

Координационная химия, 2022, T. 48, № 1, стр. 11-17
Синтез и кристаллическая структура хелата кобальта(III) с тридентантным азометиновым лигандом, включающим бензимидазольный фрагмент
Л. Д. Попов 1, *, С. А. Бородкин 1, М. А. Кискин 2, А. А. Павлов 3, П. А. Князев 4, В. В. Чернявина 1, И. Н. Щербаков 1
1 Южный федеральный университет
Ростов-на-Дону, Россия
2 Институт общей и неорганической химии им. Н.С. Курнакова РАН
Москва, Россия
3 Институт элементорганической химии им. А.Н. Несмеянова РАН
Москва, Россия
4 Научно-исследовательский институт физической и органической химии
Южного федерального университета
Ростов-на-Дону, Россия
* E-mail: ldpopov@mail.ru
Поступила в редакцию 07.04.2021
После доработки 14.05.2021
Принята к публикации 15.05.2021
- EDN: EHOYFM
- DOI: 10.31857/S0132344X21110037
Аннотация
Синтезирован азометин HL путем взаимодействия о-аминофенола с 2-ацетилбензимидазолом, на его основе получен комплекс кобальта(III) состава [CoL2]2(ClO4)2 ⋅ 3H2O (I). Строение комплекса I установлено методами спектроскопии ЯМР в растворе и РСА в кристалле (CIF file CCDC № 2051279). Ион кобальта имеет октаэдрическое окружение в степени окисления +3. Кристаллы моноклинные, пр. гр. C2/c, a = 12.405(7), b = 13.946(11), c = 18.907(13) Å, α = 109.87(3)°, β = = 94.534(12)°, γ = 104.564(19)°, V = 2928(3) Å3, ρ(выч.) = 1.521 г/см 3, Z = 2. Комплекс диамагнитен. Изучение электрохимического поведения комплекса I в ацетонитриле показало возможность его восстановления до нейтральной и анион-радикальной формы.
Основания Шиффа (азометины) занимают одно из ведущих мест в различных областях химии – биоорганической [1, 2], медицинской [3, 4], супрамолекулярной [5, 6], координационной [7, 8], фотохимии [9–12] и т.д. Особый интерес представляют гетероциклические основания Шиффа, поскольку многие гетероциклы проявляют фармакофорные свойства и содержат в своем составе гетероатомы, способные образовывать устойчивые связи с ионами металлов.
В настоящей работе в качестве карбонильной компоненты для синтеза азометина был выбран 2-ацетилбензимидазол. Хорошо известно, что производные бензимидазола обладают разнообразной биологической активностью: антимикробной [13], антираковой [14], антигистаминной [15], антитуберкулезной [16], антималярийной [17], антивирусной [18], анти-ВИЧ [19] и т.д. Кроме того, бен-зимидазольный фрагмент входит в состав хромогенных хемосенсоров [20–22], OLED-материалов [23], катализаторов [24] и др. В литературе описаны единичные случаи азометинов 2-ацетилбензимидазола и комплексы на их основе [25–30], однако известны многочисленные примеры “обратных” азометинов на основе 2-аминобезимидазола и их комплексы. Из литературы известно, что близкие по строению комплексы Co(II) на основе сходного по природе лиганда – продукта конденсации о-аминофенола и пиридин-2-альдегида – обладают спин-кроссовером [31]. Кроме того, некоторые комплексы кобальта(II) способны проявлять аномальные магнитные свойства, связанные с замедленной релаксацией магнитной восприимчивости [32, 33].
Цель настоящей работы заключалась в исследовании комплексообразующей способности 2-((1-(1Н-бензо[d]имидазол-2-ил)этилиден)амино)фенол (HL) – продукта конденсации 2-ацетилбензимидазола и о-аминофенола – по отношению к иону кобальта(II) и изучении его свойств.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Для синтеза целевых соединений использовали коммерчески доступные реактивы: 2-аминофенол, перхлорат кобальта(II) гексагидрат, метанол, толуол, ацетонитрил фирмы Acros.
Синтез HL. К горячему раствору 0.600 г (3.80 ммоль) 2-ацетилбензимидазола [34] в 10 мл толуола добавляли раствор 0.400 г (3.80 ммоль) 2‑аминофенола в 5 мл толуола. К полученному оранжевому раствору добавляли 0.3 мл муравьиной кислоты и кипятили смесь 4.5 ч. Выпавший осадок отфильтровывали, промывали горячим толуолом, диэтиловым эфиром и перекристаллизовывали из этанола. Выход HL 0.650 г (69%). Тпл = = 224–225°C.
ИК (ν, см–1): 2200–3200 ш.о.с, 1634 с, 1586 с, 1484 ср, 1449 о.с, 1435 о.с, 1345 ср, 1320 с, 1292 с, 1223 с, 1149 сл, 1103 сл, 972 сл, 827 сл, 860 сл, 757 о.с, 742 ср, 699 сл, 608 сл, 573 сл. ЯМР 1Н (600 МГц; ДМСО-d6; δ, м.д.): 12.97 (с., 1H, NH), 9.17 (с., 1H, OH), 7.75 (д., 3JHH = 8.0 Гц, 1Н, Bz), 7.53 (д., 3JHH = 8.0 Гц, 1Н, Bz), 7.30 (т., 3JHH = 8.0 Гц, 1Н, Bz), 7.23 (т., 3JHH = = 8.0 Гц, 1Н, Bz), 6.98–7.02 (м., 1H, Ph), 6.92 (д., 3JHH = 7.9 Гц, 1Н, Ph), 6.85 (м., 2Н, Ph), 2.33 (с., 3Н, Me). ЯМР 13C{1H} (151 МГц; ДМСО-d6; δ, м.д.): 160.09 (c., Bz), 151.22 (c., C-Me), 147.08 (c., Ph), 143.28 (c., C–NH), 136.93 (c., Ph), 134.80 (c., C–N (Bz)), 125.30 (c., Ph), 124.14 (c., Bz), 121.95 (c., Bz), 120.80 (c., Ph), 119.99 (c., Bz), 119.28 (c., Ph), 116.03 (c., Ph), 112.12 (c., Bz), 16.96 (c., Me).
Синтез I. К горячему раствору 0.150 г (0.29 ммоль) HL в 5 мл метанола добавляли горячий раствор 0.100 г (0.145 ммоль) Co(ClO4)2 ⋅ 6H2O в 4 мл метанола. Полученный темно-красный раствор оставляли на 3 сут. Выпавший кристаллический продукт отфильтровывали, промывали метанолом и высушивали в вакууме. Выход I 0.100 г (56%). По данным РСА, полученное соединение отвечает составу [CoL2]2(ClO4)2 · 3H2O.
ИК (ν, см–1): 3236 ш.сл, 1588 ср, 1568 ср, 1537 ср, 1322 с, 1256 о.с, 1218 сл, 1148 ср, 1050–1150 ш. о.с, 1030 сл, 994 сл, 845 сл, 746 с, 623 ср, 564 ср. ЯМР 1Н (300 МГц; ДМСО-d6; δ, м.д.): 14.93 (уш.с., 1H, NH), 8.16 (д., 3JHH = 7.4 Гц, 1Н, Bz), 7.60 (д., 3JHH = = 7.4 Гц, 1Н, Bz), 7.33 (м., 1Н, Bz), 7.26 (м., 1Н, Bz), 7,05 (м., 2H, Ph), 6.71 (м., 1Н, Ph), 6.60 (д., 3JHH = 8.0 Гц, 1Н, Ph), 2.33 (с., 3Н, Me).
РСА монокристалла соединения I выполнен на дифрактометре Bruker Apex II, оборудованном CCD-детектором (MoKα, λ = 0.71073 Å, графитовый монохроматор) [35], по программе SADABS [36] введена полуэмпирическая поправка на поглощение (Tmin/Tmax = 0.6320/0.7461). Структуры расшифрованы прямыми методами и уточнены полноматричным МНК в анизотропном приближении для всех неводородных атомов. Атомы водорода при атомах азота и углерода органических лигандов и молекул воды генерированы геометрически и уточнены в модели “наездника”. Уточнение структуры выполнено с учетом разупорядочения ClO4-аниона по двум позициям с заселенностями 0.733(6) и 0.267(6). Расчеты проведены по комплексу программ SHELX [37] с использованием OLEX2 [38]. Кристаллографические параметры и детали уточнения структуры I при T = 150(2) K: C60H54Cl2Co2N12O15, M = 1371.91 г/моль, пр. гр. $P\bar {1}$, черный призматический кристалл, 0.15 × 0.15 × × 0.12 мм, a = 12.405(7), b = 13.946(11), c = = 18.907(13) Å, α = 109.87(3)°, β = 94.534(12)°, γ = = 104.564(19)°, V = 2928(3) Å3, Z = 2, ρ(выч.) = = 1.521 г см–3, μ = 0.735 мм–1, 1.90° ≤ θ ≤ 28.28°, сегмент сферы –16 ≤ h ≤ 15, –18 ≤ k ≤ 18, –25 ≤ l ≤ 25, 25075 измеренных рефлексов, 14383 независимых рефлексов, 10 026 отражений с I > 2σ(I), Rint = = 0.0321, GООF = 1.021, R1 (I > 2σ(I)) = 0.0593, wR2 (I > 2σ(I)) = 0.1412, R1 (все данные) = 0.0950, wR2 (все данные) = 0.1603, Δρmin/Δρmax, е Å–3 = = ‒0.664/1.228.
Структурные данные депонированы в Кембриджском банке структурных данных (CCDC № 2051279; deposit@ccdc.cam.ac.uk или http:// www.ccdc.cam.ac.uk/data_request/cif).
Элементный анализ выполнен на приборе Perkin-Elmer 240C в лаборатории микроанализа Южного федерального университета.
Спектры ЯМР зарегистрированы на спектрометрах Bruker Avance 300 и 600 (рабочие частоты по ядрам 1Н – 300.13 и 600.22 МГц). Значения химических сдвигов (δ, м.д.) определяли относительно остаточного сигнала растворителя ДМСО (1H – 2.50 м.д., 13С – 39.52 м.д.). ИК-спектры регистрировали на приборе Varian Scimitar 1000 FT-IR в вазелиновом масле в области 400–4000 см–1. Электронные спектры поглощения (ЭСП) регистрировали на приборе Varian Cary 5000 в области 200–800 нм. Спектры диффузного отражения регистрировали на приборе Varian Cary 5000 с использованием приставки Internal DRA 2500 в области 400–1500 нм.
Магнитную восприимчивость комплекса I определяли относительным методом Фарадея при 295 К. Измерение проводили при напряженности магнитного поля 7.16 × 105 А/м. В качестве этанола для калибровки использовали Hg[Co(CNS)4].
Циклические вольтамперограммы ацетонитрильных растворов соединений были получены на потенциостате-гальваностате Elins-P45X в трехэлектродной электрохимической ячейке. В качестве электродов использовали стеклоуглеродный рабочий электрод, платиновый вспомогательный электрод, электрод сравнения Ag/Ag+ 0.01 M AgNO3. Скорость развертки потенциала v = 50 мВ/с, в качестве фонового электролита использовали 0.1 M Bu4NPF6. Ацетонитрил предварительно очищали. Для удаления низкомолекулярных органических примесей и следов воды его подвергали фракционной перегонке. Для этого ацетонитрил перемешивали в течение 2 сут в контакте с гидридом кальция (10 г/л), после чего декантировали и подвергали фракционной перегонке над P2O5 (5 г/л). Образующийся продукт в течение нескольких часов нагревали с обратным холодильником над гидридом кальция (5 г/л), а затем подвергали медленной фракционной перегонке. Cоединение NBu4PF6 (Alfa Aesar) предварительно перекристаллизовывали из этанола, высушивали и прокаливали при 80°C в течение 4 ч.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Взаимодействие 2-ацетилбензимидазола с о‑аминофенолом в толуоле при кипячении приводит к образованию основания Шиффа (HL) с выходом 69% (схема 1 ):
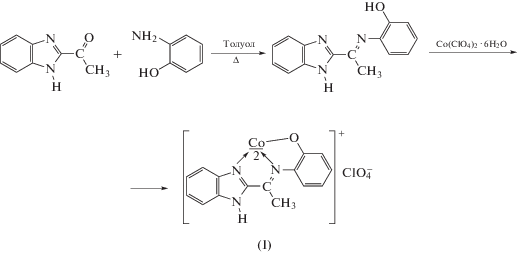
Схема 1 .
В ИК-спектре полученного соединения можно выделить интенсивную полосу в области 1634 см–1, отвечающую валентным колебаниям связи C=N азометинового фрагмента, а также полосу в области 1586 см–1, которую можно отнести к колебаниям эндоциклических связей C=N и C=С. Кроме того, в области 2200–3200 см–1 наблюдается широкая несимметричная интенсивная полоса поглощения, обусловленная образованием водородных связей с участием групп O–H и N–H.
В спектрах ЯМР 1Н и 13С{1Н} HL характеристики сигналов (положение в шкале химического сдвига, интегральные интенсивности и мультиплетности) полностью соответствуют предполагаемому строению. Полное соотнесение сигналов было сделано при помощи двумерных методик ЯМР: 1Н–1Н COSY, 1Н–13С HMQC, 1Н–13С HMBC.
В ЭСП раствора HL в этаноле наблюдается полоса поглощения с максимумом при λ = 300 нм (lg ε = 4.00) и плечом при λмакс = 358 нм (lg ε = 3.36), отвечающая π → π*-переходам в бензольном и бензимидазольном фрагментах соответственно (рис. 1).
Рис. 1.
Электронные спектры поглощения HL (с = 1 × × 10–4 М) (1) и I (с = 2 × 10–5 М) (2) в этаноле.

Реакция Co(ClO4)2 · 6H2O с HL в метаноле (схема 1 ) ведет к формированию комплексного соединения I. В ИК-спектре комплекса I, по сравнению со спектром лиганда, наблюдается только одна полоса поглощения в области 3236 см–1, отвечающая валентным колебаниям группы N–H бензимидазольного фрагмента. Одновременно с этим происходит понижение частот колебаний связей C=N на 20–50 см–1, что свидетельствует об участии атомов азота этих групп в координации к иону кобальта. Наличие широкой высокоинтенсивной полосы в области 1050–1150 см–1 указывает на присутствие в комплексе I перхлорат-иона.
В ЭСП раствора I в этаноле можно выделить три полосы поглощения (рис. 1). При сравнении со спектром HL, можно заметить, что оба π → π*-перехода претерпевают батохромный сдвиг на 50–100 нм. Третья полоса поглощения при λмакс = = 505 нм, наблюдаемая в спектре, обусловлена, вероятно, переносом заряда M→L.
Строение комплекса I было установлено на основании данных РСА. Соединение I кристаллизуется в триклинной пространственной группе $P\bar {1}$ в виде водного сольвата. Независимая часть элементарной ячейки состоит из двух катионов [CoL2]+ близкого строения, двух анионов ClO4– и трех молекул воды ([CoL2]2(ClO4)2 ⋅ 3H2O). Каждый атом кобальта координирует по два хелатных N,N,O-тридентатных лиганда с образованием конденсированных пятичленных металлоциклов (Co(1)–O 1.881(2), 1.903(3), Co(1)–N 1.917(3)–1.944(3) Å, XCo(1)X 82.68(12)°–100.94(12)°; Co(2)–O 1.878(3), 1.888(3), Co(2)–N 1.900(3)–1.935(3) Å, XCo(2)X 82.66(13)°–99.91(12)°) (рис. 2). Геометрия полиэдров CoN4O2 соответствует искаженному октаэдру. Длины связей металл–донорные атомы в обоих катионах [CoL2]+ сопоставимы и характерны для иона кобальта(III) в низкоспиновом состоянии. Ароматические фрагменты лигандов лежат в одной плоскости, выход из усредненной плоскости не превышает 0.25 Å, углы между плоскостями лигандов в катионах [CoL2]+ близки к прямым углам, 86.04(6)° и 88.95(6)°. В кристалле наблюдаются межмолекулярные водородные связи между группами N–H лиганда и атомами кислорода лиганда соседнего катиона, внешнесферных ClO4-анионов и молекул воды. Молекулы воды образуют водородные связи с атомами кислорода лиганда и анионов ${\text{ClO}}_{4}^{ - }$. Основные параметры водородных связей приведены в табл. 1.

Таблица 1.
Взаимодействия D–H…A в кристалле I
| H-связь | D–H, Å | H…A, Å | D…A, Å | D–H…A, град | Код симметрии атома A |
|---|---|---|---|---|---|
| N(3)–H(3)…O(4S) | 0.88 | 2.08 | 2.899 | 154.3 | –x + 2, –y + 2, –z + 2 |
| N(3)–H(3)…O(4SA) | 0.88 | 2.11 | 2.972 | 166.4 | –x + 2, –y + 2, –z + 2 |
| N(12)–H(12A)…O(1) | 0.88 | 1.90 | 2.648 | 141.7 | |
| N(6)–H(6)…O(1w) | 0.88 | 1.92 | 2.766 | 160.1 | |
| N(6)–H(6)…O(2SA) | 0.88 | 2.19 | 2.852 | 131.4 | |
| N(9)–H(9)…O(2w) | 0.88 | 1.86 | 2.735 | 175.9 | –x, –y, –z + 1 |
| O(2w)–H(2wA)…O(8S) | 0.85 | 2.21 | 3.008 | 157.6 | |
| O(2w)–H(2wB)…O(4) | 0.86 | 1.95 | 2.777 | 153.1 |
Исследование магнитной восприимчивости комплекса I в твердой фазе показало, что он диамагнитен. Это характерно для соединений кобальта(III) в низкоспиновом состоянии. Подтверждением октаэдрического окружения иона кобальта в комплексе I является наличие двух полос поглощения в спектре диффузного отражения в области 820 и 1080 нм, отвечающиx переходам 1А1g → 1Т1g и 1А1g → 1Т2g соответственно (рис. 3), которые также характерны для иона Co3+, находящегося в сильном поле лигандов [39].
Химические сдвиги в спектре ЯМР 1Н комплекса I демонстрируют значения, отличные от сдвигов для свободного лиганда HL. При этом наибольшие изменения наблюдаются для протонов, располагающихся наиболее близко к местам образования координационных связей с ионом кобальта, что подтверждает образование комплекса в растворе. Интегральная интенсивность сигналов и их мультиплетность также соответствуют строению комплекса I. Кроме того, сравнительно небольшая ширина сигналов и неизменность значений химических сдвигов при изменении температуры говорит о диамагнитной природе комплекса I в растворе.
Наиболее вероятными механизмами образования комплекса Со(III) представляется окисление иона Со2+ либо за счет лиганда, либо кислородом воздуха, присутствующим в растворителе. Для проверки этого предположения был проведен синтез комплекса в инертной атмосфере (аргон) и в присутствии восстановителя (аскорбиновая кислота). В результате взаимодействия был выделен комплекс, абсолютно идентичный I по строению, что подтверждается данными его РСА. Таким образом, причина окисления иона кобальта, очевидно, заключается в природе используемой лигандной системы.
Поскольку в результате синтеза комплекса I мы обнаружили, что происходит окисление кобальта до трехвалентного состояния, логично было бы исследовать электрохимические свойства комплекса I. Методом циклической вольтамперометрии (ЦВА) изучено электрохимическое поведение HL и комплекса I в ацетонитриле. Свободный лиганд восстанавливается в одноэлектронную квазиобратимую стадию при потенциале $E_{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}^{{{\text{red}}}}$ = –1.16 В с образованием соответствующего анион-радикала. Отметим, что в диапазоне развертки потенциала до –2.50 В не наблюдается дополнительных редокс-переходов, свидетельствующих о формировании дианионной формы (схема 2 ).
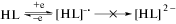
Схема 2 .
В анодной области для HL не наблюдается выраженных пиков окисления (рис. 4).
Рис. 4.
Циклические вольтамперограммы окисления (справа) и восстановления (слева) раствора HL (MeCN, c = 3 ммоль/л, 0.1 M Bu4NPF6, v = 50 мВ/с).
Для комплекса кобальта на ЦВА (рис. 5) фиксируются два катодных пика при потенциалах $E_{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}^{{{\text{red1}}}}$ = –1.27 В и $E_{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}^{{{\text{red2}}}}$ = –1.92 В (отн. Ag/Ag+), отвечающие первоначальному восстановлению до нейтральной формы и далее до анион-радикала, за счет участия в электродном процессе одного из лигандов (схема 3 ).
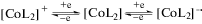
Рис. 5.
Циклические вольтамперограммы окисления (справа) и восстановления (слева) раствора комплекса I (MeCN, c = 3 ммоль/л, 0.1 M Bu4NPF6, v = = 50 мВ/с).
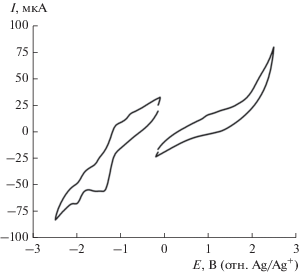
Схема 3 .
Величина потенциала первого катодного пика для комплекса I коррелирует со значением потенциала восстановления кобальтового металлохелата со сходным окружением [40]. Вид ЦВА-кривых указывает на квазиобратимость процессов восстановления (схема 3 ), протекающих на электродах. В области положительных потенциалов окислительно-восстановительные процессы слабо различимы.
Таким образом, в работе было показано, что полученное новое основание Шиффа, представляющее собой тридентантную азометиновую систему, обладает окислительными свойствами в отношении иона Co2+, поэтому кобальтовый комплекс с данного типа лигандом спин-кроссовером обладать не может. Геометрия полиэдров CoN4O2 в комплексе I соответствует искаженному октаэдру. В кристалле наблюдаются межмолекулярные водородные связи между группами N–H лиганда и атомами кислорода лиганда соседнего катиона, внешнесферных анионов ${\text{ClO}}_{4}^{ - }$ и молекул воды.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Chattopadhyay A., Meier M., Ivaninskii S. et al. // Biochemistry. 2007. V. 46. № 28. P. 8315.
Montagner C., de Souza S.M., Groposo C. et al. // Z. Naturforsch. C. 2008. V. 63. № 1–2. P. 21.
Hui M.B.V., Lien E.J., Trousdale M.D. // Antivir. Res. 1994. V. 24. № 4. P. 261.
Krause M., Rouleau A., Stark H. et al. // J. Med. Chem. 1995. V. 38. № 20. P. 4070.
Lewinski J., Zachara J., Justyniak I., Dranka M. // Coord. Chem. Rev. 2005. V. 249. № 11–12. P. 1185.
Kleij A.W. // Chem. Eur. J. 2008. V. 14. № 34. P. 10520.
Garnovskii A.D., Nivorozhkin A.L., Minkin V.I. // Coord. Chem. Rev. 1993. V. 126. № 1–2. P. 1.
Гарновский А.Д., Бурлов А.С., Васильченко И.С. и др. // Коорд. химия. 2010. Т. 36. № 2. С. 83 (Garnovskii A.D., Burlov A.S., Vasil’chenko I.S. et al. // Russ. J. Coord. Chem. 2010. V. 36. № 2. P. 81). https://doi.org/10.1134/S1070328410020016
Harada J., Uekusa H., Ohashi Y. // J. Am. Chem. Soc. 1999. V. 121. № 24. P. 5809.
Ohshima A., Momotake A., Arai T. // J. Photochem. Photobiol. A. 2004. V. 162. № 2–3. P. 473.
Ужинов Б.М., Химич М.Н. // Успехи химии. 2011. Т. 80. № 6. С. 580.
Minkin V.I., Tsukanov A.V., Dubonosov A.D., Bren V.A. // J. Mol. Struc. 2011. V. 998. № 1–3. P. 179.
Tahlan S., Ramasamy K., Lim S.M. et al. // BMC Chem. 2019. V. 13. № 12. P. 1.
Tahlan S., Ramasamy K., Lim S.M. et al. // Chem. Sent. J. 2019. V. 12. № 139. P. 1.
Wang X.J., Xi M.Y., Fu J.H. et al. // Chin. Chem. Lett. 2012. V. 23. № 6. P. 707.
Desai N.C., Shihory N.R., Kotadiya G.M., Desai P. // Eur. J. Med. Chem. 2014. V. 82. P. 480.
Camacho J., Barazarte A., Gamboa N. et al. // Bioorg. Med. Chem. 2011. V. 19. № 6. P. 2023.
Starcevic K., Kralj M., Ester K. et al. // Bioorg. Med. Chem. 2007. V. 15. № 13. P. 4419.
Monforte A.M., Ferro S. Luca L.D. et al. // Bioorg. Med. Chem. 2014. V. 22. № 4. P. 1459.
Ghosh K., Tarafdar D., Samadder A., Khuda-Bukhsh A.R. // RSC Adv. 2014. V. 4. № 102. P. 58530.
Lee D.Y., Singh N., Satyender A., Jang D.O. // Tetrahedron Lett. 2011. V. 52. № 51. P. 6919.
Тихомирова К.С., Толпыгин И.Е., Стариков А.Г., Казьмина М.А. // ХГС. 2017. V. 53. № 2. P. 179.
Kulhanek J., Bures F. // Beilstein J. Org. Chem. 2012. V. 8. P. 25.
Zhang L., Wu J., Shi L. et al. // Tetrahedron Lett. 2011. V. 52. № 30. P. 3897.
Liu J.-C., Chen L.-W., Song X.-Y. et al. // Chinese J. Struct. Chem. 2011. V. 30. № 5. P. 764.
Chang H.C., Son B.C., Song G.Y. et al. // Macromolec. Res. 2013. V. 21. № 1. P. 118.
Du J., Li L.-J., Li Y. // Inorg. Chem. Commun. 2005. V. 8. № 3. P. 246.
Lahneche Y.D., Boulebd H., Benslimane M. et al. // J. Coord. Chem. 2019. V. 72. № 18. P. 3156.
Луков В.В., Коган В.А., Левченков С.И. и др. // Коорд. химия. 2015. Т. 41. № 1. С. 3 (Lukov V.V., Kogan V.A., Levchenkov S.I. et al. // Russ. J. Coord. Chem. 2015. V. 41. № 1. P. 1). https://doi.org/10.1134/S1070328415010054
Попов Л.Д., Левченков С.И., Щербаков И.Н. и др. // Журн. общ. химии. 2010. Т. 80. № 12. С. 2040.
Pavlov A.A., Denisov G.L., Kiskin M.A. et al. // Inorg. Chem. 2017. V. 56. P. 14759.
Tupolova Y.P., Shcherbakov I.N., Popov L.D. et al. // Dalton. Trans. 2019. V. 48. № 20. P. 6960.
Tupolova Y.P., Shcherbakov I.N., Korchagin D.V. et al. // J. Phys. Chem. C. 2020. V. 124. № 47. P. 25957.
Cheeseman W.H. // J. Chem. Soc. 1964. V. 11. P. 4695.
SMART (control) and SAINT (integration) Software. Version 5.0. Madison (WI, USA): Bruker AXS Inc., 1997.
Krause L., Herbst-Irmer R., Sheldrick G.M., Stalke D. // J. Appl. Crystallogr. 2015. V. 48. P. 3.
Sheldrick G. // Acta Crystallogr. A. 2007. V. 64. P. 112.
Dolomanov O.V., Bourhis L.J., Gildea R.J. et al. // J. Appl. Crystallogr. 2009. V. 42. P. 339.
Ливер Э. Электронная спектроскопия неорганических соединений. М.: Мир, 1987. Т. 2. С. 154.
Casellato U., Fregona D., Sitran S. et al. // Inorg. Chim. Acta. 1984. V. 95. № 6. P. 309.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия