I, находятся в достаточно хорошем согласии. Разница в значениях ρ(r) и ∇2ρ(r) не превышает 0.005 и 0.022 а.е. соответственно. Таким образом, значения основных топологических характеристик для невалентных взаимодействий, которые удается локализовать в рамках как теоретического, так и экспериментально-теоретического подходов, лежат в пределах индексов переносимости этих величин [42] (рис. 5). В строгом соответствии с разницей в длинах связей наименьшее среднее отклонение в значениях ρ(r) и ∇2ρ(r) наблюдается для взаимодействий F···O, тогда как наибольшее – для взаимодействий F···H. Следует отметить, что абсолютные значения топологических характеристик, соответствующих невалентным взаимодействиям атомов фтора, находятся в хорошем согласии с литературными данными о взаимодействиях F···F [43].
Рис. 5.
Графическое представление модулей отклонения в значениях ρ(r) и ∇2ρ(r), в критических точках (3, –1), соответствующих внутримолекулярным взаимодействиям F···X (X = F, H, O), между Iisol и Iinv. Серая область демонстрирует границы индекса переносимости.
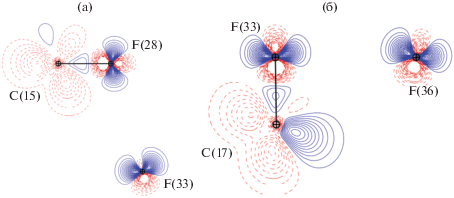
Исследования экспериментального [44] и теоретического [25] распределения ДЭП в области галогенных контактов Fδ–···Fδ– показали, что реализация взаимодействий фтор···фтор возможна за счет донирования ЭП с области ее концентрации на одном из атомов на область разрежения ЭП на втором. В рамках исследования применимости экспериментально-теоретического подхода к изучению взаимодействий с участием атомов фтора в структуре I построена карта ДЭП для модели Iinv. Как видно из рис. 6а, в области реализации взаимодействия F(28)···F(33) область накопления ЭП на F(28) строго соответствует области разрежения ЭП на атоме F(33). Отметим, что в молекуле I наблюдаются укороченные пары атомов F···F, для которых не удается локализовать критические точки (3, –1) ни в случае теоретического, ни при использовании экспериментально-теоретического подхода к получению распределения ЭП. Например, расстояние F(33)···F(36) в Iinv составляет 2.774(4) Å. Такое значение входит в интервал характерных для F···F взаимодействий (табл. 4), однако критическая точка и связевый путь для этой пары атомов не были локализованы (рис. 4б). С целью определения принципиального отличия таких невзаимодействующих пар атомов было построено распределение ДЭП в Iinv в плоскости C(17)–F(33)···F(36) (рис. 6б). В отличие от взаимодействия F(28)···F(33), атомы F(33) и F(36) располагаются друг относительно друга таким образом, что область накопления ЭП на одном из атомов соответствует области накопления ЭП на втором атоме. Именно это является причиной отсутствия взаимодействия между атомами F(33)···F(36). Распределение теоретической ДЭП в области невалентных взаимодействий фтора в молекуле Iisol отлично воспроизводит данные модели Iinv.
Рис. 6.
Распределение ДЭП (0.05 а.е.) в комплексе Iinv в плоскости С(15)–F(28)···F(33) (а) и плоскости C(17) –F(33)···F(36) (б). Сплошными линиями отмечены области концентрации ЭП, пунктирными линиями – области разрежения ЭП.
Помимо невалентных взаимодействий с участием атомов фтора F···F, F···O, F···H и C–F → Yb в структуре Iisol локализованы также внутримолекулярные контакты: три O···H и два O···O. Энергия взаимодействий O···H составляет 0.93, 1.33 и 1.65 ккал/моль, O···O – 3.36 и 4.24 ккал/моль. Анализ топологии ЭП в Iinv не позволил локализовать взаимодействие O···H в молекуле. Однако одно из двух обнаруженных в Iisol взаимодействий O···O воспроизводится в Iinv. Критическая точка (3, –1) локализована между атомами кислорода μ3-мостиковых гексафторизопропоксидных лигандов. Рассчитанная, согласно корреляции Эспинозы – Моллинса – Лекомта [41], энергия этого взаимодействия составляет 4.24 и 4.96 ккал/моль в Iisol и Iinv соответственно.
Таким образом, было проведено исследование топологии распределения ЭП в комплексе I, полученного методами: теоретическим (DFT квантово-химический расчет) и экспериментально-теоретическим (молекулярный инвариом). Показано, что основные топологические характеристики, соответствующие взаимодействиям в координационной сфере атомов иттербия, находятся в пределах индекса переносимости между Iisol и Iinv. Это утверждение справедливо, в частности для внутримолекулярных взаимодействий C–F → Yb в I. Оптимизация геометрии приводит к более сильному изменению геометрии по сравнению с использованием модели молекулярного инвариома. В результате количество внутримолекулярных взаимодействий с участием атомов фтора и их энергия в Iisol оказывается существенно выше, чем в Iinv. Однако значения основных топологических характеристик (ρ(r) и ∇2ρ(r)), соответствующих взаимодействиям F···X (X = F, H, O), которые удается локализовать в рамках обоих исследованных подходов, лежат в пределах индексов переносимости этих величин. Исследование распределения деформационной электронной плотности в Iinv показало, что реализация взаимодействий Fδ–···Fδ– возможна за счет соответствия области концентрации ЭП на одном из атомов области разрежения ЭП на другом независимо от способа получения распределения электронной плотности в молекуле.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Gatti C., Matta C.F. Modern Charge-Density Analysis. Heidelberg: Springer, 2012.
Dittrich B., Matta C.F. // IUCrJ. 2014. V. 1. P. 457.
Abramov Yu.A. // Acta Crystallogr. A. 1997. V. 53. P. 264.
Цирельсон В.Г. // Соросовский образовательный журн. 2000. Т. 6. № 6. С. 94.
Santos G.M., Catellani I.B., Bini R.D. et al. // Ferroelectrics. 2016. V. 500. № 1. P. 26.
Жигулин Г.Ю., Забродина Г.С., Каткова М.А. и др. // Изв. АН. Сер. хим. 2019. № 4. С. 743.
Sangwan R., Saini M., Verma R. et al. // J. Mol. Struct. 2020. V. 1208. ID 127786.
Dittrich B., Koritsnszky T., Luger P. // Angew. Chem. Int. Ed. 2004. V. 43. P. 2718.
Dittrich B., Hubschle C.B., Luger P., Spackman M.A. // Acta Crystallogr. D. 2006. V. 62. P. 1325.
Wandtke C.M., Weil M., Simpson J. et al. // Acta Crystallogr. B. 2017. V. 73. P. 794.
Dittrich B., Wandtke C.M., Meents A. et al. // ChemPhysChem. 2015. V. 16. P. 412.
Malischewski M., Seppelt K., Sutter J. et al. // Angew. Chem. Int. Ed. 2017. V. 56. P. 13372.
Фукин Г.К., Баранов Е.В., Черкасов А.В. и др. // Коорд. химия. 2019. Т. 45. № 10. С. 584 (Fukin G.K., Baranov E.V., Cherkasov A.V. et al. // Russ. J. Coord. Chem. 2019. V. 45. № 10. P. 680). https://doi.org/10.1134/S1070328419090045
Nelyubina Y.V., Korlyukov A.A., Lyssenko K.A. et al. // Inorg. Chem. 2017. V. 56. P. 4688.
Фукин Г.К., Баранов Е.В., Черкасов А.В. и др. // Изв. АН. Сер. хим. 2019. № 9. С. 1650.
Fukin G.K., Cherkasov A.V., Baranov E.V. et al. // ChemistrySelect. 2019. V. 4. P. 10976.
Kovalenko A.A., Nelyubina Y.V., Korlyukov A.A. et al. // Z. Kristallogr. Cryst. Mater. 2018. V. 233. № 5. P. 317.
Проблема белка. Т. 3: Структурная организация белка / Под ред. Попова Е.М. М.: Наука, 1997. 604 с.
Evans W.J., Forrestal K.J., Ansari M.A. et al. // J. Am. Chem. Soc. 1998. V. 120. P. 2180.
Liu B., Roisnel T., Maron L. et al. // Chem. Eur. J. 2013. V. 19. P. 3986.
Maleev A.A., Fagin A.A., Ilichev V.A. et al. // J. Organomet. Chem. 2013. V. 747. P. 126.
Melman J.H., Rohde C., Emge T.J. et al. // Inorg. Chem. 2002. V. 41. P. 28.
Кузяев Д.М., Румянцев Р.В., Фукин Г.К. и др. // Изв. АН. Сер. хим. 2014. № 4. С. 848.
Kuzyaev D.M., Balashova T.V., Burin M.E. et al. // Dalton Trans. 2016. V. 45. P. 3464.
Румянцев Р.В., Фукин Г.К. // Коорд. химия. 2019. Т. 45. № 11. С. 663 (Rumyantsev R.V., Fukin G.K. // Russ. J. Coord. Chem. 2019. V. 45. № 11. P. 767). https://doi.org/10.1134/S1070328419110058
Frisch M.J., Trucks G.W., Schlegel H.B. et al. Gaussian09. Revision D.01. Wallingford (CT, USA): Gaussian, Inc., 2009.
Schuchardt K.L., Didier B.T., Elsethagen T. et al. // J. Chem. Inf. Model. 2007. V. 47. № 3. P. 1045.
Ditchfield R., Hehre W.J., Pople J.A. // J. Chem. Phys. 1971. V. 54. P. 724.
Hehre W.J., Ditchfield R., Pople J.A. // J. Chem. Phys. 1972. V. 56. P. 2257.
Hariharan P.C., Pople J.A. // Theor. Chim. Acta. 1973. V. 28. P. 213.
Clark T., Chandrasekhar J., Spitznagel G.W. et al. // J. Comput. Chem. 1983. V. 4. P. 294.
Dolg M., Stoll H., Preuss H. // J. Chem. Phys. 1989. V. 90. P. 1730.
AIMAll (version 17.11.14) / Todd A. Keith. Overland Park (KS, USA): TK Gristmill Software, 2017 (aim.tkgristmill.com).
Jorge F.E., Martins L.S.C., Franco M.L. // Chem. Phys. Lett. 2016. V. 643. P. 84.
Jayatilaka D., Grimwood D.J. // Comput. Sci. ICCS. 2003. V. 2660. P. 142.
Jelsch C., Guillot B., Lagoutte A. et al. // J. Appl. Crystallogr. 2005. V. 38. P. 38.
Stash A., Tsirelson V. // J. Appl. Crystallogr. 2002. V. 35. P. 371.
Lu T., Chen F. // J. Comp. Chem. 2012. V. 33. P. 580.
Fukin G.K., Baranov E.V., Jelsch C. // J. Phys. Chem. A. 2011. V. 115. P. 8271.
Bader R.F. Atoms in Molecules: A Quantum Theory. Oxford: Oxford Univ. Press, 1990.
Espinosa E., Molins E., Lecomte C. // Chem. Phys. Lett. 1998. V. 285. P. 170.
Checińska L., Mebs S., Hubschle C.B. et al. // Org. Biomol. Chem. 2006. V. 4. P. 3242.
Karnoukhova V.A., Fedyanin I.V., Lyssenko K.A. // Struct. Chem. 2016. V. 27. P. 17.
Hathwar V.R., Guru Row T.N. // Cryst. Growth Des. 2011. V. 11. P. 1338.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия