

Координационная химия, 2022, T. 48, № 11, стр. 647-668
Хелатирующие агенты для циркония-89 в синтезе радиофармацевтических препаратов: текущее состояние и перспективы развития
В. Б. Бубенщиков 1, *, А. А. Ларенков 1
1 Федеральный медицинский биофизический центр им. А.И. Бурназяна ФМБА России
Москва, Россия
* E-mail: bubenschikov2011@yandex.ru
Поступила в редакцию 03.02.2022
После доработки 11.04.2022
Принята к публикации 14.04.2022
- EDN: ERTYHD
- DOI: 10.31857/S0132344X22110020
Аннотация
Цирконий является крайне востребованным элементом и широко применяется в различных сферах науки и техники. Для циркония известно 39 изотопов, один из которых – цирконий-89 (T1/2 = 78.42 ч) привлекает сегодня все больше внимания научного сообщества в аспекте радионуклидной диагностики различных заболеваний. В настоящее время проводится активная разработка и внедрение в клиническую практику радиофармацевтических препаратов на основе 89Zr. Наиболее востребован 89Zr при разработке препаратов на основе моноклональных антител, основной областью применения которых является диагностика и мониторинг терапии онкологических заболеваний. Для введения 89Zr в структуру векторных молекул, позволяющих ему селективно накапливаться в различных очагах, необходимо использовать подходящий хелатирующий агент. При этом особенности использования комплексов циркония-89 в составе радиофармацевтических препаратов накладывает дополнительныe требования к свойствам самого хелатора: требуется высокая эффективность комплексообразования без нагревания реакционной смеси при значениях pH, близких к нейтральному. Кроме того, полученные комплексы должны быть стабильны не только in vitro, но и in vivo. Основной трудностью дизайна новых хелаторов является сложная координационная химия циркония, выраженная в образовании полиядерных форм с различной степенью полимеризации, состав которых меняется в зависимости от pH и времени, из-за чего классические методы, такие как EXAFS, рентгеноструктурный анализ и потенциометрическое титрование в водных средах являются зачастую малоинформативными. Данный факт вынуждает исследователей прибегать к ряду альтернативных методов изучения полученных соединений. В свою очередь, обобщение экспериментальных данных различных исследований имеет большое практическое значение. В обзоре обсуждаются проблемы, связанные с использованием популярного хелатора дефероксамина, последние достижения в области поиска и синтеза новых хелаторов, а также применение различных методов оценки полученных комплексов для использования в высокотехнологичных процедурах ядерной медицины.
ВВЕДЕНИЕ
Позитронно-эмиссионная компьютерная томография (ПЭТ) является одним из наиболее востребованных методов молекулярной визуализации для диагностики различных заболеваний. В последнее время проводится активная разработка и внедрение в клиническую практику визуализирующих агентов на основе моноклональных антител (mAb). Сочетание высокой селективности моноклональных антител и высокой чувствительности ПЭТ способствовало развитию отдельного направления ПЭТ – иммуноПЭТ, направленного на диагностику, планирование и мониторинг терапии онкологических заболеваний [1]. Отличительной особенностью mAb является медленная фармакокинетика препаратов на их основе, в результате чего оптимальное биораспределение обычно достигается не ранее, чем через сутки после введения. Для создания радиофармацевтических лекарственных препаратов (РФЛП) на основе mAb наиболее подходящими ПЭТ радионуклидами (РН) по совокупности ядерно-физических характеристик являются 124I (Т1/2 = 4.17 сут), 64Cu (Т1/2 = = 12.7 ч), 86Y (Т1/2 = 14.7 ч), 89Zr (Т1/2 = 78.42 ч). В данном ряду выгодно выделяется 89Zr, который имеет более подходящий период полураспада, чем 64Cu и 86Y и образует более стабильные меченные формы по сравнению с 124I in vitro и in vivo [2, 3]. 89Zr имеет практически идеальные ядерно-физические характеристики для иммуноПЭТ. Длительный период полураспада 89Zr (Т1/2 = 78.42 ч) позволяет проводить оценку биораспределения вплоть до 30 сут после введения [4], а низкая энергия испускаемых позитронов позволяет получать ПЭТ-изображения с высоким разрешением [5–7].
Сегодня 89Zr наиболее широко используется при разработке РФЛП на основе меченных клеток, моноклональных антител и их фрагментов. Последние достижения в данной области, связанные с клиническим применением циркония-89, подробно освeщены в недавних обзорах [8–12]. Для введения 89Zr в структуру векторной молекулы обычно используются бифункциональные хелаторы, способные образовывать кинетически инертный и термодинамически стабильный комплекс с РН и ковалентную связь с векторной молекулой. Однако при проведении синтеза также следует учитывать, что многие векторы (особенно mAb) не способны выдержать достаточно жесткие условия, необходимые обычно для образования комплекса (повышенная температура, рН отличный от нейтрального). Настоящий обзор посвящен разработке и оценке различных перспективных хелаторов для циркония-89 в рамках развития диагностического потенциала иммуноПЭТ.
ЯДЕРНО-ФИЗИЧЕСКИЕ ХАРАКТЕРИСТИКИ, ПОЛУЧЕНИЕ И ВЫДЕЛЕНИЕ ЦИРКОНИЯ-89
89Zr (Т1/2 = 78.42 ч) распадается путем электронного захвата (77.2%) и испускания позитронов (22.8%, Еβ+ = 902 кэВ) в метастабильный 89mY (Т1/2 = = 15.84 c), который распадается в стабильный 89Y (ИП, Еγ = 909 кэВ, Gγ = 99.0%) (схема 1 ) [13].
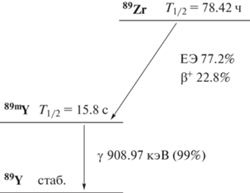
Схема 1 .
В настоящее время существует несколько способов получения 89Zr, из которых наиболее популярным является протонное облучение иттриевой мишени 89Y(p, n) 89Zr. Основным преимуществом данного метода является возможность получения 89Zr на низкоэнергетических циклотронах облучением мишени из моноизотопного природного иттрия [14–16]. Для выделения 89Zr из облученной мишени были предложены различные методы, среди которых наиболее популярным стал метод твердофазной экстракции на гидроксаматно-модифицированных смолах [15, 17, 18]. Данный метод позволяет получать 89Zr в форме [89Zr]Zr-оксалата с высоким выходом и высокой объемной активностью [18, 19]. Альтернативным вариантом является получение 89Zr в форме [89Zr]Zr-хлорида [20, 21]. Данный способ является менее популярным, поскольку [89Zr]Zr-хлорид не стабилен при нейтральных значениях pH и используется в основном при проведении научных исследований, а для синтеза 89Zr-mAb РФЛП применяют [89Zr]Zr-оксалат [22].
КООРДИНАЦИОННАЯ ХИМИЯ Zr
Как элемент цирконий имеет сложную химию в водных растворах, связанную с многообразием возможных химических форм. Zr существует в степенях окисления +2, +3, +4 и образует комплексы с различной геометрией и координационными числами (КЧ) от 4 до 12 [23, 24]. При этом наиболее предпочтительной является степень окисления +4 и КЧ 8. Часть знаний о химии циркония была получена из химии гафния, поскольку данные элементы образуют соединения со схожими химическими свойствами. Подобие образуемых соединений связанно со сжатием атомных (Hf = = 1.44 Å и Zr = 1.45 Å) и ионных радиусов (Hf = = 0.85 Å и Zr = 0.86 Å) при переходе от La к Lu и понижения радиуса Hf до значения Zr [25]. Катион Zr4+ благодаря высокой плотности заряда и маленькому ионному радиусу является жесткой кислотой Льюиса, поэтому при комплексообразовании предпочтительными являются группы, содержащие жесткие основания Льюиса (т.е. содержащие атомы азота и кислорода).
Первоначально для хелатирования Zr4+ были использованы такие известные хелаторы как этилендиаминтетрауксусная кислота (EDTA) и диэтилентриаминпентауксусная кислота (DTPA) (cхема 2). Исследования кристаллических структур Zr-EDTA и Zr-DTPA показали, что в обоих случаях Zr4+ образует восьмикоординированные комплексы, а в формировании комплекса Zr-EDTA дополнительно участвуют две молекулы воды [26, 27]. Образование полиядерных комплексов в данном случае авторами не рассматривалось. Несмотря на достаточно высокие константы термодинамической стабильности (Zr(EDTA) lgβ = 27.9 ± 0.1; Zr(DTPA) lgβ = 35.3 ± 0.3 [28]) на практике комплекс Zr(DTPA) оказался недостаточно стабильным. При инкубации в плазме крови в течении 24 ч комплекс 88Zr c DTPA частично диссоциировал (сохранялось 80% комплекса), тогда как комплекс с дефероксамином (DFO) оставался стабильным (сохранялось 99.8% комплекса) [29]. Дефероксамин был впервые выделен в 1960 году из Streptomyces pilosus [30]. Гексадентатный хелатор DFO является бифункциональным сидерофором, содержащим три последовательно связанные гидроксаматные группы (схема 2 ). Учитывая склонность Zr4+ к взаимодействию с жeсткими основаниями Льюиса и его способность образовывать комплексы с моногидроксаматами авторы [29] в 1992 году показали, что DFO образует стабильные комплексы с 89Zr и может использоваться в качестве хелатора при синтезе радиоиммуноконъюгатов. Структуры хелаторов DTPA, EDTA, DFO представлены на схеме 2 .

Схема 2 .
Согласно квантовохимическим расчетам, высокая стабильность комплекса [89Zr]Zr-DFO обусловлена хелатированием 89Zr4+ тремя гидроксаматными группами молекулы DFO. Образование комплекса происходит тремя нейтральными и тремя отрицательно заряженными атомами кислорода, которые совместно с двумя молекулами воды координируют ион циркония (рис. 1) [31]. В дальнейших теоретических исследованиях было показано, что комплексообразование Zr с DFO в водном растворе сопровождается образованием комплексов [Zr(DFO)(H2O)n]+ (n = 0–2), среди которых наиболее стабильным является комплекс [ZrDFO(H2O)]+ с КЧ 7 (lgβ = 41.51) [32]. Одновременно с этим образование комплекса [ZrDFO(H2O)]+ было подтверждено экспериментально [33]. Впоследствии было проведено еще одно исследование (квантовохимические расчеты, аппроксимация результатов EXAFS) в результате которого авторы пришли к выводу, что координационная сфера комплекса Zr-DFO, наиболее вероятно, завершается двумя ионами гидроксила, а не двумя молекулами воды [34]. Таким образом, несмотря на активные исследования, в настоящее время точная структура комплекса Zr-DFO остается невыясненной.
Рис. 1.
Упрощенная структурная формула комплекса [ZrDFO(H2O)2]+ (а), структура комплекса [ZrDFO(H2O)2]+, вычисленная с помощью теории функционала плотности (б). Построено на основании данных [32].

Определение точной структуры и констант устойчивости комплексов Zr в водных растворах значительно осложнено образованием малорастворимых полиядерных соединений с различной степенью полимеризации, состав которых меняется в зависимости от pH и времени [35]. Затрудняет точную интерпретацию результатов и определение адекватных структурных характеристик комплексов циркония невозможность экстраполяции данных, полученных с использованием циркония в весовых количествах, на системы, где цирконий присутствует в качестве микрокомпонента (89Zr4+ без носителя). Недавно различными группами ученых были опубликованы результаты исследований по определению термодинамических констант стабильности для комплекса Zr-DFO [36–38]. Наиболее высокое значение константы (lg β = 49.1) было получено для комплекса [Zr(HDFO)]2+, который, согласно полученным результатам, является преобладающей формой при проведении реакции радиомечения (СDFO = 1 мкМ, pH 7) [38]. В [36] сообщается о значении константы (lg β = = 46.4–47.7 и 40.4 для [Zr(HDFO)]2+ и Zr(DFO)]+ соответственно), при этом депротонированная форма становится преобладающей при значении pH ~ 6.4 (СZr(IV) = 1 мМ, СDFO = 1 мМ). Наиболее низкие значения константы комплекса [Zr(DFO)]+ (lg β = 36.14) были получены в [37]. При этом авторы отмечают, что стехиометрия комплексов [Zr(DFO)]+ значительно меняется в зависимости от pH и преобладающими формами в растворе при концентрациях циркония и DFO, равных 1 мМ, являются биядерные комплексы с мольным соотношением 2 : 2 и 2 : 3 (Zr : DFO). Присутствие таких форм в системах с пикомолярным содержанием циркония (как в случае растворов радионуклида циркония-89 без носителя) крайне маловероятно.
Необходимо отметить, что при выборе хелатора для РФЛП кинетическая стабильность является более важной, чем термодинамическая стабильность комплекса металл–хелат. Термодинамические константы стабильности могут быть полезны при предварительном сравнении различных хелатирующих агентов, но не отражают устойчивость комплекса in vivo, тогда как стабильность комплекса in vivo является главным критерием для применения полученных комплексов в области ядерной медицины [39]. При введении РФЛП в организм целевой комплекс подвергается ряду негативных воздействий. Присутствующие в организме ионы металлов имеют значительно большую концентрацию и могут вытеснять цирконий как микрокомпонент из его комплекса. Еще больше усложняет ситуацию присутствие в организме сильных нативных металлсвязывающих транспортных белков, способных перелигандировать радионуклид [39]. Например, было показано, что Zr связывается с трансферином, фракцией альбумина и α-, β- и γ-глобулином [40, 41]. Указанные факторы, с одной стороны, вынуждают исследователей к применению разнообразных косвенных методов определения свойств комплексов циркония-89, но в то же время дают более практически значимую оценку приемлемости данных соединений непосредственно in vitro и in vivo.
В настоящее время дефероксамин, благодаря своей способности образовывать прочные комплексы со многими металлами (включая Zr), а также коммерческой доступности, является “золотым стандартом” в области синтеза [89Zr]Zr-mAb радиофармпрепаратов. Для присоединения DFO к mAb были предложены различные методы, главным недостатком которых остается сложный многоступенчатый синтез. Эти методы подробно освещены в ряде обзоров [8, 42, 43]. Авторы [44, 45] предложили более простой одностадийный метод конъюгации p-Bn-NSC-DFO с аминогруппой лизина немодифицированного антитела (рис. 2). Реакция конъюгации основана на образовании тиомочевинной связи и проводится в мягких условиях (37°C, 30 мин). В качестве буфера используется, как правило, карбонат натрия с pH 9.
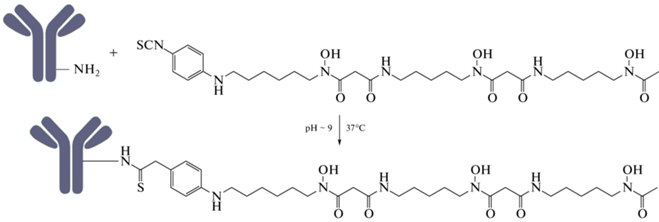
Однако гексадентатный DFO не является оптимальным хелатором для Zr, поскольку Zr предпочтительно образует комплексы с КЧ 8. Данное несоответствие особенно ярко проявляется при изучении биораспределения препаратов на основе [89Zr]Zr-DFO. В ряде работ отмечено высокое накопление 89Zr (~10% от введенной дозы на грамм (%ID/г) в скелете мышей после внутривенного введения (в/в) 89Zr-DFO-mAb [31, 46–49]. Наиболее правдоподобным объяснением данного факта является высвобождение циркония-89 из структуры комплекса с DFO. Высокая остеотропность иона циркония-89 приводит к его накоплению в костной ткани, что искажает реальное биораспределение препарата и может приводить к ложноположительным или ложноотрицательным результатам, а также к недооценке дозы облучения, поглощенной костным мозгом [50, 51]. Данные результаты стали отправной точкой для разработки более совершенных, главным образом октадентатных хелаторов для циркония-89.
ЛИНЕЙНЫЕ ГИДРОКСАМАТЫ
Авторы [52] модифицировали хелатор DFO добавлением дополнительной гидроксаматной группы и получили хелаторы DFO* и DFOcyclo*, структуры которых представлены на схеме 3 .
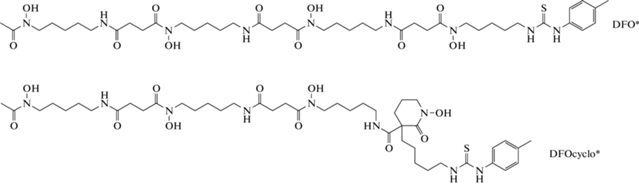
Схема 3 .
Полученный октадентатный хелатор DFO* был конъюгирован с янтарным ангидридом для дальнейшего использования в качестве бифункционального хелатирующего агента (БХА). Далее полученный хелатор был конъюгирован с бомбезином и помечен 89Zr. Для оценки устойчивости [89Zr]Zr-DFO*-бомбезин инкубировали в избытке DFO (образец сравнения [89Zr]Zr-DFO-бомбезин инкубировали в избытке DFO*). Препарат на основе DFO* был более стабилен к перелигандированию по сравнению с препаратом на основе исходного DFO. Впоследствии данная работа была продолжена авторами [53]. DFO* был конъюгирован с 1,4‑фенилендиизотиоцианатом, a далее с mAb (трастузумабом, ритуксимабом, цетуксимабом). Интересной особенностью данного исследования является предварительное мечение p-Bn-NCS-DFO* и p-Bn-NCS-DFO (prelabeling) 89Zr. В данном случае последующая реакция конъюгации комплекса с mAb была возможна только для [89Zr]Zr-p-Bn-DFO*. Авторы связывают данный факт с участием изотиоцианатного линкера в формировании комплекса [89Zr]Zr-DFO, в результате чего он становится недоступным для последующей реакции конъюгации с mAb. Оценка стабильности препаратов [89Zr]Zr-DFO-трастузумаб (I) и [89Zr]Zr-DFO*-трастузумаб (II) была выполнена in vitro в сыворотке крови, изотоническом растворе NaCl и растворе, содержащем 20 мМ гистидина и 240 мМ сахарозы. По результатам экспериментов препарат I показал более высокую иммунореактивность и стабильность во всех трех средах. Данные по стабильности в сыворотке крови представлены в табл. 1. Сравнительное исследование in vivo показало схожее биораспределение (через 24 ч после введения). При дальнейшем наблюдении (72 ч, 144 ч) авторами было отмечено постепенное уменьшение накопления в скелете для препарата I и увеличение для II. В целом препарат I показал более низкое неспецифическое накопление, главным образом в костной ткани (табл. 1).
Таблица 1.
Результаты in vitro и in vivo исследований для линейных гидроксаматов
| Хелатор | Вектор | Стабильность in vitro | Стабильность in vivo | Литература | ||
|---|---|---|---|---|---|---|
| метод | % интактного комплекса | накопление в кости, % от введенной дозы на грамм (%ID/г) | временная точка | |||
| DFO | Tрастузумаб | Сыворотка крови (7 сут) |
72.0 | 3.9 ± 0.8 | 144 ч | [53] |
| DFO* | 96.3 | 0.8 ± 0.1 | ||||
| DFO | T-DM1 | 9.6 ± 0.4 | 96 ч | [54] | ||
| DFO* | 6.6 ± 0.6 | |||||
| DFO | Tрастузумаб | 1000-кратный избыток EDTA (7 cут) |
~50% | 4.5 ± 0.6 | 168 ч | [55] |
| DFO* | >98% | 2.0 ± 0.3 | ||||
| DFOcyclo* | >98% | 1.5 ± 0.3 | ||||
| DFO | Tрастузумаб | 7.1 ± 0.8 | 96 ч | [56] | ||
| Orn3-hx | 10.0 ± 1.5 | |||||
| Orn4-hx | 7.0 ± 2.2 | |||||
| DFO | Трастузумаб | Плазма крови (3 сут) |
55 | 7.6 ± 0.4 | 72 ч | [57] |
| C1 | 3 | 19.5 ± 3.6 | ||||
| C2 | 16 | 18.3 ± 2.9 | ||||
| DFO* | Tрастузумаб | 375-кратный
избыток EDTA (1 сут) |
>90 | 0.8 ± 0.3 | 144 ч | [58] |
| DFOSq | 82 | 8.2 ± 0.8 | ||||
| DFO | 68 | 4.6 ± 2.3 | ||||
| DFO*Sq | >90 | 1.2 ± 0.4 | ||||
| DFO | HPG | 97 | 3.3 ± 0.4 | |||
| DFO* | HPG | >99 | 3.1 ± 0.7 | |||
Аналогичное исследование было выполнено в [54]. Авторы конъюгировали DFO и DFO* с человеческим иммуноглобулином-G (hIgG) и трастузумаб-эмтанзином (T-DM1), который согласно предыдущим исследованиям позволяет проводить более точную визуализацию HER2+ рака молочной железы, чем обычный трастузумаб [59]. По результатам оценки стабильности in vitro [89Zr]Zr-DFO*-hIgG является стабильным в плазме крови и 0.1 мМ растворе EDTA в течении 5 сут. Инкубирование [89Zr]Zr-DFO*-hIgG в избытке DFO приводит к небольшому высвобождению 89Zr (11.0 ± 1.9% через 1 сут и 26.0 ± 2.1% через 5 сут). Сравнительное исследование in vivo (мыши линии NOD/SCID с ксенотрансплантами рака яичников человека SKOV-3) показало более низкое неспецифическое накопление [89Zr]Zr-DFO*-T-DM1 во всех тканях (табл. 1). Необходимо отметить, что при аналогичном исследовании на здоровых мышах линии BALB/c происходило значительно более высокое неспецифическое накопление во всех тканях и органах: накопление [89Zr]Zr-DFO*-T-DM1 в крови через 96 ч после введения – 8.4 ± 0.7% ID/г, тогда как для мышей линии NOD/ SCID – 0.1 ± 0.0% ID/г. По мнению авторов, данное различие связано с более низкой концентрацией IgG у мышей линии NOD/SCID, что приводит к более высокому клиренсу mAb из крови. В целом полученные данные хорошо согласуются с ранее опубликованными результатами [52, 53].
Еще одно сравнительное исследование с DFO* было выполнено в [55]. Помимо сравнения DFO c DFO* авторами был проведен синтез нового октадентатного хелатора DFOcyclo*-pPhe-NCS (cхема 3). Условия синтеза комплексов 89Zr для всех рассматриваемых хелаторов, а также значения коэффициента распределения в системе окатонол-1–вода представлены в табл. 2. Оценка комплексов 89Zr с DFO, DFO*, DFOcyclo* была выполнена в плазме крови, 1000-кратном избытке EDTA и DFO. Авторы отмечают высокую стабильность всех исследуемых комплексов в плазме крови в течение 7 сут (>98%). Наиболее высокую стабильность в избытке EDTA показали комплексы [89Zr]Zr-DFOcyclo* и [89Zr]Zr-DFO* (>98%), в то время как после 7 сут инкубирования только 53% [89Zr]Zr-DFO осталось в неизменном виде. Аналогичные результаты в присутствии избытка EDTA были получены для препаратов [89Zr]Zr‑DFOcyclo*-трастузумаб (III), [89Zr]Zr-DFO*-трастузумаб (IV), [89Zr]Zr-DFO-трастузумаб (V). В присутствии избытка DFO препарат III имеет значительно более высокую стабильность (50% интактного комплекса через 48 ч) по сравнению с IV (50% через 4 ч) и V (<50% через 1 ч). Сравнительное исследование in vivo показало примерно одинаковую фармакокинетику для препаратов III–V, а также более низкое накопление в костной ткани для препаратов на основе DFO* и DFOcyclo* по сравнению с DFO (табл. 1). Важно отметить, что рассматриваемый хелатор DFO* имеет более высокую лиофильность (lg P = −0.44 [60]), и как следствие при синтезе РФЛП 89Zr на основе mAb возникает необходимость использования более высокой концентрации ДМСО, что, в свою очередь, может привести к агрегации белков [53].
Таблица 2.
Условия мечения различных хелаторов и значение lg P для полученных комплексов1
| Хелатор | Условия мечения | Выход реакции комплексообразования, % | lg P 89Zr-L |
Литература |
|---|---|---|---|---|
| DFO* | RT, 15 мин | 100 | −3.5 | [55] |
| DFOcyclo* | RT, 15 мин | 100 | −2.1 | [55] |
| Orn3-hx | RT, 20 мин | 100 | [56] | |
| Orn4-hx | RT, 20 мин | 100 | [56] | |
| oxoDFO* | RT, 120 мин | 100 | −1.5 | [60] |
| 4HMS | RT, 7 мин | 100 | [61] | |
| DFO | RT, 90 мин | 100 | −3.0 | [62] |
| TAFC | RT, 90 мин | 100 | −2.0 | [62] |
| FSC(succ-RGD)2AA | RT, 90 мин | 100 | −3.3 | [63] |
| FSC(succ-RGD)3 | RT, 90 мин | 100 | −3.5 | [63] |
| L1 | RT, 5 мин | 100 | −1.4 | [64] |
| L2 | RT, 5 мин | 100 | −2.3 | [64] |
| L3 | RT, 5 мин | 100 | −2.1 | [64] |
| L4 | RT, 5 мин | 100 | −3.4 | [64] |
| CTH36 | RT, 60 мин | 100 | [65] | |
| DFO-HOPO | RT, 60 мин | 100 | −0.9 | [66] |
| BPDET-LysH22,2-3-HOPO | RT, 15 мин | 100 | −1.5 | [67] |
| THPN | RT, 10 мин | 100 | −3.1 | [68] |
| IAM-1 | 95°C, 120 мин | 100 | −3.0 | [69] |
| IAM-2 | 50°C, 60 мин | 100 | −1.5 | [69] |
| TAM-1 | RT, 15 мин | 100 | −3.4 | [70] |
| TAM-2 | RT, 15 мин | 100 | −3.4 | [70] |
| DOTA | 99°C, 120 мин (90°C, 45 мин*) | 65 ± 9.6 (100*) | −3.8 | [71] |
| DOTP | 99°C, 120 мин (90°C, 45 мин*) | 70 ± 10.6 (100*) | −3.9 | [71] |
| DOTAM | 99°C, 120 мин (90°C, 45 мин*) | 9 ± 1.3 (100*) | −1.4 | [71] |
| PTCA | 37°C, 60 мин | 100 | −3.1 | [72] |
| NOTA | 37°C, 60 мин | 100 | −2.5 | [72] |
| TRITA | 99°C, 120 мин | 100 | −3.1 | [72] |
Для решения данной проблемы авторы [73] модифицировали молекулу DFO посредством введения в структуру молекулы эфирных мостиков. В результате биоорганического синтеза авторами был получен ряд аналогов DFO, содержащих один (DFO-O1), два (DFO-O2) или три (DFO-O3) эфирных атома кислорода (схема 4). Измерения коэффициента распределения в системе н-октанол–вода (lg P) показали, что DFO-O3 является примерно в 45 раз более гидрофильным, чем исходный DFO. Впоследствии авторы синтезировали и провели сравнительное исследование нескольких октадентатных хелаторов, аналогичных DFO [74]. Среди исследуемых хелаторов (схема 4 ) наиболее нестабильным оказался комплекс Zr(IV) с хелатором, содержащим в своей структуре эфирный мостик (ряд хелаторов по уменьшению стабильности: DFOB-PBH ≈ ≈ DFOB-PPH > DFOB-PPHNOCO $ \gg $ DFO), в то время как введение дополнительной метиленовой группы в углеводородный скелет не повлияло на стабильность, но привело к увеличению скорости реакции комплексообразования.

Схема 4 .
Аналогичный подход использовали в [60]. Методом твердофазного синтеза было получено окдадентатное, растворимое в воде, производное DFO*, содержащее четыре эфирных мостика – oxoDFO* (cхема 4, lgP = −1.5 ± 0.2). Оценка стабильности комплексов 89Zr с DFO, DFO* и oxoDFO* была выполнена в 5–50 мМ избытке DTPA (pH 6). В данных условиях комплексы [89Zr]Zr-oxoDFO* ≥ ≥ [89Zr]Zr-DFO* были более стабильны, чем [89Zr]Zr-DFO [75]. При проведении аналогичного исследования с 68Ga количественный выход был достигнут только при нагревании, при этом полученные комплексы были менее стабильны в присутствии 5 мМ DTPA ([68Ga]Ga-DFO ≈ [68Ga]Ga-DFO* > [68Ga]Ga-oxoDFO*), чем аналогичные комплексы с 89Zr.
В альтернативном исследовании авторы [56] синтезировали разветвленные хелаторы Orn4-hx и Orn3-hx (схема 5 ), которые являются аналогом циклического хелатора десферрихрома (desferrichrome, DFC). Оценка стабильности была выполнена в 1000-кратном избытке EDTA. Стабильность исследуемых комплексов убывает в следующем ряду: [89Zr]Zr-Orn4-hx ~ [89Zr]Zr-DFO > [89Zr]Zr-DFC > [89Zr]Zr-Orn3-hx, что хорошо согласуются с квантовохимическими расчетами. Впоследствии исследуемые хелаторы были конъюгированы с 1,4‑фенилендиизотиоцианатом, a далее – с трастузумабом и помечены 89Zr. Сравнительное исследование in vivo на здоровых мышах показало одинаковое накопление в костной ткани через 96 ч после введения для препаратов [89Zr]Zr-DFO-трастузумаба и [89Zr]Zr-Orn4-hx-трастузумаба, а также более высокое накопление для [89Zr]Zr-Orn3-hx-трастузумаба (табл. 1). Таким образом, можно отметить, что введение дополнительной четвертой гидроксаматной группы в структуру хелатора (Orn3-hx → Orn4-hx) является необходимым для образования стабильных комплексов. Тем не менее комплекс 89Zr с Orn4-hx по стабильности in vitro и in vivo был идентичен комплексу с DFO.

Схема 5 .
Еще одним исследованием по синтезу перспективных ациклических хелаторов является работа [57]. Авторы синтезировали два разветвленных ациклических бифункциональных хелатора С1 и С2 на основе тетрагидроксамата (схема 5). Полученные хелаторы, а также p-Bn-NCS-DFO были конъюгированы с трастузумабом и помечены 89Zr. Оценка стабильности полученных препаратов была выполнена в плазме крови мышей (инкубирование в течение 3 сут при 37°C). Препараты на основе С1 и С2 оказались менее стабильны по сравнению с [89Zr]Zr-DFO-трастузумабом (табл. 1). Сравнительное исследование in vivo показало постепенное увеличение накопления 89Zr в костной ткани для всех трех препаратов. Через 3 сут после введения накопление в костных тканях для препаратов [89Zr]Zr-С1-трастузумаб и [89Zr]Zr-С2-трастузумаб было значительно выше, чем для [89Zr]Zr-DFO-трастузумаба (табл. 1). Неудовлетворительную стабильность препаратов на основе С1 и С2 авторы объясняют стерическими ограничениями, возникающими за счет недостаточной длины углеводородных цепей в скелете хелатора.
Более удачные результаты были получены с ациклическим хелатором 4HMS (схема 5 ) [61]. Образование комплекса с цирконием в соотношении 1 : 1 было подтверждено с помощью масс-спектрометрии. Оценка стабильности комплекса была выполнена в избытке DTPA (100- и 1000-кратный избыток, pH 5; 7; 8.5), в плазме крови и в присутствии избытка солей металлов (10-кратный избыток Fe3+, Co2+, Cu2+, Ni2+, Mg2+, Ca2+). Во всех используемых тест-системах комплекс [89Zr]Zr‑4HMS оставался стабильным (сохраняется >97% комплекса через 7 сут) и подвергался перелигандированию только при 1000-кратном избытке DTPA (сохраняется 91.9 ± 0.1% комплекса после 7 сут инкубирования при 37°C) и 100-кратном избытке Fe3+ (сохраняется 62.3 ± 0.3% комплекса спустя 7 сут инкубирования при 37°C). При аналогичных условиях комплекс [89Zr]Zr-DFO подвергался значительному перелигандированию (сохраняется 64.1 ± 0.8 и 33.8 ± 1.6% комплекса соответственно). Сравнительное исследование in vivo показало более быстрое выведение для комплекса [89Zr]Zr-4HMS и низкое фоновое накопление во всех органах и тканях, включая скелет (0.01 ± 0.0%ID/г через 24 ч после введения) по сравнению с [89Zr]Zr-DFO (накопление в скелете через 24 ч после введения – 0.17 ± 0.13%ID/г). В целом представленные результаты свидетельствуют о высокой перспективности хелатора 4HMS, однако для дальнейшей оценки необходимы дополнительные эксперименты по синтезу бифункционального производного 4HMS и оценки его долгосрочной стабильности in vivo.
Еще одним подходом к модификации молекулы DFO является работа [76]. В данной работе были синтезированы производные эфира DFO-скварамида – DFOSqOEt (схема 6 ). Основным преимуществом данного хелатора является несложная процедура синтеза и более высокая растворимость в воде по сравнению с другими модификациями DFO. Оценка стабильности DFOSqOEt in vivo была выполнена без предварительных in vitro исследований. Было проведено два исследования, в ходе которых p-Bn-NCS-DFO и DFOSqOEt были конъюгированы с трастузумабом и chDAB4 (APOMAB®) [76, 77]. В обоих случаях препараты, синтезированные на основе DFOSqOEt показали более низкое накопление в печени и скелете, а также более высокие коэффициенты дифференциального накопления (КДН) опухоль/фон, опухоль/костная ткань, опухоль/печень по сравнению с p-Bn-NCS-DFO.

Схема 6 .
Позднее в работе [58] было проведено детальное сравнение наиболее перспективных модификаций DFO (DFO*-NCS, DFOSq). Дополнительно авторы синтезировали хелатор DFO*Sq как производное двух различных методик. Для проведения сравнительных исследований представленные хелаторы конъюгировали с трастузумабом. Оценка стабильности полученных препаратов ([89Zr]Zr-DFO*-NCS-трастузумаб (VI), [89Zr]Zr-DFOSq-трастузумаб (VII), [89Zr]Zr‑DFO‑NCS‑ трастузумаб (VIII), [89Zr]Zr-DFO*Sq-трастузумаб (IX)) in vitro была выполнена в сыворотке крови, в 375-кратном избытке EDTA, DFO и DFO*. Дополнительно была проведена оценка стабильности комплексов [89Zr]Zr-DFO* и [89Zr]Zr-DFO в присутствии потенциально конкурирующих металлов (10-кратный избыток Co2+, Zn2+, Cu2+, Mg2+, Ga3+, Gd3+, Al3+, Fe3+, Nb3+).
При инкубировании в сыворотке крови (1 : 1) в течение 7 сут при 37°С не произошло значительного снижения радиохимической чистоты (РХЧ) ни для одного из препаратов (табл. 3). В избытке EDTA комплексы на основе DFO* были более стабильны, чем аналоги на основе DFO (табл. 1). При инкубировании в присутствии избытка DFO происходило резкое падение РХЧ (сохранялось менее 10% комплекса) в течение 4 ч для препаратов на основе DFO (VII и VIII), в то время как препараты VI и IX оставались относительно стабильными в течение 24 ч (>70%). Дополнительно авторы отмечают высокую стабильность (>97%) комплексов [89Zr]Zr-DFO* и [89Zr]Zr-DFO в присутствии Co2+, Zn2+, Cu2+, Mg2+, Ga2+, Gd3+, Al3+, небольшое замещение в присутствии Fe3+ (до 93 ± 1% и 89 ± 2% для [89Zr]Zr-DFO* и [89Zr]Zr-DFO соответственно) и резкое падание в присутствии Nb3+ (сохраняется <10% через 1 ч инкубирования для обоих комплексов).
Таблица 3.
Стабильность и иммунореактивность препаратов VI–IX в сыворотке крови в течение 7 сут [58]
| Препарат | Стабильность, % | Иммунореактивность, % |
|---|---|---|
| [89Zr]Zr-DFO*-NCS-трастузумаб (VI) | 94 ± 0 | 90 ± 1 |
| [89Zr]Zr-DFOSq-трастузумаб (VII) | 87 ± 1 | 80 ± 1 |
| [89Zr]Zr‑DFO‑NCS‑трастузумаб (VIII) | 81 ± 1 | 74 ± 1 |
| [89Zr]Zr-DFO*Sq-трастузумаб (IX) | 100 ± 0 | 96 ± 1 |
Для оценки перспективности исследуемых хелаторов in vivo был выполнен ряд сравнительных исследований с препаратами трастузумаба (VI–IX) на мышах с опухолью, экспрессирующей HER2+ антигены, препаратами [89Zr]Zr-DFO*-NCS-цетуксимаб и [89Zr]Zr-DFO-NCS-цетуксимаб на мышах с опухолью A431 и препаратами [89Zr]Zr-DFO*-NCS-трастузумаб, [89Zr]Zr-DFO-NCS-трастузумаб, [89Zr]Zr‑DFO*‑NCS‑B12, [89Zr]Zr-DFO-NCS-B12 (препараты сравнения с антителом против ВИЧ) на мышах с моделью костяных метастазов [58]. В целом все полученные результаты подтверждают более высокую стабильность препаратов 89Zr на основе комплексов с DFO*. Во всех трех исследованиях препараты на основе DFO* (VI и IX) имели более низкое поглощение в скелете по сравнению с препаратами VII и VIII. При этом, несмотря на более высокую стабильность in vitro, препараты на основе DFOSq (VII и IX) имели биораспределение, аналогичное препаратам на основе DFO (VI и VIII). Данные для препаратов VI–IX представлены в табл. 1. Важно отметить, что, несмотря на низкую растворимость p-Bn-NCS-DFO* и практически аналогичный профиль биораспределения препаратов на основе DFO* и DFO*Sq, по мнению авторов, DFO*-NCS является более перспективным хелатором за счет более высокой скорости реакции конъюгации и коммерческой доступности.
ЦИКЛИЧЕСКИЕ ГИДРОКСАМАТЫ
Были исследованы процессы комплексообразования и устойчивость комплексов 89Zr с различными гидрoксаматно-модифицированными циклами L [64, 78], CTH36 [65], фуразином (FSC) и его триацетилированным аналогом (TAFC) [62].
FSC является макроциклическим хелатором, содержащим три гидроксаматные группы и три группы первичных аминов, которые могут быть использованы для биоконъюгации. TAFC является триацетилированным аналогом FSC (cхема 7) [62]. В ходе исследования авторами был синтезирован ряд соединений на основе пептида RGD с использованием различных линкеров: FSC(RGDfE)3, FSC(succ-RGD)3, FSC(Mal-RGD). Сравнительное исследование [89Zr]Zr-TAFC, [89Zr]Zr-FSC(succ-RGD)3, и [89Zr]Zr-DFO in vitro (инкубирование в избытке EDTA в течение 7 сут) показало более высокую стабильность для [89Zr]Zr-TAFC (97.2 ± 0.2% интактного комплекса) и [89Zr]Zr-FSC(succ-RGD)3 (93.9 ± 0.7%) по сравнению с [89Zr]Zr-DFO (42.2 ± 2.3%). При инкубировании [89Zr]Zr-DFO в избытке TAFC авторы отмечают количественное перелигандирование уже через 1 ч после смешивания, тогда как в обратном случае (инкубирование [89Zr]Zr-TAFC в избытке DFO) комплекс [89Zr]Zr-TAFC оставался относительно стабильным (сохранялось 74.2 и 39.8% комплекса спустя 1 и 7 сут соответственно). Исследование [89Zr]Zr-FSC(succ-RGD)3in vivo показало высокий клиренс из крови, низкое поглощение в скелете и преобладающую почечную экскрецию спустя 6 ч после инъекции. Схожие результаты in vivo были получены и для комплекса [89Zr]Zr-TAFC, однако необходимо отметить, что сравнительное in vivo исследование с комплексами [89Zr]Zr-DFO или [89Zr]Zr-DFO-RGD не проводилось.

Схема 7 .
Впоследствии авторы конъюгировали FSC с янтарным и уксусным ангидридом для добавления дополнительных координационных групп [63]. В результаты были получены хелаторы FSC(succ)2, FSC(succ)2AA и FSC(succ)3 содержащие две или три дополнительные сукцинатные группы (схема 7 ). Согласно результатам in vitro исследований, все исследуемые комплексы с 89Zr были более стабильны, чем аналогичный комплекс с DFO. По уменьшению стабильности комплексы можно расположить в следующем ряду: [89Zr]Zr-FSC(succ)3 > > [89Zr]Zr-TAFC > [89Zr]Zr-FSC(succ)2AA $ \gg $ ⪢ [89Zr]Zr-DFO. Сравнительное in vivo исследование комплексов [89Zr]Zr-FSC(succ)3 и [89Zr]Zr-TAFC показало более медленный клиренс [89Zr]Zr-FSC(succ)3 из крови и более высокое неспецифическое накопление во всех основных органах за исключением печени, кишечника и селезенки, в которых из-за гепатобилиарной экскреции отмечается более высокое накопление для [89Zr]Zr-TAFC.
Непосредственное сравнение FSC и DFO было выполнено в работе [79]. В качестве вектора был использован фрагмент антитела, специфичный к рецепторам эпидермального фактора роста (ZEGFR:2377). Оценка стабильности препаратов [89Zr]Zr-FSC-ZEGFR:2377 (X) и [89Zr]Zr-DFO-ZEGFR:2377 (XI) была выполнена в PBS и в 1000-кратном избытке EDTA. Препарат на основе FSC был более стабилен в обеих средах по сравнению с аналогичным препаратом на основе DFO (данные по стабильности в EDTA представлены в табл. 4, что хорошо согласуется с ранее описанными результатами по сравнению комплексов [89Zr]Zr-FSC и [89Zr]Zr-DFO [63]).
Таблица 4.
Результаты in vitro и in vivo исследований для циклических гидроксаматов и хелаторов на основе гидроксипиридона
| Хелатор | Вектор | Стабильность in vitro | Стабильность in vivo | Литература | ||
|---|---|---|---|---|---|---|
| метод | % интактного комплекса | накопление в кости, % от введенной дозы на грамм (%ID/г) | временная точка | |||
| BF4 | Трастузумаб | 18.9 ± 1.1 | 96 ч | [64] | ||
| DFO | 2.9 ± 1.0 | |||||
| BPDET-LysH22,2-3-HOPO | Трастузумаб | Сыворотка крови (4 сут) | ~40 | 15.1 ± 2.7 | 144 ч | [67] |
| DFO | 10.6 ± 1.0 | |||||
| FSC | ZEGFR:2377 | 1000-кратный избыткок EDTA (1сут) | 85.7 ± 0.4 | 0.1 ± 0.3 | 24 ч | [79] |
| DFO | 67.7 ± 3.6 | 1.0 ± 0.3 | ||||
| p-Bn-NCS-HOPO | Трастузумаб | Сыворотка крови (7 сут) |
89.2 ± 0.9 | 2.4 ± 0.3 | 366 ч | [80] |
| DFO | 94.7 ± 0.7 | 17.0 ± 4.1 | ||||
| 3,2-HOPO | MSLN-mAb | Сыворотка крови (4 сут) |
23 | 15.40 ± 2.40 | 144 ч | [81] |
| DFO | MSLN-mAb | 46 | 6.51 ± 1.82 | |||
| YM103 | Трастузумаб | Сыворотка крови (7 сут) |
>95 | 25.9 ± 0.6 | 168 ч | [82] |
| DFO | >95 | 6.5 ± 0.4 | ||||
| THPN | HPG | Плазма крови (5 сут) |
96 ± 1 | 8.4 ± 2.2 | 144 ч | [83] |
| DFO | 97 ± 0 | 3.3 ± 0.4 | ||||
| DFO* | >99 | 3.1 ± 0.7 | ||||
Дополнительно авторы сообщают, что проведение реакции мечения при 85°C приводит к небольшому увеличению стабильности обоих комплексов как in vitro, так и in vivo. Сравнительное исследование in vivo показало более низкий клиренс из крови, эквивалентное накопление в скелете (табл. 4) и более высокое накопление во всех основных органах и в опухоли для препарата X (через 24 ч после введения КДН опухоль/кровь – 17.5 ± 5.6; опухоль/мышца – 21.9 ± 2.2), по сравнению с XI (КДН опухоль/кровь – 13.7 ± 2.0; опухоль/мышца – 18.6 ± 3.2).
Еще одним примером синтеза макроциклических хелаторов для циркония является работа [64]. Авторы синтезировали различные гидроксаматно-модфицированные макроциклические хелаторы L1–L3 (схема 7 ). Согласно результатам по оценке стабильности (инкубирование в 50 мМ EDTA), все полученные комплексы [89Zr]Zr-L1–3 являются менее стабильными, чем [89Zr]Zr-DFO. Более низкую стабильность комплексов с синтезированными хелаторами авторы объясняют при помощи квантовохимических расчетов: при комплексообразовании 89Zr с хелаторами L1 и L3 из-за стерических ограничений происходило образование семикоординированных (для L2 – шестикоординированных), а не восьмикоординированных комплексов. Впоследствии, после оптимизации структур квантовохимическим методом, был синтезирован новый хелатор L4 (схема 7 ) и его бифункциональное производное BF4. Оценка стабильности in vitro показала примерно одинаковую устойчивость комплексов [89Zr]Zr-L4 (87% интактного комплекса) и [89Zr]Zr-DFO (91%) в присутствии избытка EDTA и более высокую стабильность в плазме крови для комплекса [89Zr]Zr-L4 (94%) по сравнению с [89Zr]Zr-DFO (53%). Сравнительное исследование in vivo проводилось в две стадии: первоначально сравнивали биораспределение комплексов [89Zr]Zr-DFO и [89Zr]Zr-L4 в организмах здоровых мышей. Для обоих исследуемых комплексов наблюдался высокий почечный клиренс (>98% от введенной активности выводилось в течение 30 мин). При этом можно отметить заметно более высокое накопление в костной ткани для [89Zr]Zr-L4 (0.60 ± 0.19%ID/г) по сравнению с [89Zr]Zr-DFO (0.05 ± 0.02%ID/г). Величины накопления в остальных тканях различались незначительно. На второй стадии была проведена оценка клеточного связывания и сравнительное исследование биораспределения препаратов [89Zr]Zr-BF4-трастузумаб и [89Zr]Zr-DFO-трастузумаб в организмах мышей с опухолями HER2+ и HER2–. По результатам исследования оба препарата показали эквивалентную аффинность, иммунореактивность и одинаковое накопление во всех основных тканях и органах за исключением скелета. В костной ткани отмечается значительно более высокое накопление для [89Zr]Zr-BF4-трастузумаба по сравнению с [89Zr]Zr-DFO-трастузумабом (табл. 4).
Дополнительно отметим работу [65]. Авторами были выполнены предварительные квантовохимические расчеты для определения оптимальной структуры, синтезирован макроциклический хелатор CTH36 (схема 8 ), содержащий четыре гидроксаматные группы и тетразиновые производные tCTH36 и tDFO. Эти производные в дальнейшем были конъюгированы с транс-циклооктен-модифицированным аналогом RGDfK (TCO-c(RGDfK)) и использованы для синтеза комплексов [89Zr]Zr-CTH36-c(RGDfK) и [89Zr]Zr-DFO-c(RGDfK). Полученный комплекс на основе хелатора CTH36 показал более высокую стабильность при инкубировании в избытке EDTA, чем аналогичный комплекс на основе DFO. В работе также сообщается о продолжении исследований и переходе к оценке стабильности полученных комплексов in vivo.

Схема 8 .
Сравнительное исследование линейных и циклических гидроксаматов было выполнено в работе [78]. Авторы синтезировали три макроцикла, различающихся по размеру полости (28–36 атомов в цикле), а также их ациклические аналоги (схема 8 ). По сравнению с DFO все синтезированные хелаторы показали более низкую скорость комплексообразования, при этом наиболее высокий радиохимический выход (>99%) был достигнут с хелаторами C7 и L7 (схема 8 ) после 120 мин инкубирования при 20°С. Исследования стабильности полученных комплексов проводили в 0.1 М фосфатном буфере (рН 6.5) и в 1750-кратном избытке EDTA (pH 7). Наиболее стабильными оставались комплексы 89Zr с хелаторами (сохранялось >70% комплекса после 7 сут инкубирования в присутствии избытка EDTA). Для комплексов 89Zr с C6, L6 и DFO наблюдалось заметное разложение (сохранялось менее 50%). В случае 89Zr[Zr]-C5 и 89Zr[Zr]-L5 происходило полное перелигандирование для в течение нескольких минут. Дополнительно можно отметить, что комплексы с циклическими хелаторами (n = 5 и 6) были менее стабильными, чем аналогичные комплексы с их ациклическими аналогами. Полученные результаты хорошо согласуются с результатами квантовомеханических расчетов [78].
Таким образом, использование циклических хелаторов является оправданным только при оптимальном соотношении размеров полости и катиона, поскольку в ином случае возникающие стерические затруднения негативно сказываются на стабильности полученных комплексов. В этом аспекте наиболее предпочтительным сегодня является использование ациклических хелаторов, что позволяет получать, как правило, более стабильные комплексы с высоким выходом при более мягких условиях синтеза.
ХЕЛАТОРЫ НА ОСНОВЕ ГИДРОКСИПИРИДОНА
Изначально различные варианты хелаторов на основе гидроксипиридона (HOPO) были предложены в качестве эффективного хелатора при лечении отравления плутонием-238 [84]. Среди исследованных хелаторов наиболее перспективными оказались 3,4,3-(LI-1,2-HOPO), и DFO-HOPO. Дальнейшие исследования данных хелаторов показали, что они также подходят для хелатирования 89Zr. Aвторы [66] синтезировали и провели исследование гибридного хелетора DFO-HOPO (схема 9 ). Комплекс [89Zr]Zr-DFO-HOPO оставался стабильным в сыворотке крови, растворах EDTA и DFO в течение 7 сут. Дополнительно сообщается, что DFO-HOPO способен перелигандировать 89Zr из комплекса с DFO с высокой эффективностью (>60% за 1 ч). Результаты сравнительного исследования in vivo показали быстрый клиренс и более низкое накопление в скелете для [89Zr]Zr-DFO-HOPO по сравнению c [89Zr]Zr-DFO (0.004 ± 0.001 и 0.037 ± 0.002%ID/г через 24 ч после в/в для [89Zr]Zr-DFO-HOPO и [89Zr]Zr-DFO соответственно). Дополнительно можно отметить заметно более высокое накопление в кишечнике для [89Zr]Zr-DFO-HOPO, связанное с высокой липофильностью исследуемого комплекса (табл. 2). Несмотря на полученные многообещающие результаты, необходимо отметить, что в данном исследовании представлены результаты экспериментов с комплексами, характеризующимися гораздо более быстрой фармакокинетикой по сравнению c синтезируемыми на их основе конъюгатами. Для объективной оценки долгосрочной стабильности необходимы дополнительные эксперименты с такими векторами, как mAb.

Схема 9 .
Синтез и подробное исследование хелатора (3,4,3-(LI-1,2-HOPO) (схема 9 ) был выполнен в работе [85]. Образование комплекса Zr-HOPO в соотношении 1 : 1 было подтверждено при помощи масс-спектрометрии высокого разрешения (HRMS). Сравнительное исследование комплексов показало, что комплекс [89Zr]Zr-3,4,3-(LI-1,2-HOPO) оставался стабильным в сыворотке крови, растворах EDTA (pH 5–8) и DFO в течение 7 сут, в то время как аналогичный комплекс с DFO подвергался значительному перелигандированию, особенно при понижении pH (с 8 до 5). Дополнительно авторами была проведена оценка стабильности комплексов в присутствии конкурирующих металлов (10-кратный избыток). В данных условиях оба комплекса оставались относительно стабильными (сохраняется более 95% комплекса) за исключением эксперимента в присутствии Fe3+. При инкубации в избытке Fe3+ происходило более сильное замещение для [89Zr]Zr-DFO (сохраняется 39% комплекса) по сравнению с [89Zr]Zr-HOPO (сохраняется 83% комплекса). Полученные результаты являются вполне закономерными, поскольку DFO относится к сидерофорам и имеет большое сродство к Fe3+. Сравнительное исследование комплексов [89Zr]Zr-HOPO и [89Zr]Zr-DFO in vivo показало более медленное выведение из организма и незначительно более высокое накопление в костях для [89Zr]Zr‑3,4,3‑(LI-1,2-HOPO) (0.17 ± 0.03%ID/г) по сравнению с [89Zr]Zr-DFO (0.06 ± 0.01%ID/г). В дальнейших исследованиях 3,4,3-(LI-1,2-HO-PO) был модифицирован добавлением бензилизотиоцианатной группы (схема 9 ) и конъюгирован с трастузумабом [80]. Несмотря на небольшое снижение стабильности для [89Zr]Zr-HOPO-трастузумаба относительно [89Zr]Zr-DFO-трастузумаба in vitro (табл. 4), исследование in vivo показало более низкое накопление для [89Zr]Zr-HOPO-трастузумаба в скелете (табл. 4) и более высокое значение КДН опухоль/скелет (~26) по сравнению с аналогичным препаратом на основе DFO (КДН опухоль/скелет ~8).
Другие хелаторы на основе гидроксипиридона, к сожалению, показали менее впечатляющие результаты. Несмотря на более высокую устойчивость комплекса [89Zr]Zr-BPDET-LysH22,2-3-H-OPO (схема 10 ) к перелигандированию в растворе DTPA, исследование данного комплекса и синтезированного на его основе препарата [89Zr]Zr-2,3-HOPO-p-Phe-NCS-трастузумаба in vivo показало более высокое накопление в почках, печени и скелете по сравнению с [89Zr]Zr-DFO и [89Zr]Zr-DFO-трастузумабом (табл. 4) [67]. Данные результаты хорошо согласуются с работой [81]. Авторы сиентизировали радиоиммуноконъюгаты на основе 3,2-HOPO (схема 9 ) и DFO с mAb, нацеленным на мезотелин (MSLN-mAb, анетумаб). Оценка стабильности полученных препаратов в сыворотке крови показала низкую устойчивость для препарата [89Zr]Zr-3,2-HOPO-MSLN-mAb по сравнению с [89Zr]Zr-DFO-MSLN-mAb (табл. 4). Последующее исследование in vivo также подтвердило более низкую стабильность препарата на основе 3,2-HOPO. Для [89Zr]Zr-3,2-HOPO-MSLN-mAb наблюдалось значительно более высокое, возрастающее накопление в бедренной кости (табл. 4). Дополнительно можно отметить заметно более высокое накопление в опухоли и, соответственно, более высокие КДН для препарата на основе DFO по сравнению с 3,2-HOPO (накопление в опухоли – 28.49 ± 3.78 и 7.97 ± 0.77%ID/г для [89Zr]Zr-DFO-MSLN-mAb и [89Zr]Zr-3,2-H-OPO-MSLN-mAb соответственно). Схожие результаты были получены для хелаторов CP256, YM103 и THPN(p-SCN-Bn-THPN) (схема 10 ).
Синтез хелатора CP256 и его бифункционального производного YM103 (схема 10 ) описан в работе [82]. Образование комплекса CP256 с Zr в соотношении 1 : 1 было подтверждено при помощи масс-спектрометрии. Сравнительное исследование стабильности комплексов in vitro проводилось в присутствии 1, 10, 100 эквивалентов DFO или CP256 для [89Zr]Zr-CP256 и [89Zr]Zr-DFO соответственно. При данных условиях комплекс [89Zr]Zr-CP256 оставался более стабильным, чем комплекс [89Zr]Zr-DFO. Добавление DFO к [89Zr]Zr-CP256 сопровождалось существенным перелигандированием [89Zr]Zr-CP256 (в 10- и 100-кратном избытке DFO сохранялось ~80 и ~15% комплекса соответственно), в то время как при добавлении 10- и 100-кратного избытка CP256 к [89Zr]Zr-DFO происходило полное замещение. Необходимо отметить, что несмотря на более высокую стабильность в присутствии ионов Fe3+ (1 мМ) комплекс [89Zr]Zr-CP256 (концентрация CP256 0.1 мМ) оказался менее стабильным (сохранятся ~14% комплекса), чем аналогичный комплекс с DFO (сохранятся ~93% интактного комплекса). Далее хелаторы YM103 и p‑Bn-NCS-DFO были конъюгированы с трастузумабом и помечены 89Zr. Оба синтезированных препарата были получены с высоким радиохимическим выходом (>98%) и оставались стабильными в сыворотке крови в течение 7 сут (табл. 4). Исследование in vivo показало практически одинаковое биораспределение для препаратов [89Zr]Zr‑YM103-трастузумаба и [89Zr]Zr-DFO-трастузумаба через 6 ч после введения. Однако при дальнейшем наблюдении авторы отмечают значительный рост накопления в костной ткани для препарата на основе YM103 (8.3 ± 0.1%ID/г через 6 ч; 25.9 ± 0.6%ID/г через 7 сут), тогда как для препарата на основе DFO такая динамика отсутствовала (7.7 ± 0.7%ID/г через 6 ч; 6.5 ± 0.4% ID/г через 7 cут).

Схема 10 .
Похожие результаты были получены и для хелатора THPN (схема 10 ). Сравнительное исследование стабильности комплексов [89Zr]Zr-THPN и [89Zr]Zr-DFO in vitro показало одинаковую устойчивость комплексов в сыворотке крови и более высокую устойчивость комплекса [89Zr]Zr-THPN в 100-кратном избытке EDTA [68]. Исследование in vivo показало схожее биологическое поведение для [89Zr]Zr-THPN и [89Zr]Zr-DFO через 24 ч после введения. На следующей стадии исследования THPN был конъюгирован с 1,4‑фенилендиизотиоцианатом [83]. Для проведения сравнительного исследования p-SCN-Bn-THPN, DFO и DFO* были конъюгированы с высокомолекулярным (800 кДа) полимерным гиперразветвленным полиглицерином (HPG) и помечены 89Zr. При исследовании на здоровых мышах in vivo наибольшее накопление в скелете отмечается для препарата [89Zr]Zr-THPN-HPG по сравнению с [89Zr]Zr-DFO-HPG и [89Zr]Zr-DFO*-HPG (табл. 4). Таким образом, несмотря нa высокую термодинамическую стабильность комплекса [89Zr]Zr-THPN (lg β = 50.3) и более высокую стабильность по сравнению с [89Zr]Zr-DFO in vitro, при проведении исследований in vivo препарат на основе THPN показал более низкую стабильность и соответствующее более высокое накопление в скелете. В качестве одной из возможных причин неудовлетворительной стабильности указывается введение п-фенилендиизотиоцианатной группы, что согласно квантовохимическим расчетам приводит к небольшому нарушению координационной геометрии.
ПРОЧИЕ ХЕЛАТОРЫ
Были проведены исследования комплексов 89Zr с хелаторами DOTA, DOTAM, DOTP, NOTA, TETA, TRITA, PCTA (схема 11 ) [71, 72] и с новыми хелаторами, содержащими четыре гидроксиизофталамидые (IAM-1 и IAM-2) [69] или дигидрокситерефталамидные (TAM-1 иTAM-2) группы [70].

Схема 11 .
При рассмотрении данных хелаторов важно учитывать возможность проведения реакции комплексообразования без дополнительного нагревания (табл. 2). В данном случае только комплексы [89Zr]Zr-TAM-1 и [89Zr]Zr-TAM-2 (схема 12 ) могут быть получены в тех же условиях, что и комплекс [89Zr]Zr-DFO.
Эксперименты по оценке стабильности комплексов [89Zr]Zr-IAM-1 и [89Zr]Zr-IAM-2 in vitro показали, что [89Zr]Zr‑IAM-1 является более стабильным (сохраняется 72% комплекса) в присутствии избытка DTPA (50 мМ, инкубирования в течение 7 сут) по сравнению с [89Zr]Zr-DFO (сохраняется 41% комплекса) и [89Zr]Zr-IAM-2 (сохраняется 26% комплекса) [69]. Однако при инкубировании в сыворотке были получены противоположные результаты: комплекс [89Zr]Zr-DFO оставался стабильным, тогда как в случае [89Zr]Zr-IAM-1 и [89Zr]Zr-IAM-2 сохранялось только 75 и 17% исходного комплекса соответственно. Последующие эксперименты in vivo подтвердили более низкую стабильность синтезированных комплексов. Комплексы [89Zr]Zr-IAM-1 и [89Zr]Zr-IAM-2 показали значительно более высокое накопление в печени и скелете (накопление в скелете через 72 ч после введения: [89Zr]Zr-DFO – 0.08 ± 0.01%ID/г; [89Zr]Zr-IAM-1 – 0.11 ± 0.01%ID/г; [89Zr]Zr-IAM-2 – 0.68 ± 0.33%ID/г). При этом можно отметить различную динамику накопления в костной ткани для комплексов [89Zr]Zr-IAM-1 и [89Zr]Zr-IAM-2: для [89Zr]Zr-IAM-1 накопление постепенно уменьшается, что может быть связано с перфузей и низким клиренсом, тогда как для [89Zr]Zr-IAM-2 накопление увеличивается, что обусловлено более низкой стабильностью исследуемого комплекса. В данном случае различие в стабильности [89Zr]Zr-IAM-1 и [89Zr]Zr-IAM-2 можно объяснить менее жесткой структурой хелатора IAM-2, который образует с 89Zr более лабильные комплексы.
Схожие результаты были получены с хелаторами TAM-1 и TAM-2 в [70]. В присутствии избытка DTPA (50 мМ, инкубирование в течении 7 сут при 37°С) комплексы [89Zr]Zr-TAM-1 и [89Zr]Zr-TAM-2 были более стабильны (сохраняется 100% комплексов), по сравнению с [89Zr]Zr-DFO (сохраняется 41% комплекса). Исследование комплексов [89Zr]Zr-TAM-1 и [89Zr]Zr-TAM-2 in vivo выявило более высокую устойчивость и более высокий клиренс для [89Zr]Zr-TAM-1. При сравнительном исследовании [89Zr]Zr-TAM-1 с [89Zr]Zr-DFO отмечается несущественное различие в накоплении в скелете ([89Zr]Zr‑DFO – 0.078 ± 0.014%ID/г; [89Zr]Zr-TAM-1 – 0.074 ± 0.022%ID/г) и более высокое накопление в печени и почках. Таким образом, несмотря на более высокую стабильность in vitro, полученные комплексы не обладают достаточной кинетической стабильностью.

Схема 12 .
Необходимо отметить, что поскольку цирконий-89 используется в основном для создания препаратов на основе моноклональных антител с использованием в качестве хелатора DFO, вопрос комплексообразования и стабильности с азамакроциклическими хелаторами подробно не исследовался. Относительно недавно подробное исследование комплексообразования 89Zr с DOTA, DOTP, DOTAM было представлено в работе [71]. Согласно представленным данным 89Zr образует очень стабильные комплексы с DOTA (схема 11 ). При этом стабильность комплексов 89Zr с DOTA, DOTP, DOTAM убывает в следующем ряду: [89Zr]Zr-DOTA $ \gg $ [89Zr]Zr-DOTP > [89Zr]Zr-DO-TAM > [89Zr]Zr‑DFO. Авторы отмечают, что комплекс [89Zr]Zr-DOTA не подвергается перелигандированию даже в присутствии 1000-кратного избытка EDTA в течение 7 сут. В аналогичных условиях лишь 20% 89Zr остается в комплексе [89Zr]Zr-DFO. Последующее сравнительное исследование in vivo показало, что среди исследуемых комплексов наиболее низкое неспецифическое накопление (кровь, скелет, печень, почки) достигается для комплекса [89Zr]Zr-DOTА. Однако необходимо учитывать, что возможность использования DOTA для синтеза 89Zr-РФЛП значительно ограничена: для образования комплекса [89Zr]Zr-DOTA требуется нагревание (>70°C в течение 30 и более минут), что делает невозможным использование предварительно конъюгированных антител. Более того, даже в случае реализации концепции предварительного мечения и последующей конъюгации (prelabeling) область применения DOTA в качестве хелатора значительно ограничена. Комплекс [89Zr]Zr-DOTА не может быть конъюгирован с векторной молекулой, поскольку не имеет свободных карбоксильных групп.
Позднее авторами опубликовали еще одно подробное исследование с хелаторами TETA, TR-ITA, PCTA и NOTA (схема 11 ) [72]. Комплексы Zr-TRITA, Zr-PCTA и Zr-NOTA были получены с высоким выходом и охарактеризованы различными методами (ВЭЖХ, ЯМР, масс-спектрометрия высокого разрешения). Дополнительно авторами представлены результаты рентгеноструктурного анализа полученных комплексов, которые указывают на образование биядерных комплексов [Zr(PCTA)]2O · 8H2O и [Zr(NOTA)(OH)]2 · 6H2O, что является следствием недостаточной дентатности используемых хелаторов. Комплексы [89Zr]Zr-PCTA, [89Zr]Zr‑NOTA и [89Zr]Zr‑TRITA были получены с количественным выходом (табл. 2). Комплекс с TETA синтезировать не удалось. Для оценки стабильности полученные комплексы инкубировали в присутствии избытка солей металлов, EDTA и в сыворотке крови. Наиболее устойчивым к перелигандированию в присутствии 1000-кратного избытка EDTA (pH 5–7) был комплекс [89Zr]Zr-PCTA (сохраняется 100% комплекса после 7 сут инкубирования при 37°С), комплекс [89Zr]Zr-NOTA подвергался заметному перелигандированию (сохраняется 70.7% комплекса), а [89Zr]Zr-TRITA полностью диссоциировал (сохраняется 9.6% комплекса). Аналогичные результаты были получены при инкубировании в сыворотке крови и в присутствии солей металлов. Также необходимо отметить, что несмотря на введение пиридинового кольца в структуру хелатора PCTA, комплекс [89Zr]Zr-PCTA является более гидрофильным, чем [89Zr]Zr-NOTA и [89Zr]Zr-DFO (табл. 2). Сравнительное исследование комплексов in vivo показало более низкий клиренс для [89Zr]Zr-TRITA и соответствующее более высокое накопление во всех органах и тканях. Более того, по прошествии 48 ч отмечается четырехкратное увеличение накопления в костной ткани, что подтверждает неудовлетворительную стабильность комплекса [89Zr]Zr-TRITA. В целом биораспределение комплексов [89Zr]Zr-PCTA и [89Zr]Zr-NOTA было аналогично [89Zr]Zr-DFO: исследуемые комплексы имеют низкий клиренс и соответствующее высокое неспецифическое накопление по сравнению с ранее представленными результатами для [89Zr]Zr‑DOTA.
Подводя итог, необходимо отметить недавнее исследование авторов [86], посвященное непосредственному сравнению перспективных хелаторов из различных групп: DFO*, CTH36, 3,4,3-(LI-1,2-HOPO) и DOTA-GA. Авторами были синтезированы тетразин-модифицированные хелаторы, которые в последствии были конъюгированы с пептидом, c(RGDfK), и помечены 89Zr. Комплексы 89Zr с DFO-c(RGDfK), DFO*-c(RGDfK) и CTH36-c(RGDfK) были получены с высоким выходом (>96%) в течение часа инкубации при 37°С, для синтеза [89Zr]Zr-3,4,3-(LI-1,2-HOPO)-c(RGDfK) потребовалось нагревание в течение 5 ч, при этом предполагается образование нескольких форм комплекса. Синтезировать комплекс с DOTA-GA-c(RGDfK) не удалось даже при инкубировании при 99°С в течение нескольких часов. Авторы отмечают, что согласно квантовохимическим расчетам модификация молекулы хелатора тетразином оказывает незначительное влияние на геометрию комплексов, за исключением комплекса с DOTA-GA, в котором происходит удлинение связей Zr–N по сравнению с комплексом Zr-DOTA. Тем не менее данный факт не объясняет нулевой выход реакции комплексобразования. Для оценки стабильности полученные комплексы инкубировали в 10 000-кратном избытке EDTA в течение 54 ч. В данных условиях комплекс [89Zr]Zr-DFO-c(RGDfK) быстро диссоциировал (сохраняется <10% комплекса через 24 ч). Комплексы [89Zr]Zr-DFO*-c(RGDfK) и [89Zr]Zr-3,4,3-(LI-1,2-HOPO)-c(RGDfK) оставались достаточно стабильными в течение всего эксперимента (сохраняется >80 и >95% соответственно). Наиболее интересным является факт, что комплекс на основе хелатора CTH36 был немного стабильнее, чем [89Zr]Zr-DFO-c(RGDfK) (сохраняется ~10% комплекса через 24 ч). Согласно квантовохимическим расчетам (B3LYP, базис DGDZVP), хелатор CTH36 образует с Zr комплекс, характеризующийся одной из самых высоких констант термодинамической стабильности (табл. 5) [32]. Помимо CTH36 89Zr образует так же стабильные комплексы с oxoDFO*, DFO-HOPO, DFO*, CTH36 и THPN (табл. 5).
Таблица 5.
Константы термодинамической стабильности для некоторых комплексов Zr [32]
| Комплекс | lg β |
|---|---|
| Zr(oxoDFO*) | 54.16 |
| Zr(DFO-HOPO) | 53.51 |
| Zr(DFO*) | 51.56 |
| Zr(CTH36) | 52.84 |
| Zr(THPN) | 47.28 |
| [ZrDFO(H2O)]+ | 41.41 |
| [Zr(DFO-O3)]+ | 43.37 |
Более высокая термодинамическая стабильность комплекса Zr(oxoDFO*) в данном случае объясняется более высокой гибкостью структуры oxoDFO* за счет наличия в структуре хелатора эфирных мостиков, что приводит к снижению стерических затруднений. Данный эффект также можно наблюдать при сравнении комплексов [ZrDFO(H2O)]+ и [Zr(DFO-O3)]+ (табл. 5). Таким образом, введение эфирных мостиков, вероятно, является одним из наиболее удачных подходов к модификации хелатора, поскольку одновременно увеличивает и термодинамическую стабильность, и гидрофильность исходной молекулы. Помимо CTH36 и производных DFO можно отметить высокую термодинамическую константу стабильности для комплекса Zr с THPN, однако на практике препараты на основе хелатора THPN были менее стабильны in vivo по сравнению с аналогичными препаратами на основе DFO и DFO* [83]. Данные результаты еще раз подтверждают, что термодинамические константы являются полезными при предварительном сравнении различных хелаторов, но не могут быть использованы для оценки стабильности комплексов in vivo.
В заключение отметим, что последнее десятилетие в мире был достигнут значительный прогресс в области радиофармацевтической химии 89Zr. Сегодня происходит активное внедрение различных препаратов циркония-89 в клиническую практику. Несмотря на многообещающие результаты, полученные при проведении клинических и доклинических исследований новых 89Zr-РФЛП, препараты на основе хелатора дефероксамина, оказались склонны к диссоциации in vivo. Это послужило толчком к разработке и детальной оценке целого ряда новых хелаторов для 89Zr. Основной сложностью при создании нового “идеального” хелатора стало заметное различие между термодинамической и кинетической стабильностью полученного комплекса. В результате чего, несмотря на более высокую стабильность большинства новых хелаторов in vitro, полученные комплексы так же, как и комплекс с DFO, диссоциировали in vivo. Кроме того, одним из требований к хелатору является возможность быстрой конъюгации с векторной молекулой и проведение реакции комплексообразования без дополнительного нагрева. Сегодня по сумме опубликованных результатов в качестве наиболее перспективного хелатора можно выделить DFO*, который хорошо зарекомендовал себя в ряде различных исследований, а также его производное oxoDFO*, которое является более гидрофильным и, согласно предварительным оценкам, позволяет образовывать более стабильные комплексы с 89Zr по сравнению DFO*.
К настоящему времени в мире запланированы или уже находятся в активной фазе более 30 клинических исследований, а завершено более 20 [87]. Большинство из представленных РФЛП на основе циркония-89 являются препаратами на основе моноклональных антител (например, широко применяемый для диагностики рака молочной железы [89Zr]Zr-трастузумаб). В России, к сожалению, препараты циркония-89 находятся на начальном этапе разработки и не находят пока рутинного применения в медицинских учреждениях. Основными сложностями в данном случае является как отсутствие отлаженного производства самого 89Zr, ограничивающее проведение дальнейших исследований различными научными группами, так и отсутствие четких и проработанных юридических механизмов клинического применения незарегистрированных новых РФЛП. Поскольку сегодня применение РФЛП стало неотъемлемой частью оказания качественной и высокотехнологичной медицинской помощи в развитых странах, есть все основания предполагать, что данные сложности будет преодолены в ближайшее время.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
van Dongen G.A.M.S., Beaino W., Windhorst A.D. et al. // J. Nucl. Med. 2021. V. 62. № 4. P. 438.
Mendler C.T., Gehring T., Wester H.-J. et al. // J. Nucl. Med. 2015. V. 56. № 7. P. 1112.
Cheal S.M., Punzalan B., Doran M.G. et al. // Eur. J. Nucl. Med. Mol. Imaging. 2014. V. 41. № 5. P. 985.
Berg E., Gill H., Marik J. et al. // J. Nucl. Med. 2020. V. 61. № 3. P. 453.
Disselhorst J.A., Brom M., Laverman P. et al. // J. Nucl. Med. 2010. V. 51. № 4. P. 610.
Conti M., Eriksson L. // EJNMMI Phys. 2016. V. 3. № 1.
Sánchez-Crespo A., Andreo P., Larsson S.A. // Eur. J. Nucl. Med. Mol. Imaging. 2004. V. 31. № 1. P. 44.
Marquez-Nostra B.V., Viola N. // Radiopharmaceutical Chemistry / Eds. Lewis J.S., Windhorst A.D., Zeglis B.M. Springer. Cham, 2019. P. 371.
McKnight B.N., Viola-Villegas N.T. // J. Label. Compd. Radiopharm. 2018. V. 61. № 9. P. 727.
Yoon J.-K., Park B.-N., Ryu E.-K. et al. // Int. J. Mol. Sci. 2020. V. 21. № 12. P. 4309.
Kurebayashi Y., Choyke P.L., Sato N. // Nanotheranostics. 2021. V. 5. № 1. P. 27.
De Feo M.S., Pontico M., Frantellizzi V. et al. // Clin. Transl. Imaging. 2022. V. 10. № 1. P. 23.
Laboratoire National Henri Becquerel. URL: http://www.lnhb.fr/en/
Link J.M., Krohn K.A., O’Hara M.J. // Appl. Radiat. Isot. 2017. V. 122. P. 211.
Queern S.L., Aweda T.A., Massicano A.V.F. et al. // N-ucl. Med. Biol. 2017. V. 50. P. 11.
Alnahwi A.H., Tremblay S., Guérin B. // Appl. Sci. 2018. V. 8. № 9. P. 1.
Meijs W.E., Herscheid J.D.M., Haisma H.J. et al. // A-ppl. Radiat. Isot. 1994. V. 45. № 12. P. 1143.
Verel I., Visser G.W.M., Boellaard R. et al. // J. Nucl. Med. 2003. V. 44. № 8. P. 1271.
Holland J.P., Sheh Y., Lewis J.S. // Nucl. Med. Biol. 2009. V. 36. № 7. P. 729.
Graves S.A., Kutyreff C., Barrett K.E. et al. // Nucl. Med. Biol. 2018. V. 64–65. P. 1.
Pandya D.N., Bhatt N.B., Almaguel F. et al. // J. Nucl. Med. 2019. V. 60. № 5. P. 696.
Bubenshchikov V.B., Larenkov A.A., Kodina G.E. // Radiochemistry. 2021. V. 63. № 3. P. 369.
Intorre B.J., Martell A.E. // J. Am. Chem. Soc. 1961. V. 83. № 17. P. 3618.
Intorre B.I., Martell A.E. // J. Am. Chem. Soc. 1960. V. 82. № 2. P. 358.
Shannon R.D. // Acta Crystallogr. A. 1976. V. 32. № 5. P. 751.
Pozhidaev A.I., Porai-Koshits M.A., Polynova T.N. // J. Struct. Chem. 1974. V. 15. № 4. P. 548.
Ilyukhin A.B., Davidovich R.L., Samsonova I.N. et al. // Crystallogr. Reports. 2000. V. 45. № 1. P. 39.
Friend M.T., Wall N.A. // Inorg. Chim. Acta. 2019. V. 484. P. 357.
Meijs W.E., Herscheid J.D.M., Haisma H.J. et al. // Int. J. Radiat. Appl. Instrumentation. A. 1992. V. 43. № 12. P. 1443.
Bickel H., Gäumann E., Keller-Schierlein W. et al. // Experientia. 1960. V. 16. № 4. P. 129.
Holland J.P., Divilov V., Bander N.H. et al. // J. Nucl. Med. 2010. V. 51. № 8. P. 1293.
Holland J.P. // Inorg. Chem. 2020. V. 59. № 3. P. 2070.
Racow E.E., Kreinbihl J.J., Cosby A.G. et al. // J. Am. Chem. Soc. 2019. V. 141. № 37. P. 14650.
Summers K.L., Sarbisheh E.K., Zimmerling A. et al. // Inorg. Chem. 2020. V. 59. № 23. P. 17443.
Ekberg C., Källvenius G., Albinsson Y. et al. // J. Solution Chem. 2004. V. 33. № 1. P. 47.
Toporivska Y., Gumienna-Kontecka E. // J. Inorg. Biochem. 2019. V. 198. P. 110753.
Savastano M., Bazzicalupi C., Ferraro G. et al. // Molecules. 2019. V. 24. № 11. P. 2098.
Imura R., Ida H., Sasaki I. et al. // Molecules. 2021. V. 26. № 16. P. 4977.
Price E.W., Orvig C. // Chem. Soc. Rev. 2014. V. 43. № 1. P. 260.
Mealey J. // Nature. 1957. V. 179. № 4561. P. 673.
Zhong W., Parkinson J.A., Guo M. et al. // JBIC J. Biol. Inorg. Chem. 2002. V. 7. № 6. P. 589.
Deri M.A., Zeglis B.M., Francesconi L.C. et al. // Nucl. Med. Biol. 2013. V. 40. № 1. P. 3.
Bhatt N., Pandya D., Wadas T. // Molecules. 2018. V. 23. № 3. P. 638.
Vosjan M.J.W.D., Perk L.R., Visser G.W.M. et al. // Nat. Protoc. 2010. V. 5. № 4. P. 739.
Perk L.R., Vosjan M.J.W.D., Visser G.W.M. et al. // Eur. J. Nucl. Med. Mol. Imaging. 2010. V. 37. № 2. P. 250.
Viola-Villegas N.T., Rice S.L., Carlin S. et al. // J. Nucl. Med. 2013. V. 54. № 11. P. 1876.
Perk L.R., Visser G.W.M., Vosjan M.J.W.D. et al. // J. Nucl. Med. 2005. V. 46. № 11. P. 1898.
Oude Munnink T.H., Korte M.A. de, Nagengast W.B. et al. // Eur. J. Cancer. 2010. V. 46. № 3. P. 678.
Nayak T.K., Garmestani K., Milenic D.E. et al. // J. -Nucl. Med. 2012. V. 53. № 1. P. 113.
Meijs W.E., Haisma H.J., Klok R.P. et al. // J. Nucl. Med. 1997. V. 38. № 1. P. 112.
Abou D.S., Ku T., Smith-Jones P.M. // Nucl. Med. Biol. 2011. V. 38. № 5. P. 675.
Patra M., Bauman A., Mari C. et al. // Chem. Commun. 2014. V. 50. № 78. P. 11523.
Vugts D.J., Klaver C., Sewing C. et al. // Eur. J. Nucl. Med. Mol. Imaging. 2017. V. 44. № 2. P. 286.
Cho H., Al-saden N., Lam H. et al. // Nucl. Med. Biol. 2020. V. 84–85. P. 11.
Raavé R., Sandker G., Adumeau P. et al. // Eur. J. Nucl. Med. Mol. Imaging. 2020. V. 47. № 2. P. 505.
Adams C.J., Wilson J.J., Boros E. // Mol. Pharm. 2017. V. 14. № 8. P. 2831.
Rousseau J., Zhang Z., Dias G.M. et al. // Bioorganic Med. Chem. Lett. 2017. V. 27. № 4. P. 708.
Chomet M., Schreurs M., Bolijn M.J. et al. // Eur. J. N-ucl. Med. Mol. Imaging. 2021. V. 48. № 3. P. 694.
Al-Saden N., Lam K., Chan C. et al. // Mol. Pharm. 2018. V. 15. № 8. P. 3383.
Briand M., Aulsebrook M.L., Mindt T.L. et al. // Dalton Trans. 2017. V. 46. № 47. P. 16387.
Alnahwi A.H., Ait-Mohand S., Dumulon-Perreault V. et al. // ACS Omega. 2020. V. 5. № 19. P. 10731.
Zhai C., Summer D., Rangger C. et al. // Mol. Pharm. 2015. V. 12. № 6. P. 2142.
Zhai C., He S., Ye Y. et al. // Biomolecules. 2019. V. 9. № 3. P. 91.
Boros E., Holland J.P., Kenton N. et al. // Chempluschem. 2016. V. 81. № 3. P. 274.
Seibold U., Wängler B., Wängler C. // ChemMedChem. 2017. V. 12. № 18. P. 1555.
Allott L., Da Pieve C., Meyers J. et al. // Chem. Commun. 2017. V. 53. № 61. P. 8529.
Tinianow J.N., Pandya D.N., Pailloux S.L. et al. // Theranostics. 2016. V. 6. № 4. P. 511.
Buchwalder C., Rodríguez-Rodríguez C., Schaffer P. et al. // Dalton Trans. 2017. V. 46. № 29. P. 9654.
Bhatt N.B., Pandya D.N., Xu J. et al. // PLoS One. 2017. V. 12. № 6. P. e0178767.
Pandya D.N., Pailloux S., Tatum D. et al. // Chem. Commun. 2015. V. 51. № 12. P. 2301.
Pandya D.N., Bhatt N., Yuan H. et al. // Chem. Sci. 2017. V. 8. № 3. P. 2309.
Pandya D.N., Henry K.E., Day C.S. et al. // Inorg. Chem. 2020. V. 59. № 23. P. 17473.
Richardson-Sanchez T., Tieu W., Gotsbacher M.P. et al. // Org. Biomol. Chem. 2017. V. 15. № 27. P. 5719.
Brown C.J.M., Gotsbacher M.P., Codd R. // Aust. J. Chem. 2020. V. 73. № 10. P. 969.
Brandt M., Cowell J., Aulsebrook M.L. et al. // JBIC J. Biol. Inorg. Chem. 2020. V. 25. № 5. P. 789.
Rudd S.E., Roselt P., Cullinane C. et al. // Chem. Commun. 2016. V. 52. № 80. P. 11889.
Liapis V., Tieu W., Rudd S.E. et al. // EJNMMI Radiopharm. Chem. 2020. V. 5. № 1. P. 27.
Guérard F., Lee Y.-S., Brechbiel M.W. // Chem. - A Eur. J. 2014. V. 20. № 19. P. 5584.
Summer D., Garousi J., Oroujeni M. et al. // Mol. Pharm. 2018. V. 15. № 1. P. 175.
Deri M.A., Ponnala S., Kozlowski P. et al. // Bioconjug. Chem. 2015. V. 26. № 12. P. 2579.
Roy J., Jagoda E.M., Basuli F. et al. // Cancer Biother. Radiopharm. 2021. V. 36. № 4. P. 316.
Ma M.T., Meszaros L.K., Paterson B.M. et al. // Dalton Trans. 2015. V. 44. № 11. P. 4884.
Buchwalder C., Jaraquemada-Peláez M.D.G., Rousseau J. et al. // Inorg. Chem. 2019. V. 58. № 21. P. 14667.
White D.L., Durbin P.W., Jeung N. et al. // J. Med. Chem. 1988. V. 31. № 1. P. 11.
Deri M.A., Ponnala S., Zeglis B.M. et al. // J. Med. Chem. 2014. V. 57. № 11. P. 4849.
Damerow H., Hübner R., Judmann B. et al. // Cancers (Basel). 2021. V. 13. № 24. P. 6349.
ClinicalTrials.gov. URL: http://www.clinicaltrials.gov
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия