

Координационная химия, 2022, T. 48, № 1, стр. 3-10
Исследование катионных порфиринов и их металлокомплексов методами ЭПР
Н. Э. Санникова 1, К. А. Жданова 2, А. С. Спицына 1, 3, Н. А. Брагина 2, М. В. Федин 1, *, О. А. Крумкачева 1, **
1 Международный томографический центр СО РАН
Новосибирск, Россия
2 МИРЭА – Российский технологический университет
Москва, Россия
3 Новосибирский институт органической химии им. Н.Н. Ворожцова СО РАН
Новосибирск, Россия
* E-mail: mfedin@tomo.nsc.ru
** E-mail: olesya@tomo.nsc.ru
Поступила в редакцию 30.04.2021
После доработки 29.06.2021
Принята к публикации 30.07.2021
- EDN: GSTRDA
- DOI: 10.31857/S0132344X22010030
Аннотация
В работе методами электронного парамагнитного резонанса (ЭПР) проведено исследование катионных мезо-арилзамещенных порфиринов и их комплексов с металлами в качестве модельных соединений для фотодинамической терапии. Синтезированы симметричные катионные порфирины с терминальными пиридиниевыми группировками на коротких алкильных спейсерах и их комплексы с Zn(II) и Cu(II). Методом ЭПР с временным разрешением определены параметры расщепления в нулевом магнитном поле фотовозбужденных триплетных состояний. Для комплексов Cu(II) измерена величина сверхтонкого взаимодействия между парамагнитным лигандом и ядрами азота кольца порфирина. На основе полученных данных проанализировано влияние пиридиниевых заместителей и лиганда на магнитно-резонансные параметры порфиринов.
Производные порфиринов играют важную роль в различных фотохимических и фотобиологических процессах, протекающих в природе. Амфифильные мезо-арилзамещенные порфирины и их комплексы с металлами представляют значительный интерес с точки зрения медицинского применения [1–3]. Данные соединения обладают особыми свойствами – высокими коэффициентами молярного поглощения, относительно высокими квантовыми выходами триплетного состояния и флуоресценции, их комплексы с парамагнитными металлами обеспечивают применение порфиринов в медицине в качестве активных соединений в радиологической [4] и магнитно-резонансной [5] диагностике рака и в качестве фотосенсибилизаторов в фотодинамической терапии (ФДТ) рака [6–8].
Известно, что небольшие изменения в структуре порфиринов позволяют модифицировать их физико-химические свойства и, следовательно, изменять область приложения данных веществ [9]. Такие трансформации были успешно использованы для разработки нескольких активных и селективных биомиметических каталитических систем, а также в медицинской химии, особенно для создания контрастных веществ, применяемых в медицинской визуализации, ФДТ и фотодинамической инактивации бактерий (АФДТ) [10]. Кроме того, некоторые фотосенсибилизаторы (ФС), относящиеся к этой группе молекул, уже присутствуют на фармацевтическом рынке и одобрены для клинического использования [11].
Учитывая перспективность мезо-арилзамещенных порфиринов в качестве ФС, исследование свойств их триплетного состояния является актуальной задачей. Важным вопросом также является исследование роли лигандов и структуры порфиринов в связывании с биомолекулами [12]. Предполагается, что лиганд изменяет геометрическую структуру порфирина, делая кольцо порфирина более или, наоборот, менее плоским, что оказывает влияние на степень связывания с ДНК [13]. Измерение величины сверхтонкого взаимодействия между парамагнитным лигандом и ядрами азота центрального кольца порфирина позволяет получить информацию о геометрии центрального остова порфирина и отследить структурные изменения, связанные с введением лиганда и заместителей.
Метод ЭПР с временным разрешением (ВР ЭПР) – это прямой метод детектирования короткоживущих радикалов и триплетных молекул [14–16]. Метод основан на возбуждении молекул образца с помощью лазерного излучения и дальнейшей регистрации эволюции сигнала намагниченности во времени при фиксированном значении магнитного поля. Форма ЭПР спектров триплетного состояния определяется параметрами расщепления триплетных подуровней (D и E) в нулевом магнитном поле и относительной заселенностью этих подуровней (схема 1 , рис. 1). Известно, что параметры D и E зависят от структуры фотосенсибилизатора и природы центрального металла [17–19]. Сравнение этих данных для различных ФС позволяет получить информацию об изменении пространственного распределения спиновой плотности и симметрии триплетного состояния [17–19].


Схема 1 .
В связи с этим в данной работе были синтезированы новые симметричные катионные порфирины с терминальными пиридиниевыми группировками на коротких алкильных спейсерах и их комплексы с Zn(II) и Сu(II). Методом ВР ЭПР исследовано влияние пиридиниевых заместителей и Zn(II) на магнитно-резонансные параметры фотовозбужденных триплетных порфиринов. Для комплексов Cu(II) измерена величина сверхтонкого взаимодействия (СТВ) между парамагнитным лигандом и ядрами азота центрального кольца порфирина. Полученные результаты сравнивались с данными для аналогичного, коммерчески доступного катионного порфирина (5,10,15,20-тетракис(N-метил-4-пиридил))порфирина (TMPyP4), не имеющего объемных заместителей.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы и методы. Все химические реагенты были аналитической чистоты и получены от фирмы Sigma-Aldrich. Растворители очищены согласно стандартным процедурам. Спектры ЯМР 1H и 13C исследуемых растворов в CDCl3 или CD3OD были записаны на Bruker MSL-300 pulse Fourier transform спектрометре. Тетраметилсилан или эфират трехфтористого бора использовали в качестве внешнего стандарта. Элементный анализ на C, H, N, S проводили анализаторе FLASH EA 112 фирмы Termo Finnigan (Италия). Масс-спектры регистрировали с помощью жидкостного хроматографа. ЖХ-система состояла из двух LC-20AD насосов (Shimadzu, Япония) и автосамплера, подключенного онлайн к масс-спектрометру LCMS-IT-TOF, оснащенному источником ионизации электрораспылением (Shimadzu, Япония). Спектры получали в режиме прямого ввода без колонки. Масс-спектры получали в диапазоне m/z от 120 до 700 Да (режим отрицательной ионизации) и 100–700 Да для положительной. Условия ионизации: напряжение ESI 4.50 кВ; скорость потока газа-носителя 1.50 л/мин; температура 200.0°С. Тюнинг оборудования (калибровка масс и проверка чувствительности) проводили перед анализом. Электронные спектры поглощения (ЭСП) растворов порфиринов записывали на приборе HACH DR-4000V (Hach-Lange, США) в диапазоне длин волн 320–1100 нм с шагом в 1 нм в кварцевых кюветах с длиной оптического пути 10 мм при комнатной температуре. Соединения C5Py и его комплексы с металлами описаны ранее [10]. Порфирин TMPyP4 (323497) приобретали у фирмы Sigma-Aldrich Chem. Структуры исследуемых порфиринов представлены на схеме 2.
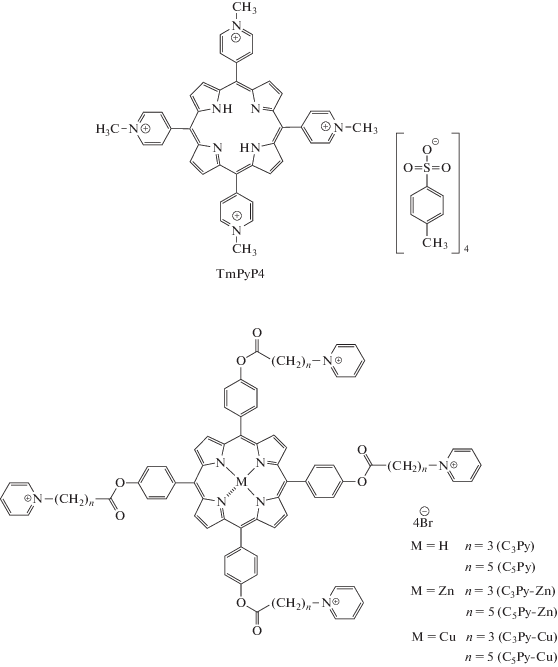
Схема 2 .
Cинтез 5,10,15,20-тетракис(4-(4-бром-н-бутаноил)оксифенил)порфирин. Навески 100 мг (1.50 ммоль) пиррола и 406.3 мг (1.50 ммоль) 4-(4-бром-н-бутаноил)оксибензальдегида растворяли в 100 мл хлористого метилена. Реакционную массу перемешивали при комнатной температуре в течение 15 мин, насыщая колбу инертным газом. Затем добавляли 20 мкл (0.15 ммоль) BF3 Et2O и 200 мкл абсолютного этанола. Реакционную смесь перемешивали 1 ч в токе аргона, после чего вводили 341 мг (1.50 ммоль) DDQ. Спустя 3 ч реакционную массу концентрировали при пониженном давлении на роторном испарителе. Целевое соединение выделяли методом колоночной хроматографии на силикагеле G60. Элюирование проводили хлористым метиленом с последующим увеличением полярности до системы хлористый метилен–этилацетат = 40 : 1. Вещество сушили при пониженном давлении над P2O5. Выход 0.16 г (33%). Rf = 0.7 (хлористый метилен/этилацетат = 40 : 1). ЯМР 1Н (CDCl3; δ, м.д.): 2.74 (2Н, с., NH-пиррол), 2.35–2.58 (8H, м., OCOСН2СН2), 2.90–3.15 (8Н, м., OCOСН2), 3.60–3.75 (8Н, м., СН2Br), 7.52 (8Н, д., J = 8.25 Гц, 3,5-(ArH)), 8.32 (8Н, д., J = 8.18 Гц, 2,6-(ArH)), 8.92 (8Н, с., CH-пиррол).
Синтез 5,10,15,20-тетракис(4-(4-пиридилбутаноил)оксифенил)порфирин тетрабромид (C3Py). В 15 мл безводного пиридина растворяли 40.0 мг (0.031 ммоль) 5,10,15,20-тетракис(4-(4-бром-н-бутаноил)оксифенил)порфирина. Реакционную массу кипятили с обратным холодильником в течение 3 ч. Образовавшийся в ходе синтеза осадок промывали хлороформом и отфильтровывали. Растворившуюся в хлороформе реакционную массу концентрировали при пониженном давлении. Затем растворяли в метаноле и перекристаллизовывали из медицинского эфира. Вещество сушили при пониженном давлении над P2O5.
Rf 0.2 (CH2Cl2 : EtOAc = 1 : 1); ЭСП (λmax, нм (lg ε)): 415 (5.50); 513 (4.24); 547 (3.73); 589 (3.57); 645 (3.53). ЯМР 1Н (CD3OD; δ, м.д.): –2.73 (2Н, с., NH), 2.53 (8Н, м., ОСОСН2СН2СН2), 2.99 (8Н, м., ОСОСН2СН2СН2), 3.34 (8Н, м., СН2Py), 7.41 (8Н, д., J = 8.13 Гц, 3,5-(ArH)), 8.04 (8Н, д., J = 8 Гц, 2,6-(ArH)), 8.20 (8Н, т., J = 7.72 Гц, 3,5-Py), 8.65–8.76 (12Н, m., 4-Py + CH), 9.12 (8Н, м., 2,6-пиррол). ЯМР 13C (CD3OD; δ, м.д.): 172.72.
Синтез тетрабромида 5,10,15,20-тетракис(4-(4-пиридилбутаноил)оксифенил)порфиринцинка (C3Py‑Zn). Навeску 30.0 мг (0.024 ммоль) 5,10,15,20-тетракис(4-(4-бром-н-бутаноил)оксифенил)порфирина растворяли в 15 мл хлористого метилена. Затем к этому раствору добавляли 15 мл раствора 26.3 мг (0.12 ммоль) двуводного ацетата цинка. Реакционную массу перемешивали при комнатной температуре в течение 3 ч. Степень протекания реакции оценивали по данным ЭСП. Далее реакционную смесь концентрировали при пониженном давлении и экстрагировали в системе хлористый метилен/вода. Целевое соединение выделяли методом колоночной хроматографии на силикагеле G60. Элюирование проводили в системе хлористый метилен–этилацетат = 40 : 1. Вещество сушили при пониженном давлении над P2O5. Выход 30 мг (94%). Rf = 0.50 (хлористый метилен). ЭСП (λmax, нм (lgε)): 425 (5.50), 556.2 (4.18), 598.5 (3.77).
Навеску 30 мг (0.022 ммоль) 5,10,15,20-тетракис(4-(4-бром-н-бутаноил)оксифенил)порфиринцинка растворяли в 15 мл безводного пиридина. Реакционную массу кипятили с обратным холодильником в течение 3 ч. Осадок, выпавший в ходе синтеза, отфильтровывали и промывали хлористым метиленом. Растворившуюся в неполярном растворителе реакционную массу концентрировали под вакуумом. Затем растворяли в метаноле и перекристаллизовывали из медицинского эфира. Целевое соединение сушили при пониженном давлении над P2O5. Выход 34.0 мг (73%).
Rf = 0.35 (метанол). Rf 0.45 (CH3OH). Rf 0.2 (CH2Cl2 : EtOAc = 1 : 1); ЭСП, λmax, нм (lgε)): 419 (5.28); 562 (4.61); 603 (3.96). ЯМР 1Н (CD3OD; δ, м.д.): 2.03 (8Н, м., ОСОСН2СН2СН2), 3.34 (8Н, м., ОСОСН2СН2СН2), 4.80 (8Н, м., СН2СН2Py), 7.22 (8Н, д., J = 8.26 Гц, 3,5-(ArH)), 7.60 (8Н, д., J = 8 Гц, 2,6-(ArH)), 8.01 (8Н, т., J = 7.76 Гц, 3,5-Py), 8.59–8.69 (12Н, м., 4-Py + CH пиррол), 8.89 (8Н, д., J = 5.66 Гц, 2,6-Py). ЯМР 13C (CD3OD; δ, м.д.): 172.57, 151.59, 145.82, 144.78, 139.39, 134.96, 128.43, 119.68, 119.23, 61.14, 30.21, 26.12. ESI-MS 827.23 [M]2+.
Синтез тетрабромидa 5,10,15,20-тетракис(4-(4-пиридилбутаноил)оксифенил)порфиринмеди (C3Py-Cu). 30.0 мг (0.024 ммоль) 5,10,15,20-тетракис(4-(4-бром-н-бутаноил)оксифенил)порфирина растворяли в 15 мл хлористого метилена. Затем к этому раствору добавляли 15 мл раствора 45.7 мг (0.251 ммоль) ацетата меди. Реакционную массу перемешивали при комнатной температуре в течение 3 ч. Степень протекания реакции оценивали по данным ЭСП. Далее реакционную смесь концентрировали при пониженном давлении и экстрагировали в системе хлористый метилен–вода. Целевое соединение выделяли методом колоночной хроматографии на силикагеле G60. Элюирование проводили в системе хлористый метилен–этилацетат = 40 : 1. Вещество сушили при пониженном давлении над P2O5. Выход 29.1 мг (93%). Rf = 0.50 (хлористый метилен). ЭСП (λmax, нм (lg ε)): 413 (5.45), 531(4.51).
Навеску 20 мг (0.015 ммоль) 5,10,15,20-тетракис(4-(4-бром-н-бутаноил)оксифенил)порфиринмеди растворяли в 15 мл безводного пиридина. Реакционную массу кипятили с обратным холодильником в течение 3 ч. Осадок, выпавший в ходе синтеза, отфильтровывали и промывали хлористым метиленом. Растворившуюся в неполярном растворителе реакционную массу концентрировали под вакуумом. Затем растворяли в метаноле и перекристаллизовывали из медицинского эфира. Целевое соединение сушили при пониженном давлении над P2O5. Выход 21 мг (85%).
Rf = 0.35 (метанол). Rf 0.60 (CH3OH). ЭСП (λmax, нм (lg ε)): 413 (5.31), 537 (4.54). ESI-MS 826.7 [M]2+.
Приготовление образцов для ЭПР проводили при температурах 80 К, при этом катионные порфирины растворяли в ДМФА в концентрации 15 мМ. Перед каждым измерением концентрированные растворы порфиринов разбавляли до концентрации 250 мкМ и помещали в кварцевые ампулы (внешний диаметр 3.8 мм, внутренний диаметр 2.8 мм) в объеме 40 мкл. Перед помещением в резонатор ЭПР спектрометра образцы подвергали шоковой заморозке в жидком азоте. Лазерное возбуждение проводили с использованием второй гармоники импульсного лазера Nd:YAG Lotis TII (532 нм) с частотой повторения 10 Гц. Излучение на выходе лазера с помощью системы поворотных призм заводили через окно резонатора. Средняя мощность лазерного импульса перед окном составляла 15–20 мДж; этого было достаточно, чтобы возбудить все молекулы порфирина в образце.
Эксперименты ЭПР с временным разрешением проводили на самодельном спектрометре, собранном на базе Bruker EMX, оборудованном системой охлаждения жидким азотом (77–300 К). Шаг по полю для всех ВР ЭПР спектров составлял 0.4 мТл, количество измерений в каждой точке 30, сканов 3, мощность 13 мкВт. Из каждой временной зависимости вычитали записанную в нерезонансном поле (100 мТл). зависимость. Моделирование ВР ЭПР выполняли с помощью пакета Easyspin для Matlab [20].
Спектры стационарного ЭПР получали на ЭПР спектрометре Bruker Elexsys E580 X-диапазона, оснащенном резонатором ER 4118XMD5 (9.75 ГГц) и температурным контроллером Oxford Instruments для проведения измерений в температурном диапазоне 20–277 К.
Моделирование спектров ЭПР выполняли с помощью пакета Easyspin для Matlab [20].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Катионные мезо-арилзамещенныe порфирины (схема 2 ) с заряженными пиридиниевыми группами были синтезированы согласно подходу, разработанному нами ранее [21]. Схема синтеза целевых катионных порфиринов включает две стадии. На первой конденсацией замещенных остатками жирных кислот бензальдегидов и пиррола в мягких условиях по методу Линдсея [22] с использованием эфирата трехфтористого бора (BF3 ⋅ Et2O) в качестве кислотного катализатора получают бромзамещенные порфирины-предшественники. Второй этап заключается в получении катионных соединений реакцией кватернизации пиридина. Использование подобного подхода уменьшает количество стадий получения целевых порфиринов продуктов, а также облегчает их выделение и хроматографическую очистку. Комплексы Zn(II), Cu(II) получали на стадии порфиринов-предшественников по стандартным методам с выходами, близкими к количественным. Образование металлокомплекса отслеживали с помощью электронной спектроскопии. Строение полученных продуктов на всех стадиях синтеза подтверждали данными методов мультиядерной спектроскопии ЯМР, УФ-спектроскопии, масс-спектрометрии, элементным анализом.
На рис. 2 представлены спектры ВР ЭПР, записанные при 80 К через 2 мкс после лазерного импульса. Все спектры фотовозбужденных триплетных состояний для порфиринов без металлов имеют одинаковый тип электронной спиновой поляризации (EEE/AAA, где A – абсорбция, E – эмиссия), однако положение линий и, следовательно, параметры D и E, для C3Py и С5Py отличаются от данных для TMPyP4.
Рис. 2.
ВР ЭПР спектры порфиринов, записанные при 80 K в ДМФА: TmPyP4 (а), C3Py (б), C5Py (в), C3Py-Zn (г), C5Py-Zn (д). Черным цветом показаны экспериментальные данные, красным – моделирование.

Параметры D и E для катионных порфиринов, определенные из моделирования ЭПР спектров, представлены в табл. 1. Согласно литературным данным, для тетрафенилпорфирина (ТPP) без заместителей параметры D и E составляют 1120 и 221 МГц соответственно [17, 19]. Анализ полученных данных показывает, что максимальное изменение параметров D и E по сравнению с TPP наблюдается для порфирина TMPyP4, в котором пиридиниевые группы введены непосредственно в макроцикл порфирина. В данном случае наблюдается увеличение |D| на 10% и уменьшение |E| в 1.5 раза, что, наиболее вероятно, связано с уменьшением делокализации спиновой плотности в триплетном состоянии. Введение пиридиниевых заместителей через алкильные спейсеры существенно не изменяет параметры D и E и, следовательно, не влияет на конфигурацию триплетного состояния. Этот результат важен, поскольку катионные группы вводятся для обеспечения растворимости и не должны вызывать нежелательных изменений электронной структуры.
Таблица 1.
Параметры D и E для триплетных состояний порфиринов, определенные из моделирования спектров ВР ЭПР*
Введение цинка в порфирины C3Py и C5Py приводит к изменению формы спектра и смене типа поляризации на AAA/EEE (cм. выше). Такое изменение формы спектра характерно для Zn-содержащих порфиринов и связано с изменением населенностей триплетных подуровней при введении Zn. Появление спин-орбитального взаимодействия с ионом цинка, приводит к смешиванию d-орбиталей цинка c π-системой порфирина, и в результате в ходе синглет-триплетной (S-T) интеркомбинационной конверсии в фотовозбужденном триплетном состоянии преимущественно заселяется подуровень Tz. Как и для порфиринов без металлов, введение пиридиниевых заместителей через алкильные спейсеры не оказывает значительного влияния на электронную структуру триплетного состояния порфирина в комплексе с цинком.
Комплексы порфиринов с Cu(II) невозможно исследовать методом ЭПР с временным разрешением из-за короткого времени жизни триплетного состояния. Поэтому при исследовании комплексов C3Py-Cu(II) и C5Py-Cu(II) мы использовали метод стационарного ЭПР для измерения величины СТВ между парамагнитным центром Cu(II) и ядрами азота центрального кольца порфирина.
На рис. 3 представлены спектры стационарного ЭПР, записанные при 80 К для C3Py-Cu и C5Py-Cu. Для содержащих Cu(II) порфиринов взаимодействие неспаренного электрона меди с его ядерным спином I = 3/2 (СТВ) приводит к расщеплению каждой линии, соответствующей определенной ориентации в спектре ЭПР, на четыре. Кроме того, ионы меди(II) характеризуются значительной анизотропией g-тензора, что приводит к достаточно сложным порошковым спектрам с перекрывающимися линиями в Х-диапазоне. Дополнительное взаимодействие неспаренного электронного спина с четырьмя атомами азота с I = 1 приводит к появлению дополнительной сверхтонкой структуры. Для полученных экспериментальных данных было проведено моделирование с учетом анизотропии g- и СТВ-тензоров (A), результаты которого показаны в табл. 2. Параметры уширения линий ЭПР имеют близкие значения для обоих комплексов. СТВ на азотах макроцикла порфирина зависит от спиновой плотности парамагнитного центра на этих ядрах и в приближении точечных диполей определяется расстоянием между парамагнитным центром и азотами. Моделирование спектров ЭПР показало, что азоты в макроцикле C3Py-Cu(II) и C5Py-Cu(II) эквивалентны, и тензор СТВ имеет близкие значения к данным для TMPyP4-Cu(II) [23].
Рис. 3.
Стационарные спектры ЭПР в X-диапазоне в ДМФА при 80 K: C3Py-Cu (а), C5Py-Cu (б). Черным цветом показаны экспериментальные данные, красным – моделированные.

Таблица 2.
Параметры g- и СТВ-тензоров, а также уширение линий для комплексов порфиринов с медью, определенные из моделирования стационарных спектров ЭПР с помощью программного пакета EasySpin
| Порфирин | g-Тензор | СТВ-тензор с Cu (А_Cu), МГц | СТВ-тензор с N (А_N), МГц | Уширение линий от пика до пика, мТл |
|---|---|---|---|---|
| C3Py-Cu | gxx = 2.054 gyy = 2.072 gzz = 2.211 | A_Cuxx = 48 A_Cuyy = 56 A_Cuzz = 604 | A_Nxx = A_Nyy = 52 A_Nzz = 43 | Неоднородное (0.10), однородное (0.45) |
| C5Py-Cu | Неоднородное (0.20), однородное (0.51) |
На основании полученных результатов для C3Py-Cu(II) и C5Py-Cu(II) можно предположить, что пространственное расположение азотов внутри макроцикла порфирина в комплексе Cu(II) существенно не изменяется при введении пиридиниевых заместителей в порфириновый остов, то есть не нарушается их эквивалентность.
Таким образом, в результате выполнения данной работы были синтезированы катионные ме-зо-замещенные порфирины и их комплексы с Zn(II) и Cu(II). Полученные соединения охарактеризованы современными физико-химическими методами анализа. Методом ЭПР с временным разрешением определены параметры расщепления в нулевом магнитном поле фотовозбужденных триплетных состояний. Обнаружено влияние пиридиниевых групп, введенных непосредственно в макроцикл порфирина, на параметры триплетного состояния. Однако введение пиридиниевых заместителей через алкильные спейсеры существенно не изменяет конфигурацию триплетного состояния. Показано, что пиридиниевые заместители также не нарушают эквивалентность азотов внутри макроцикла порфирина в комплексе Cu(II).
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Shi Y., Zhang F., Linhardt R.J. // Dye. Pigment. 2021. V. 188. P. 109136.
Wang Z., Sun, Q., Liu, B. et al. // Coord. Chem. Rev. 2021. V. 439. P. 213945.
Kawczyk-Krupka A., Pucelik B., Międzybrodzka A. et al. // Photodiagnosis Photodyn. Ther. 2018. V. 23. P. 132.
Lee J.-H., Shao S., Cheng K. T. et al. // J. Liposome Res. 2015. V. 25. № 2. P. 101.
He M., Chen Y., Tao C. et al. // ACS Appl. Mater. Interfaces. 2019. V. 11. № 45. P. 41946.
Annunzio S.R.D., Costa N.C.S., Mezzina R.D. et al. // Int. J. Mol. Sci. 2019. V. 20. № 16. P. 3861.
Mironov A.F., Zhdanova K.A., Bragina N.A. // Russ. Chem. Rev. 2018. V. 87. № 9. P. 859.
Sengupta D., Timilsina U., Mazumder Z.H. et al. // Eur. J. Med. Chem. 2019. V. 174. P. 66.
Yi X., Wang F., Qin W. et al. // Int. J. Nanomedicine. 2014. P. 1347.
Gao D., Guo X., Zhang X. et al. // Mater. Today Bio. 2020. V. 5. P. 100035.
Habermeyer B., Guilard R. // Photochem. Photobiol. Sci. 2018. V. 17. № 11. P. 1675.
Sannikova N.E., Timofeev I.O., Chubarov A.S. et al. // J. Photochem. Photobiol. B. 2020. V. 211. P. 112008.
DuPont J.I., Henderson K.L., Metz A. et al. // Biochim. Biophys. Acta Gen. Subj. 2016. V. 1860. № 5. P. 902.
Malcolm D.E., Forbes M.D.E., Lauren E. et al. // Adv. Phys. Org. Chem. 2013. P. 1.
Wang Z., Ivanov M., Gao Y. et al. // Chem. – A Eur. J. 2020. V. 26. № 5. P. 1091.
Ivanov M.Y., Veber S.L., Prikhod’ko S.A. et al. // J. Phys. Chem. B. 2015. V. 119. № 42. P. 13440.
Kay C.W.M. // J. Am. Chem. Soc. 2003. V. 125. № 45. P. 13861.
Langhoff S.R. // J. Chem. Phys. 1975. V. 62. № 1. P. 169.
Yamauchi A., Fujiwara S. Nishimura K. et al. // J. Phys. Chem. A. 2021. P. 4334.
Stoll S., Schweiger A. // J. Magn. Reson. 2006. V. 178. № 1. P. 42.
Zhdanova K.A., Savelyeva I.O., Ignatova A. A. et al. // Dye. Pigment. 2020. V. 181. P. 108561.
Lindsey J.S., Hsu H.C. // Tetrahedron Lett. 1986. V. 27. № 41. P. 4969.
Greiner S.P., Rowlands D.L., Kreilick R.W. // J. Phys. Chem. 1992. V. 96. № 23. P. 9132.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия