

Координационная химия, 2022, T. 48, № 2, стр. 67-73
1,1'-Дифенил-бис-силатран – первый пример структурно-охарактеризованного бис-силатрана
Ю. Ван 1, Д. М. Вахрушева 2, 3, И. В. Крылова 2, А. Я. Козьменкова 2, Е. Н. Николаевская 2, Б. Н. Манкаев 4, М. Е. Миняев 2, М. А. Сыроешкин 2, *, М. П. Егоров 2, В. В. Жуйков 1
1 Институт химических наук Ренна, Университет Ренн-1
Ренн, Франция
2 Институт органической химии им. Н.Д. Зелинского
Москва, Россия
3 Российский химико-технологический университет им. Д.И. Менделеева,
Высший химический колледж РАН
Москва, Россия
4 Московский государственный университет им. М.В. Ломоносова
Москва, Россия
* E-mail: syroeshkin@ioc.ac.ru
Поступила в редакцию 20.04.2021
После доработки 27.06.2021
Принята к публикации 28.06.2021
- EDN: QLQKRY
- DOI: 10.31857/S0132344X22010078
Аннотация
Взаимодействием трис-(1,3-дигидроксипропан-2-ил)амина (L) с PhSi(OEt)3 в толуоле получен 1,1'-дифенил-бис-силатран (I). Молекулярное строение комплекса I установлено с методом РСА (CIF file CCDC № 2078347). Изучение окислительно-восстановительных свойств нового бис-силатрана I показало, что генерированные с помощью циклической вольтамперометрии его ион-радикалы стабильны благодаря тому, что радикальный центр, локализованный на атоме азота, стерически защищен внутри бис-атрановой клетки.
Атраны – широкий класс соединений, содержащих в своем составе трициклический фрагмент N–(C–C–Y)3 (Y = O, S, NR) (схема 1 ). На сегодняшний день известны атрановые структуры с элементами групп 1, 2, 12–15 периодической таблицы [1]. Исторически сложилось, что первыми представителями атранов стали силатраны (Е = Si, Y = O). Основоположником силатрановой химии по праву считается М.Г. Воронков с соавторами [2], после публикации которых общее количество работ по этой теме неустанно растет (рис. 1) [3].


Схема 1 .
Популярность данного направления обусловлена несколькими причинами: 1) относительно легкая синтетическая методика, включающая в себя взаимодействие триэтаноламина с замещенными триалкоксисиланами; 2) фундаментальный интерес – исследование длины трансаннулярной связи Si ← N (для ознакомления с интервалами длин этой связи см. [4]); 3) прикладной интерес, обусловленный проявляемыми силатранами фармакологическими [3] (противоопухолевая, противораковая [5], антибактериальная [6–9], противовоспалительная, фунгицидная активность [10–12], стимулирующее действие в животноводстве [13] и эффекты прорастания семян [14, 15]) свойствами, в том числе биологической активностью [2, 16]. Их успешно применяют в золь-гель процессах [17], для получения новых молекулярных сит [18] и даже как проявляющий ДНК компонент в атомно-силовой микроскопии [19–26].
В настоящее время интерес к химии атранов переживает очередной подъем. В значительной степени это связано с использованием новых лигандных систем. Так, получение трис-(1,3-дигидроксипропан-2-ил)амина [27] способствовало резкому скачку интереса к синтезу и исследованию свойств бис-металатрановых соединений на его основе. Предполагается, что такие бис-металлатрановые соединения будут проявлять все свойства комплексов с одним атрановым фрагментом, но по интенсивности проявляемого свойства его превосходить, что откроет новые перспективные области его применения, в частности в дизайне новых полупроводниковых элементов, а также веществ, проявляющих свойства “молекулярного мускула”. Так, уже синтезирован ряд бис-герматранов с тиенильными [28] и фенильным заместителями [29], изучены их окислительно-восстановительные свойства. Интересно, что по сравнению с металлатранами с одним атрановым фрагментом [30, 31], бис-металлатраны показывают большую стабильность катион-радикалов, образованных в редокс-процессах [29, 30].
Цель настоящей работы – синтез, изучение структуры различными, в том числе и с помощью РСА, методами и подробное исследование окислительно-восстановительных свойств нового комплекса 1,1'-дифенил-бис-силатрана (I) на основе трис-(1,3-дигидроксипропан-2-ил)амина (L).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Все операции по синтезу проводили в атмосфере аргона с использованием трис-(1,3-дигидроксипропан-2-ил)амина, полученного согласно методике, описанной в [27], и PhSi(OEt)3, полученного взаимодействием Si(OEt)4 с PhCl в присутствии избытка магния. Растворители очищали стандартными методами [32].
Синтез комплекса I. К раствору лиганда L (0.19 г, 0.8 ммоль) в метаноле прибавляли раствор PhSi(OEt)3 (0.38 г, 1.6 ммоль) в толуоле. Реакционную смесь кипятили с перемешиванием, отгоняя метанол. Далее толуол упарили с использование роторного испарителя, остаток промыли последовательно петролейным эфиром и хлороформом. Продукт получали в виде белого кристаллического порошка. Выход I 71%.
ЯМР 1Н (300 МГц; CDCl3; δ, м.д.): 7.7–7.68 (м., 4Н, СНаром), 7.44–7.33 (м., 6Н, СНаром), 3.82 (д.д., J = 4.6 Гц, 11.5 Гц, 6Н, CH2), 3.77–3.70 (м., 6Н, CH2), 3.55–3.45 (м., 3Н, CH). ЯМР 13С (75 МГц; CDCl3; δ, м.д.): 134.20, 131.68, 130.37, 127.83, 61.00, 57.47.
HRMS: для изотопа 28Si 461.155 [M + NH4]+ (рассчитано 461.1559), 466.1101 [M + Na]+ (рассчитано 466.1113), 482.0837 [M + K]+ (рассчитано 482.0852).
Электрохимические измерения. Окислительно-восстановительное поведение комплекса I и лиганда L изучали методом циклической вольтамперометрии (ЦВА). Приготовление растворов соединений и все измерения проводили в сухом перчаточном боксе с атмосферой аргона и показателями влажности и кислорода, не превышающими 1 м.д. Перед этим ацетонитрил (Acros, HPLC grade) с содержанием воды не более 100 м.д. дополнительно осушали над молекулярными ситами (4 Å), которые предварительно прокаливали в вакууме при 200–250°С в течение 4 ч. Соль фонового электролита Bu4NClO4 (Sigma Aldrich) также сушили в вакууме при 70–80°С в течение 4 ч. Содержание воды в растворе фонового электролита (0.1 M Bu4NClO4/MeCN) не превышало 20 м.д., согласно титрованию по Карлу Фишеру при использовании титратора Mettler-Toledo Titrator C10SD. Анализируемые соединения, растворенные в фоновом электролите, подвергали электрохимическим испытаниям в стандартной трехэлектродной стеклянной ячейке при скоростях развертки потенциала 0.05–1 В с–1. Рабочим электродом служил стеклоуглеродный дисковый электрод с диаметром диска 1.7 мм. Перед использованием его поверхность полировали наждачной бумагой и затем пастой ГОИ до достижения зеркального блеска. В качестве вспомогательного электрода выступала платиновая проволока, прокаленная в пламени газовой горелки для удаления с поверхности оксидов и других возможных загрязнений. Потенциалы изучаемых процессов измеряли относительно электрода сравнения, представляющего собой серебряную проволоку, покрытую слоем хлорида серебра, отделенную от основного объема электролита электролитическим мостиком, заполненным раствором фонового электролита. Электрод сравнения калибровали относительно пары ферроцен/ферроцений (Е0 = 0.400 В отн. нормального водородного электрода).
Электронные спектры (УФ- и видимый диапазон) проводили на спектрометре Agilent 8453 для 5 × 10–4 М раствора I в ацетонитриле, используя 10 мм кварцевую кювету с тефлоновой крышкой. Приготовление раствора осуществляли в сухом перчаточном боксе, перед выносом кюветы контакт между крышкой и кюветой герметизировали лентой Parafilm. Затем в течение нескольких минут записывали спектры.
РСА. Дифракционные данные для монокристалла соединения I получены при 100(2) K на дифрактометре Bruker Quest D8, оснащенном детектором Photon-III (графитовый монохроматор, φ- и ω‑сканирование) с использованием MoKα-излучения (λ = 0.71073 Å). Интенсивности отражений проинтегрированы программой SAINT [33] и скорректированы по эквивалентным отражениям программой SADABS [34] для учета поглощения излучения и разложения кристалла. Структура расшифрована прямыми методами с использованием программного пакета SHELXT [35] и уточнена МНК в анизотропном (для атомов водорода – изотропном) полноматричном приближении по F 2 с использованием программы SHELXL [36]. Позиции атомов водорода рассчитаны геометрически и уточнены по модели жесткого тела. Основные кристаллографические данные и детали дифракционных экспериментов приведены в табл. 1.
Таблица 1.
Кристаллографические данные, параметры эксперимента и уточнения структуры I
| Параметр | Значение |
|---|---|
| M | 443.60 |
| Сингония | Моноклинная |
| Пр. гр. | P21/c |
| a, Å | 10.4100(2) |
| b, Å | 11.4017(2) |
| c, Å | 17.4199(3) |
| β, град | 101.5437(7) |
| V, Å3 | 2025.77(6) |
| Z | 4 |
| ρ(выч.), г/см3 | 1.454 |
| μ, мм–1 | 0.216 |
| F(000) | 936 |
| Размер кристалла, мм | 0.15 × 0.08 × 0.05 |
| Область сканирования по θ, град | 1.997–34.350 |
| Диапазон индексов hkl | –16 ≤ h ≤ 16, –18 ≤ k ≤ 18, –27 ≤ l ≤ 27 |
| Число отражений измеренных/ независимых (Rint) | 76 446/8490 (0.0531) |
| Число отражений с I > 2σ(I) | 6292 |
| GOOF | 1.026 |
| R-факторы (I > 2σ(I)) | R1 = 0.0444, wR2 = 0.1068 |
| R-факторы (по всем отражениям) | R1 = 0.0681, wR2 = 0.1211 |
| Остаточная электронная плотность (max/min), e/Å3 | 0.585/–0.490 |
Полные таблицы межатомных расстояний и валентных углов, координаты атомов и параметры атомных смещений депонированы в Кембриджский банк структурных данных (CCDC № 2078347; https://www.ccdc.cam.ac.uk/structures/).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Взаимодействием трис-(1,3-дигидроксипропан-2-ил)амина (L) с PhSi(OEt)3 в толуоле получен 1,1'-дифенил-бис-силатран (I) (схема 2 ).

Схема 2 .
Молекулярная структура соединения I подтверждена данными рентгеноструктурных исследований (рис. 2), которые показали разупорядоченность атомов фрагментов –O–CH2–CH–CH2–O– по двум положениям в соотношении 0.8491(8) : 0.1509(8).
Рис. 2.
Молекулярная структура комплекса I (p = 50%). Разупорядоченность фрагментов –O–CH2–CH–CH2–O– не показана.
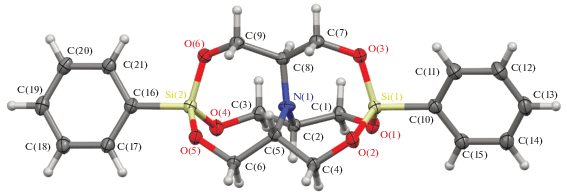
Это первый пример бис-силатрана, охарактеризованного рентгеноструктурно, поэтому сравнение геометрических характеристик длин связей и валентных углов будет вестись с соединениями, содержащими один атрановый фрагмент.
С момента первого синтеза силатранов было получено и полностью охарактеризовано достаточно большое их количество с различными заместителями как при атоме металла, так и в атрановом фрагменте [4, 37]. Накоплен значительный материал по строению таких производных, в частности длинам трансаннулярной связи N → Si в 3с–4е (трехцентровом четырехэлектронном) фрагменте С–Si ← N [38]. Ее длина может варьироваться в широком интервале: от 2.004 Å [39] до 3.19 Å [40]. При этом длина связи хорошо коррелируется с индуктивным эффектом заместителя при атоме кремния – более электроотрицательные заместители смещали электронную плотность не только с атома кремния, но и связанного с ним атома азота, тем самым укорачивая дативную связь N–Si(X). Так одни из наиболее коротких трансаннулярных связей демонстрировали галогензамещенные силатраны (Х = F: 2.042 Å [41], 2.050 Å (210 K) [42], 2.056 (100 K) [8], 2.034 [43]; X = Cl: 2.022 [44], 2.023 [45]), а в случае фенилсилатрана – ближайшего аналога нового бис-силатрана I – данная величина оценивалась в 2.193 Å [46].
Длины связей Si(1)–N(1) и Si(2)–N(1) в I (2.7579(12) и 3.0293(12) Å соответственно) значительно отличаются от таковых, которые были получены для фенилсилатрана. Тем не менее данные значения существенно меньше суммы ван-дер-ваальсовых радиусов атомов кремния и азота (3.65 Å [47]), что однозначно свидетельствует о слабой донорно-акцепторной связи между ними в случае соединения I. Таким образом, систему С(16)–Si(2)–N(1)–Si(1)–C(10) можно рассматривать как единую 5с–6е (пятицентровую шестиэлектронную) связь; причем эта центральная 5с–6е ось практически прямая – углы С(16)Si(2)N(1), Si(2)N(1)Si(1) и N(1)Si(1)C(10) близки к 180° (178.71(5)°, 179.75(4)° и 178.32(5)° соответственно).
Обращает на себя внимание еще и тот факт, что расстояния Si(1)–N(1) и Si(2)–N(1) в I значительно отличаются (Δ = 0.271 Å). По нашему мнению, данное различие может быть объяснено энергетическим компромиссом между внутримолекулярным донорным взаимодействием азота и его стерически-вынужденной пентакоординацией.
Центральный атом азота находится в sp2-гибридизации, углы С(8)N(1)С(5), С(8)N(1)С(2) и С(2)N(1)С(5) близки к 120° (119.92(11)°, 118.84(11)° и 120.00(11)° соответственно). Атом азота незначительно выходит из плоскости, образованной атомами С(2), С(5), С(8), всего на 0.095(14) Å. Каждый атом кремния в структуре имеет тетрагональное окружение (средние значения углов СSiО и OSiО в I составляют 106.2° и 112.5° соответственно), это разительно отличает соединение I от фенилсилатрана, средние значения углов СSiО в котором в среднем составляли 97.1°, что соответствовало геометрическим параметрам искаженной тригональной бипирамиды.
Окислительно-восстановительные свойства бис-силатрана I изучали методом ЦВА на стеклоуглеродном дисковом электроде в растворе 0.1 M Bu4NClO4/MeCN. Вольтамперные кривые, полученные при скоростях развертки потенциала от 0.05 до 1 В с–1 (рис. 3), соответствуют электрохимически квазиобратимому (приближающемуся к обратимому) одноэлектронному процессу окисления, приводящему к образованию катион-радикала. Сравнение электрохимического поведения данной системы с поведением ферроцена в тех же условиях позволило выявить немного заниженные значения токов прямого и обратного пиков в случае бис-силатрана I при их соотношении, равном 1 : 1. Кроме того, путем экстраполяции потенциалов пиков окисления и восстановления к нулевому току была определена величина интервала между пиками ΔEp = 0.075 В, которая немного превышает теоретическое значение для электрохимически обратимых реакций (0.059 В [48]), что, вероятно, связано с более медленным переносом электрона [49]. Потенциал полуволны бис-силатрана составил E1/2 = 1.549 В.
Рис. 3.
ЦВА-кривые, полученные для 3 × 10–3 M раствора I в 0.1 M Bu4NClO4/MeCN на стеклоуглеродном дисковом электроде при скоростях развертки потенциала 0.05, 0.1, 0.2, 0.5 и 1 В с–1.

Отметим, что в отличие от 1,1'-дифенил-бис-силатрана I фенилсилатран показывает химически необратимое поведение вплоть до скоростей развертки потенциала ≥ 1 В [30]. Это указывает на бόльшую стабильность катион-радикала бис-силатрана вследствие того, что радикальный центр, локализованный на атоме азота, стерически защищен внутри бис-атрановой клетки. Значение потенциала пика окисления $E_{{\text{p}}}^{{{\text{ox}}}}$ для фенилсилатрана на 0.63 В выше, чем для исходного триэтаноламина из-за вовлечения неподеленной пары электронов атома азота силатрана в дативное взаимодействие N → Si, что делает его менее доступным для окисления [30]. Подобное смещение потенциала пика окисления наблюдается также в случае I и L, при этом величина сдвига оказывается больше – 0.79 В (для L $E_{{\text{p}}}^{{{\text{ox}}}}$ = 0.801 B, что согласуется с данными [27]). Это свидетельствует об участии атома азота бис-силатрана в двойном дативном взаимодействии Si ← N → Si и, как следствие, еще большем снижении активности данного центра в процессах окисления.
В отличие от 1,1'-дифенил-бис-герматрана, на кривой восстановления которого не наблюдается фарадеевских процессов вплоть до разряда фонового электролита [29], бис-силатран I восстанавливается при $\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle-}$}}{E} _{{\text{p}}}^{{{\text{red}}}}$ = –2.685 В.
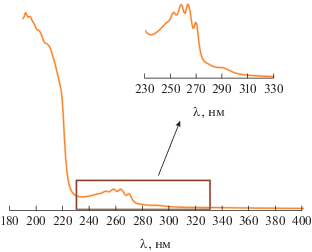
В УФ-спектре для I наиболее длинноволновая (широкая и малоинтенсивная) полоса поглощения имеет максимум при 289 нм (рис. 4, вставка), справа от тонкой структуры при 245–275 нм, характерной для бензола [50]. Величина энергии данного поглощения 4.29 эВ находится в хорошем соответствии с разницей между потенциалами пиков окисления и восстановления 4.27 В.
Таким образом, мы впервые получили и охарактеризовали различными физико-химическими методами (в том числе и рентгеноструктурно) 1,1'-дифенил-бис-силатран I, исследованы его окислительно-восстановительные свойства.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Verkade J.G. // Coord. Chem. Rev. 1994. V. 137. P. 233.
Voronkov M.G. Bioactive Organo-Silicon Compounds. Topics in Current Chemistry. Springer, Berlin Heidelberg GmbH, 1979. P. 77.
Adamovich S.N. // Appl. Organometal. Chem. 2019. V. 33. № 7. P. e4940.
Puri J.K., Singha R., Chahala V.K. // Chem. Soc. Rev. 2011. V. 40. P. 1791.
Sculimbrene B.R., Decanio R.E., Peterson B.W. et al. // Tetrahedron Lett. 2001. V. 42. P. 4979.
Li Z., Tian D., Zhu C. // Phosphorus, Sulfur Silicon Relat. Elem. 2000. V. 165. P. 99.
Chen L., Xie Q., Sun L. et al. // J. Organomet. Chem. 2003. V. 678. P. 90.
Tharmaraj P., Kodimunthiri D., Sheela C.D. et al. // J. Coord. Chem. 2009. V. 62. P. 2220.
Li Z., Tian D. // Heterocycl. Commun. 2003. V. 9. P. 605.
Li Z., Song X., Su H. et al. // Heterocycl. Commun. 2005. V. 11. P. 475.
Li Z., Su H., Gao X. et al. // Main Group Met. Chem. 2007. V. 30. P. 101.
Wan S.G., Yang X.Y., Yu Y. et al. // Phosphorus, Sulfur Silicon Relat. Elem. 2005. V. 180. P. 2813.
Popov I.V., Bolabovko A.E., Nikitiuk I.Y. et al. // Morfologiya. 2005. V. 128. P. 64.
Voronkov M.G., Dolmaa G., Tserenpil S. et al. // Dokl. Biol. Sci. 2005. V. 404. P. 367.
Макарова Л.Е., Соколова М.Г., Боровский Г.Б. и др. // Агрохимия. 2009. № 1. С. 27.
Lukevics E., Ignatovich L. Metallotherapeutic Drugs and Metal-Based Diagnostic Agents: The Use of Metals in Medicine. Chichester: John Wiley & Sons, 2005. P. 278.
Ortiz de Zarate D., Fernandez L., Beltran A. et al. // Solid State Sci. 2008. V. 10. P. 587.
Yadav G.D., Manyar H.G. // Clean Techn. Environ. Policy. 2005. V. 7. P. 162.
Shlyakhtenko L.S., Gall A.A., Filonov A. et al. // Ultramicroscopy. 2003. V. 97. P. 279.
Lyubchenko Y.L., Shlyakhtenko L.S., Binus M. et al. // Nucleic Acids Res. 2002. V. 30. P. 4902.
Liu R., Yuan B., Emadi S. et al. // Biochemistry. 2004. V. 43. P. 6959.
Yamin G., Munishkina L.A., Karymov M.A. et al. // Biochemistry. 2005. V. 44. P. 9096.
Emadi S., Liu R., Yuan B. et al. // Biochemistry. 2004. V. 43. P. 2871.
Pavlicek J.W., Oussatcheva E.A., Sinden R.R. et al. // Biochemistry. 2004. V. 43. P. 10664.
Mikheikin A.L., Lushnikov A.Y., Lyubchenko Y.L. // Biochemistry. 2006. V. 45. P. 12998.
Lyubchenko Y.L., Shlyakhtenko L.S., Gall A.A. // Methods Mol. Biol. 2009. V. 543. P. 337.
Jie Y., Livant P., Li H. et al. // J. Org. Chem. 2010. V. 75. P. 4472.
Romanovs V., Spura J., Jouikov V. // Synthesis. 2018. V. 50. P. 3679.
Romanovs V., Vakhrusheva D.M., Krylova I. et al. // Mendeleev Commun. 2020. V. 30. P. 567.
Romanovs V., Sidorkin V., Belogolova E. et al. // Dalton Trans. 2017. V. 46. P. 8849.
Sidorkin V.F., Belogolova E.F., Wang Y. et al. // Chem. Eur. J. 2017. V. 23. P. 1910.
Гордон А., Форд Р. Спутник химика. М.: Мир, 1976. С. 437.
Bruker. APEX-III. Madison (WI, USA): Bruker AXS Inc., 2019.
Krause L., Herbst-Irmer R., Sheldrick G. M. et al. // J. Appl. Cryst. 2015. V. 48. P. 3.
Sheldrick G. M. // Acta Crystallogr. A. 2015. V. 71. P. 3.
Sheldrick G.M. // Acta Crystallogr. C. 2015. V. 71. P. 3.
Lukevits É., Pudova O.A. // Chem Heterocycl. Comp. 1996. V. 32. P. 1381.
Wang Y., Romanovs V., Spura J. et al. // Chem. Heterocycl. Comp. 2016. V. 52. P. 546.
Schomburg D. // Z. Anorg. Allg. Chem. 1982. V. 493. P. 53.
Kemme A., Bleidelis J., Urtane I. et al. // J. Organomet. Chem. 1980. V. 202. P. 115.
Parkanyi L., Hencsei P., Bihatsi L. et al. // J. Organomet. Chem. 1984. V. 269. P. 1.
Korlyukov A.A., Antipin M.Yu., Buzin M.I. et al. // J. Struct. Chem. 2009. V. 50. P. 873.
Forgacs G., Kolonits M., Hargittai I. // Struct. Chem. 1990. V. 1. P. 245.
Kemme A.A., Bleidelis Ya.Ya., Pestunovich V.A. et al. // Dokl. AN SSSR. 1978. V. 243. P. 688.
Hencsei P. // Struct. Chem. 1991. V. 2. P. 21.
Turley J.W., Boer F.P. // J. Am. Chem. Soc. 1968. V. 90. P. 4026.
Bondi A. // J. Phys. Chem. 1964. V. 68. № 3. P. 441.
Bard A.J., Faulkner L.R. Electrochemical Methods. N.Y.: John Wiley and Sons, 2001. 833 p.
Nicholson R.S. // Anal. Chem. 1965. V. 37. № 11. P. 1351.
Ziegler L.D., Hudson B. // J. Chem. Phys. 1981. V. 74. P. 982.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия