

Координационная химия, 2022, T. 48, № 2, стр. 98-128
Функционализация аэрогелей координационными соединениями
Х. Э. Ёров 1, А. Е. Баранчиков 1, М. А. Кискин 1, А. А. Сидоров 1, В. К. Иванов 1, *
1 Институт общей и неорганической химии им. Н.С. Курнакова РАН
Москва, России
* E-mail: van@igic.ras.ru
Поступила в редакцию 02.06.2021
После доработки 20.06.2021
Принята к публикации 23.06.2021
- EDN: YMHHKP
- DOI: 10.31857/S0132344X22020013
Аннотация
Аэрогели представляют собой уникальные твердофазные материалы, характеризующиеся крайне высокой открытой пористостью и удельной поверхностью. Благодаря этим особенностям аэрогели широко используются в качестве тепло- и звукоизоляторов, газовых фильтров, сорбентов и др. Кроме того, аэрогели являются перспективными матрицами для создания селективных катализаторов, газовых сенсоров, люминофоров и т.д. Методы и подходы координационной химии открывают принципиально новые возможности для конструирования новых гибридных материалов, характеризующихся сочетанием практически важных физико-химических свойств, присущих аэрогелям и комплексам металлов. Модификация аэрогелей координационными соединениями позволяет получать полифункциональные материалы, обладающие высокой пористостью и удельной поверхностью в сочетании с превосходными каталитическими, фотокаталитическими, магнитными, люминесцентными и другими свойствами. В настоящем обзоре впервые обобщены и проанализированы подходы координационной химии, обеспечивающие химическое связывание координационных соединений с матрицей аэрогеля и позволяющие получать аэрогели с новыми функциональными свойствами. Рассмотрены известные примеры успешной реализации указанных подходов, в том числе предложенные ранее авторами обзора, а также приведены сведения о практическом применении получаемых таким образом материалов.
Аэрогели представляют собой высокопористые твердофазные материалы с размерами пор преимущественно в диапазоне от 2 до 50 нм. Их структура, как правило, образована соединенными друг с другом изотропными (с размером менее 10 нм) или анизотропными наночастицами или полимерными молекулами [1]. С момента получения Стивеном Кистлером первого аэрогеля в 1931 г. [2], синтезировано и изучено множество аэрогелей различных типов как на основе неорганических соединений [3, 4], так и на основе полимеров [5–7].
Аэрогели на основе оксидов металлов традиционно используются в качестве конструкционных материалов, однако, вследствие присущей им высокой термической стабильности и значительной открытой пористости они также могут выступать в качестве носителей при получении функциональных материалов. В последние годы активно проводятся исследования гибридных органо-неорганических материалов на основе аэрогелей [8]. Среди модификаторов, пригодных для функционализации аэрогелей, особое место занимают координационные соединения, обладающие широким спектром практически значимых свойств. Модифицированные аэрогели могут быть использованы в качестве катализаторов, газовых сенсоров, детекторов Черенкова и сцинтилляторов [1]; закрепленные в матрице аэрогелей каталитически активные металлокомплексы сочетают в себе преимущества гомогенных и гетерогенных катализаторов [9].
Наиболее распространенным в настоящее время подходом к получению металлооксидных аэрогелей, модифицированных координационными соединениями, является физическое связывание металлокомплексов с матрицей за счет их захвата формирующимся гелем, либо в результате их внедрения в сетку уже сформированного геля. Несмотря на простоту реализации, данный подход имеет ряд существенных недостатков, включая неравномерность распределения комплекса по объему материала, блокировку пор матрицы, частичное разрушение пористой структуры геля за счет действия капиллярных сил и др. Перечисленные проблемы могут быть в значительной степени решены с применением подходов, основанных на химическом (ковалентном) связывании координационных соединений с матрицей аэрогеля. К настоящему времени успешная реализация этих подходов продемонстрирована на достаточно большом круге систем, при этом разработка новых эффективных подходов к получению функциональных материалов на основе аэрогелей, химически модифицированных координационными соединениями, остается актуальной задачей, требующей использования подходов координационной и неорганической химии, а также неорганического материаловедения.
Настоящий обзор посвящен рассмотрению основных подходов к получению функционализированных металлооксидных аэрогелей с использованием приемов координационной химии.
ИСПОЛЬЗОВАНИЕ ПОДХОДОВ КООРДИНАЦИОННОЙ ХИМИИ ДЛЯ ПОЛУЧЕНИЯ ФУНКЦИОНАЛЬНЫХ МАТЕРИАЛОВ НА ОСНОВЕ АЭРОГЕЛЕЙ
Методы получения аэрогелей
Процедура получения аэрогелей обычно включает в себя три этапа: формирование золя, образование лиогеля в результате золь-гель перехода и последующее удаление жидкости из пор лиогеля, обычно реализуемое посредством сверхкритической сушки.
В классическом способе синтеза аэрогелей SiO2, впервые использованном Кистлером в 1931 г. [10], прекурсором являлся силикат натрия, гидролиз которого в присутствии соляной кислоты приводил к образованию гидрогеля гидратированного диоксида кремния. Промывка лиогеля от растворимых неорганических солей, замена воды на алифатический спирт с последующей сверхкритической (СК) сушкой позволяла получить монолитные аэрогели на основе SiO2. Невозможность получения аэрогелей сушкой в сверхкритической воде (Tкрит = 374°С, Ркрит = 22.1 МПа) вызвана ее высокой реакционной способностью, в этих условиях вызывающей полную пептизацию геля [11].
Одна из основных причин, по которым интерес к материалам на основе аэрогелей на протяжении длительного времени (около 30 лет) был невысок, заключался в том, что метод Кистлера требовал значительной продолжительности синтеза (около 7 сут). Использование алкоксидов металлов и металлоидов в качестве прекурсоров позволило значительно сократить продолжительность синтеза за счет устранения стадии замены растворителя [12].
В настоящее время для получения аэрогелей на основе оксидов металлов используют два основных метода: алкоксидный, включающий в себя синтез лиогелей путем гидролиза и поликонденсации соответствующих алкоксидов [4], и эпоксидный, включающий в себя синтез лиогелей путем гидролиза солей металлов в присутствии органических эпоксидов [13]. Отметим, что подходы координационной химии достаточно широко применяются в золь-гель технологии. Например, такие лиганды как этилендиаминтетрауксусная кислота, лимонная кислота и др. используются для подавления гидролиза и предотвращения формирования осадков гидроксидов (гидратированных оксидов) металлов вместо гелей. Широко известен и метод Печини, где цитратные комплексы металлов находятся в связанном виде в полимере, образованном за счет поликонденсации молекул этиленгликоля и лимонной кислоты. При этом формируется полимерная ковалентная сетка, в которой равномерно распределены ионы металла [14].
Синтез аэрогелей с использованием алкоксидов является наиболее традиционным подходом в золь-гель технологии. Он основан на гидролизе алкоксидов металлов или металлоидов в присутствии кислот или оснований с последующей конденсацией образующихся гидроксосоединений и сшивкой образующихся кластеров с формированием частиц золя [14, 15]. В дальнейшем частицы золя объединяются с образованием трехмерной сетки лиогеля [4, 16].
Структура лиогелей в значительной степени зависит от соотношения скоростей гидролиза алкоксидов и конденсации их гидроксопроизводных. При проведении гидролиза в кислой среде за счет быстрого протекания соответствующих реакций одновременно образуется огромное число гидроксопроизводных в виде мономерных частиц или короткоцепочечных олигомеров, которые впоследствии формируют лиогель с полимероподобной структурой. В щелочной среде, напротив, из-за высокой скорости конденсации и относительно медленного протекания гидролиза формирующаяся трехмерная структура характеризуется крупными частицами и большими размерами пор [15].
Для получения аэрогелей на основе SiO2 чаще всего используют Si(OCH3)4 (ТМОС) [17] или Si(OC2H5)4 (ТЭОС) [18]. В отдельных случаях в качестве прекурсоров применяют и другие алкоксиды, такие как полиэтоксидисилоксан [19] и метилтриметоксисилан [20, 21]. Использование различных прекурсоров приводит к расширению диапазона свойств получаемых аэрогелей на основе SiO2. В частности, использование метилтриметоксисилана позволяет получать эластичные и гидрофобные аэрогели [22].
Большинство алкоксидов металлов более склонны к гидролизу по сравнению с алкоксидами кремния (Si(OR)4). Так, константа скорости гидролиза Ti(OEt)4 (kh = 10–3 M–1 с–1) на 5 порядков больше, чем константа скорости гидролиза Si(OEt)4 (kh = 5 × 10–9 M–1 с–1) [23]. Это обусловлено двумя основными причинами. Первая заключается в более низкой электроотрицательности металлов по сравнению с Si, что приводит к бóльшему частичному заряду (δM) на атоме металла. Вторая причина – ненасыщенность координационного числа (КЧ) металлов в алкоксидах [24]. Скорость гидролиза для многих алкоксидов металлов настолько велика, что при их взаимодействии с водой происходит мгновенное образование осадка. Основным подходом к уменьшению скорости гидролиза алкоксидов металлов является введение в реакционную смесь лигандов, которые координируют металл, повышая его КЧ. В качестве таких лигандов часто используют уксусную кислоту (ацетат-анион) или ацетилацетон [25]. В частности, взаимодействием Ti(OiPr)4 с ацетилацетоном может быть получен комплекс состава [Ti(OiPr)3(Аcac)], а с уксусной кислотой – [Ti(OiPr)3(OAc)]2 [24].
Описанный подход позволяет получать лиогели как на основе индивидуальных металлооксидов, так и на основе смешанных (двойных и тройных) оксидов. В табл. 1 приведены примеры металлооксидных аэрогелей на основе как индивидуальных, так и смешанных оксидов, синтезируемых с применением алкоксидов в качестве прекурсоров [26–42].
Таблица 1.
Примеры аэрогелей на основе индивидуальных и смешанных оксидов, полученных с использованием алкоксидов в качестве прекурсоров
| Аэрогель | Прекурсоры | Литература |
|---|---|---|
| Индивидуальные | ||
| Al2O3 | Al(OBus)3 + Еtac | [26] |
| Fe2O3 | Fe(Аcac)3 | [27] |
| TiO2 | Ti(OiPr)4 | [35] |
| SnO2 | [Sn(OBun)4] + Аcac | [36] |
| ZrO2 | Zr(OiPr)4 + Аcac | [37] |
| V2O5 | VO(OiPr)3 | [38] |
| Nb2O5 | Nb(OEt)5 | [39] |
| Смешанные | ||
| TiO2–SiO2 | Ti(OiPr)4 + Si(OEt)4 | [40] |
| Al2O3–SiO2 | Al(OBus)3 + Si(OMe)4 | [41] |
| V2O5–MgO | VO(OiPr)3 + Mg(OMe)2 | [28, 42] |
| PbO–Al2O3 | Al(OBus)3 + Pb(OAc)2 | [29] |
| PbO–ZrO2 | Zr(OiPr)4 + Pb(OAc)2 | [30] |
| BaO–Al2O3 | Ba[Al(OBus)4]2 + Al(OBus)3 | [31] |
| PbTiO3 | Ti(OiPr)4 + Pb(OAc)2 | [32] |
| Li2O–B2O3–Al2O3 | LiOMe + B(OBun)3 + Al(OBun)3 | [33] |
| MgO–Al2O3–SiO2 | Al(OBus)3 + Si(OEt)4 + Mg(OAc)2 | [34] |
Несмотря на многочисленные преимущества алкоксидов, они являются относительно дорогостоящими прекурсорами, а необходимость их дополнительной химической модификации в конечном итоге приводит к увеличению стоимости получаемых аэрогелей, что ограничивает использование этих прекурсоров на практике. Другой существенный недостаток алкоксидного метода заключается в том, что не для всех металлов можно получить соответствующие алкоксиды. В связи с этим были разработаны альтернативные подходы к получению золь-гель материалов [43]. Среди них наиболее удачным оказался использование растворов неорганических солей металлов и органических эпоксидов, выступающих в качестве гелеобразующего агента [13].
Использовать органические эпоксиды в золь-гель технологии металлооксидов впервые предложили Ито и соавт. в 1993 г. [44]. Однако настоящую популярность этот метод приобрел благодаря работе Александра Гэша [13]. Существенным преимуществом эпоксидного метода является возможность использования в качестве прекурсоров растворов неорганических солей металлов в воде, спирте или других полярных протонных растворителях.
В эпоксид-индуцированном гелеобразовании органические эпоксиды выступают в качестве инициаторов реакций гидролиза и конденсации, связывая протоны в золь-гель системе [45]. Данный процесс включает протонирование атома кислорода в эпоксиде с последующим раскрытием эпоксидного цикла через нуклеофильную атаку со стороны сопряженного основания. Например, введение эпоксида в раствор, содержащий аква-ионы [Fe(H2O)6]3+, приводит к формированию аквагидроксокомплекса железа, при этом необратимый захват протонов эпоксидом приводит к повышению pH раствора [46]. В отличие от других оснований (OH–, ${\text{CO}}_{{\text{3}}}^{{{\text{2}}--}}$ или NH3), использование эпоксидов приводит к относительно медленному увеличению pH в системе, вследствие чего происходит образование стабильных золей оксидов металлов и, в конечном итоге, монолитного оксидного лиогеля.
В эпоксидном золь-гель методе такие параметры синтеза, как тип эпоксида, природа растворителя и тип аниона в соли металла, оказывают значительное влияние на структуру и свойства получаемого аэрогеля [46, 47]. Варьируя природу эпоксида, можно в широких пределах (до 4 порядков величины) изменять длительность гелеобразования при золь-гель переходе [48].
Эпоксид-индуцированный золь-гель метод был использован при получении аэрогелей на основе различных оксидов металлов [47], например диоксида олова, поверхность которого характеризуется суперкислотными свойствами, что делает данный материал перспективным для каталитической конверсии углеводородов [49]. Одним из ключевых преимуществ эпоксидного золь-гель подхода является то, что он обеспечивает универсальный и относительно простой способ получения смешанных (бинарных или тройных) оксидов, в том числе SiO2, оксидов редкоземельных элементов и др. [50]. Данный подход также широко используется при получении композитных аэрогелей, например, содержащих металлические частицы [51, 52]. В табл. 2 приведены избранные примеры работ по получению аэрогелей на основе как индивидуальных, так и смешанных оксидов металлов эпоксидным золь-гель методом [13, 53–69]. Необходимо отметить, что эпоксидный подход уникален в том отношении, что он позволяет получать аэрогели на основе оксидов двухзарядных катионов, в том числе смешанные.
Таблица 2.
Примеры аэрогелей на основе индивидуальных и смешанных оксидов металлов, полученных с использованием эпоксидного метода
| Аэрогель | Прекурсоры | Литература | ||
|---|---|---|---|---|
| соль | эпоксид | растворитель | ||
| Индивидуальные оксиды | ||||
| Al2O3 | AlCl3 · 6H2O, Al(NO3)3 · 9H2O | PO* | EtOH | [53] |
| SnO2 | SnCl4 · 5H2O | PO | H2O | [62] |
| Ga2O3 | Ga(NO3)3 · xH2O | PO | EtOH | [13] |
| In2O3 | In(NO3)3 · 5H2O | PO | EtOH | [13] |
| Fe2O3 | FeCl3 · 6H2O | Эпихлоргидрин | EtOH | [63] |
| Cr2O3 | Cr(NO3)3 · 9H2O | PO | EtOH | [13] |
| Ru2O3 | RuCl3 · xH2O | PO, 1,2-эпоксибутан | EtOH, MeOH | [64] |
| TiO2 | TiCl4 | PO | EtOH | [65] |
| ZrO2 | ZrOCl2 | PO | EtOH | [66] |
| CuO | CuCl2 · 2H2O | PO | iPrOH | [67] |
| ZnO | Zn(NO3)2 · 6H2O | PO | MeOH, EtOH, iPrOH, ацетон | [68] |
| NiO | NiCl2 · 6H2O | PO | EtOH | [69] |
| CoO | CoCl2 · 6H2O, Co(NO3)2 · 6H2O | PO | MeOH | [54] |
| Ce2O3 | CeCl3 · 6H2O | PO | MeOH | [55] |
| Gd2O3 | GdCl3 · 6H2O, Gd(NO3)3 · 6H2O | PO | EtOH | [56] |
| UO3 | UO2(NO3)2 · 6H2O | PO | EtOH, ацетон | [57] |
| Смешанные оксиды |
||||
| CuO/NiO | CuCl2 · 2H2O + NiCl2 · 6H2O | PO | MeOH | [58] |
| Стабилизированный иттрием оксид циркония (YSZ) | ZrCl4 + YCl3 · 6H2O | PO | H2O | [59] |
| ZnFe2O4 | Zn(NO3)2 · 6H2O + Fe(NO3)3 · 9H2O | PO | i-PrOH | [60] |
| CuO/ZnO/Al2O3 | Cu(NO3)2 · 3H2O + Zn(NO3)2 · · 6H2O + Al(NO3)3 · 9H2O | PO | EtOH | [61] |
Для получения аэрогелей из лиогелей наиболее часто используют метод сверхкритической сушки, однако определенное распространение получили и другие методы – сублимационного обезвоживания и сушки при атмосферном давлении [70, 71]. Строго говоря, материалы, получаемые этими двумя методами (криогели и амбигели соответственно), не являются аэрогелями в классическом понимании, их следует отнести к аэрогелеподобным материалам.
Для сверхкритической сушки лиогелей наиболее часто используют органические растворители (как правило, низшие алифатические спирты) и СО2 [72]. В табл. 3 приведены критические параметры для наиболее часто используемых растворителей (данные для H2O приведены для сравнения) [72]. Учитывая различия в критических температурах, зачастую различают так называемые “низкотемпературную” (в CO2) и “высокотемпературную” сверхкритические сушки (в органических растворителях) [73].
Таблица 3.
Критические параметры некоторых растворителей, применяемых для СК-сушки [72]
| Растворитель | Ткрит, °С | Pкрит, бар |
|---|---|---|
| Вода | 374.1 | 220.5 |
| Метанол | 239.4 | 81 |
| Этанол | 243.0 | 63.8 |
| Изопропанол | 235.1 | 47.6 |
| Ацетон | 235.0 | 46.0 |
| Диоксид углерода | 31.0 | 73.8 |
| трет-Бутанол | 233 | 39.7 |
| Трифторэтанол | 226 | 48.4 |
| Гексафторизопропанол | 182 | 30.2 |
| Диэтиловый эфир | 193.4 | 36.1 |
| Метил-трет-бутиловый эфир | 224.1 | 34.3 |
| Этилацетат | 250 | 38.8 |
| Пентан | 196.6 | 34.0 |
Влияние растворителя, используемого при сушке в СК-условиях, на состав и свойства аэрогелей является новой областью исследований, работы в которой только начаты [73–75]. В цикле работ, опубликованных научными коллективами из Института общей и неорганической химии им. Н.С. Курнакова РАН и Института физиологически активных веществ РАН, был предложен целый ряд новых растворителей для сверхкритической сушки, характеризующийся широким диапазоном критических параметров, в том числе гексафторизопропанол, трифторэтанол, метил-трет-бутиловый и диэтиловый эфиры, этилацетат, гексан, ацетон [76–81]. Как правило, сушка в спиртах приводит к алкоксилированию поверхности аэрогеля вследствие взаимодействия спирта с поверхностными ОН-группами. В результате поверхность аэрогелей приобретает гидрофобные свойства [80]. Гидрофобные аэрогели на основе SiO2 могут быть получены сушкой в гексафтор-изопропаноле, либо за счет использования гексафторацетона в качестве сорастворителя при получении лиогелей [80, 82]. Сравнительный анализ характеристик аэрогелей SiO2, Al2O3, ZrO2, полученных СК-сушкой в среде простых эфиров и фторированных спиртов, показал, что как состав, так и текстурные характеристики получаемых аэрогелей существенным образом зависят от природы применяемого СК-растворителя [78].
Относительно недавно появились отдельные работы, посвященные сравнительному анализу структуры аэрогелей, получаемых сушкой в спиртах в сверхкритических и докритических условиях, при температурах от 85 до 265°С [83, 84]. Примечательно, что сушка во всем исследованном диапазоне температур приводит к получению материалов, характеризующихся высокой удельной поверхностью (∼1000 м2/г), при этом увеличение температуры сушки приводит к монотонному увеличению среднего размера и удельного объема пор. В то же время температура сушки оказывает существенное влияние на характер агрегации индивидуальных частиц SiO2, что отражается в уменьшении фрактальной размерности кластеров наночастиц (от 2.6 при 85°С до 2.1 при 265°С).
Применение походов координационной химии для получения многокомпонентных аэрогелей (на примере системы SiO2–TiO2)
Подходы координационной химии широко применяются при получении аэрогелей, ксерогелей, стекол и керамик на основе смешанных оксидов металлов [85–87]. Интерес к многокомпонентным аэрогелям возник благодаря тому, что эти материалы сочетают в себе ряд важных с практической точки зрения характеристик: высокую удельную поверхность, высокую пористость, каталитическую и фотокаталитическую активность и др. [88].
Среди смешанно-оксидных аэрогелей наиболее изученными являются материалы на основе SiO2–TiO2, что обусловлено их высокой практической значимостью с точки зрения использования в составе каталитических или фотокаталитических систем [89–96]. В связи с этим рассмотрим имеющиеся в литературе сведения, касающиеся синтеза аэрогелей на основе SiO2–TiO2, в первую очередь, с использованием подходов координационной химии.
В качестве прекурсоров при синтезе аэрогелей на основе SiO2–TiO2 обычно используют алкоксиды кремния и титана. В качестве источников кремния в основном используют ТМОС [74, 97] и ТЭОС [98, 99], и даже жидкое стекло (растворы силикатов натрия) [100, 101].
В качестве источника титана применяют тетраизопропоксид титана Ti(OiPr)4 (TИП) [97, 102], тетрабутоксид титана Ti(OnC4H9)4 [89, 93] или более доступные тетрахлорид титана [103] или оксохлорид титана [104].
Необходимым условием для получения многокомпонентных аэрогелей и ксерогелей золь-гель методом является достижение высокой степени кросс-полимеризации в ходе гидролиза и поликонденсации различных алкоксидов [105, 106]. В наиболее простом случае для получения двухкомпонентных гелей (на основе SiО2 и оксида металла (М)) алкоксиды гидролизуют в органическом растворителе. После гидролиза алкоксидов должно происходить образование гетеросилоксановых групп (Si–O–M); на практике скорости гидролиза и поликонденсации различных алкоксидов сильно различаются, и это приводит к независимой поликонденсации отдельных компонентов системы и к разделению фаз. Для получения химически гомогенных материалов используют различные подходы [89, 107–109], при этом ни один из них не является универсальным. Для получения смешанных аэрогелей SiO2–TiO2, как правило, применяют следующие синтетические процедуры.
1. Предварительный гидролиз одного из компонентов [89, 90, 110]. Алкоксид, характеризующийся меньшей склонностью к гидролизу (обычно это Si(OR)4), предварительно гидролизуют с использованием минимального количества воды, а потом в реакционную систему добавляют второй алкоксид, характеризующийся более высокой склонностью к гидролизу. При этом за счет отсутствия в системе свободной воды, реакционноспособный алкоксид будет вступать в реакцию конденсации с группами ≡Si–OH с образованием связей Si–O–M.
2. Хелатирование быстрогидролизующегося алкоксида [88, 90, 91, 93, 111]. К более реакционноспособному алкоксиду добавляют хелатирующий лиганд, в результате чего образуется относительно устойчивый к гидролизу комплекс. В результате происходит выравнивание скоростей гидролиза и конденсации алкоксидов, что способствует высокой степени кросс-полимеризации компонентов и получению материалов с высоким содержанием гетеросилоксановых групп. Известным примером реализации этого подхода является использование ацетилацетона (AcacH) или уксусной кислоты (AcOH) при получении смешанно-оксидных аэрогелей SiO2–TiO2.
В зависимости от мольного отношения возможно образование двух комплексов титана с ацетилацетоном (схема 1 ), которые, очевидно, обладают различной реакционной способностью, при этом гидролиз этих комплексов протекает в первую очередь по изопропоксильным группам, не затрагивая ацетилацетонатные лиганды, которые сохраняются даже при двадцатикратном избытке воды в реакционной смеси [112]. Продукты, получаемые при добавлении различного количества ацетилацетона к ТИП [113] представлены на схеме 1 .
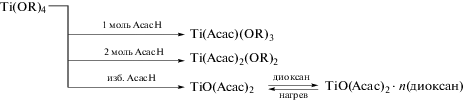
Схема 1 .
Таким образом, у комплекса Ti(Аcac)(OR)3 способны гидролизоваться три группы с формированием трехмерной сетки геля, а у комплекса Ti(Аcac)2(OR)2 [113] – только две с образованием цепочечного полимера. Имеются также сведения о возможности формирования комплекса [O=Ti(Аcac)2] в избытке ацетилацетона, однако информации о его свойствах обнаружить не удалось.
Подробное исследование состава комплекса, образующегося в эквимолярных смесях ТИП и AcacH, проведено Лостик и соавт. с использованием методов ЯМР 1H и 13C [114]. Было показано формирование пятикоординированного трис(изопропокси)ацетилацетонатотитана [Ti(Acac)-(iPrO)3], для которого обнаружена неэквивалентность атомов кислорода в ацетилацетатном лиганде (что вызвано кето-енольной таутомеризацией) и изопропоксидных групп. Необходимо подчеркнуть, что, в отличие от аналогичных этоксидных комплексов титана, изопропоксидный комплекс существует исключительно в виде мономеров.
Комплекс бис-(ацетилацетонато)-бис-(изопропокси) титана [Ti(Аcac)2(iPrO)2] был изучен Бредли [115]. Эрингтоном было показано, что данный комплекс может превращаться в димерную форму [116] с мостиковыми изопропоксидными лигандами (рис. 1). Синтез дисперсных материалов на основе SiO2–TiO2 с использованием данного комплекса описан в [117].
Рис. 1.
Перегруппировка комплекса [Ti(iPrO)2(Acac)2] в его димерную форму (а) и молекулярная структура этой формы (б) [116].

Попыток систематического анализа влияния количества вводимого в реакционную смесь ацетилацетона на свойства получаемых титансодержащих оксидных аэрогелей до настоящего времени практически не было. Накагава исследовал закономерности синтеза фотокатализаторов на основе TiO2 путем гидролиза алкоксидов титана в присутствии различного количества ацетилацетона [118]. Порошки, полученные в отсутствие ацетилацетона, состояли исключительно из изотропных наночастиц, тогда как в присутствии АcacH формировались нановолокна TiO2. При использовании мольного соотношения АcacH : ТИП = 1 : 2 образовывались более длинные и тонкие волокна, чем при использовании соотношения 1 : 1. Аналогичные результаты были получены Садехзаде с соавт. [119]. Попытки получения оксидных материалов сложного состава с использованием ацетилацетонатных комплексов титана производились лишь в единичных работах [120].
Помимо ацетилацетона, в качестве хелатирующих агентов при золь-гель синтезе используют этиленгликоль, его моноалкиловые эфиры – метил- и этилцеллозольвы, глицерин [121–123]. При взаимодействии этих соединений с алкоксидами титана происходит замещение части монодентатных алкоксидных лигандов на бидентатные производные многоатомных спиртов. Гидролиз алкоголятов титана, хелатированных целлозольвами, с последующей сверхкритической сушкой лиогелей позволил получить оптически прозрачные монолитные аэрогели диоксида титана [121].
Достаточно перспективными выглядят подходы, основанные на использовании в качестве прекурсоров гетероядерных координационных соединений, например диэтоксисилоксанэтилтитаната ([Si(OEt)2OTi(OEt)2O]) [124], а также комплексов изопропоксида титана Ti(OiPr)4 с 3-(пропилтриметоксисилил)ацетилацетоном (схема 2 , соединения 1 и 2) [125]. Недостатком использования таких подходов является строго фиксированная стехиометрия комплексов и относительно высокая стоимость прекурсоров. Синтез гетероядерных прекурсоров для получения аэрогелей на основе SiO2–TiO2 представлен на схеме 2 [125].
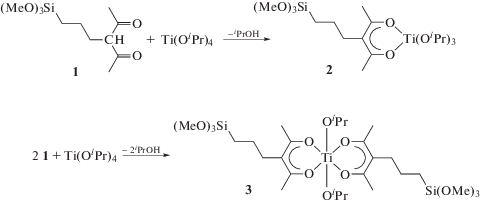
Схема 2 .
Функционализация аэрогелей с использованием подходов координационной химии
Широкое разнообразие функциональных свойств координационных соединений делают их привлекательными для использования в качестве модификаторов при функционализации аэрогелей. Иммобилизованные металлокомплексы интенсивно исследуются начиная с 1960-х гг. [126]. Закрепление изолированных и равномерно распределенных молекул комплекса на поверхности аэрогеля позволяет получать материалы, сочетающие в себе специфические свойства матрицы (прочность, пористость, высокая удельная поверхность и т.п.) с функциональными свойствами молекулярного комплекса [9, 127, 128]. При этом использование низкотемпературной сверхкритической сушки в СО2 (Tкр = 31°С), позволяет вводить в матрицу аэрогеля даже неустойчивые при высоких температурах молекулы [129].
Закрепление металлокомплексов в матрице аэрогеля позволяет осуществить гетерогенизацию гомогенных катализаторов, т.е. получить катализаторы, сочетающие в себе активность гомогенного катализатора вместе со всеми преимуществами гетерогенного катализатора [9, 130–137]. Иммобилизация люминесцентных комплексов в матрице аэрогелей и ксерогелей приводит к получению материалов, применяемых в качестве сенсоров [129], при создании дисплеев [138], сцинтилляторов [139], лазеров [140] и детекторов Черенкова [141].
В качестве пористых материалов-носителей используют как органические полимеры [142, 143], так и неорганические соединения, в первую очередь оксиды [144], получаемые с применением золь-гель технологии. Вследствие большей термостойкости (которая особенно необходима при использовании в катализе) оксидные носители применяются чаще [145]; в свою очередь, среди оксидов наиболее широко распространены диоксид кремния и оксид алюминия [1, 9, 146]. Это связано в первую очередь с тем, что материалы на основе этих оксидов сохраняют высокую пористость при высоких температурах. Кроме того, золь-гель химия SiO2 и Al2O3 достаточно хорошо изучена [1, 3, 147].
Функционализация пористых оксидных носителей осуществлятется двумя основными способами – на стадии золь-гель синтеза [9, 137] или при постсинтетической обработке предварительно полученного оксидного материала [148, 149]. К постсинтетическим относятся такие подходы как ионный обмен, пропитка, осаждение из жидкой и из газовой фазы [150]. Несмотря на кажущуюся простоту проведения постмодификации, она характеризуется рядом существенных недостатков: сложность контроля за количеством внедряемого модификатора, блокировка пор, частичное разрушение пористой структуры, многостадийность процесса и др. [150, 151]. Перечисленные сложности частично или полностью решаются при функционализации оксидного материала в процессе золь-гель перехода, например при согелировании.
Наиболее перспективным представляется подход химической иммобилизации модификатора в матрице носителя. Этот подход имеет существенные преимущества по сравнению с более традиционными. К этим преимуществам можно отнести равномерное распределение модификатора, отсутствие его агрегации и вымывания, сохранение стабильности, большая и контролируемая загрузка и др. [150].
Модификация аэрогелей на основе SiO2: основные синтетические подходы. Для химического закрепления различных молекул и групп в матрице SiO2 используется прекурсоры типа (RO)nSi–X–A, где A представляет собой функциональную органическую группу, X – гидролитически стабильный спейсер (обычно углеводородный), связывающий A и алкоксидный фрагмент (RO)nSi (схема 3 ) [152]. Следует отметить, что ключевым моментом, позволяющим значительно расширить спектр получаемых гибридных органо-неорганических функциональных материалов на основе SiO2, является тот факт, что кремний образует устойчивую связь Si–C. Благодаря этому, для получения функционализированных золь-гель материалов можно использовать различные методы органической химии. Алкоксигруппы позволяют легко закрепить синтезированные молекулы на оксидных носителях, тем самым формируя гибридный материал с требуемыми функциональными свойствами. Примеры органозамещенных алкоксисиланов, используемых в золь-гель технологии [152], представлены на схеме 3 .

Схема 3 .
Прекурсоры типа (RO)3Si(CH2)nL (L = донорная группа) могут выступать в качестве лиганда при получении координационных соединений. Получаемые из них комплексы [(RO)3Si(CH2)nL]nMXm могут быть закреплены в матрице SiO2 – либо путем постобработки SiO2, либо путем согелирования с тетраалкоксидами кремния Si(OR)4. В качестве функциональной группы L для координации катионов металлов обычно выступают группы: ‒NH2, –NHCH2CH2NH2, –CN, –SH, CH(COMe)2 и др. [153].
В целом химическая иммобилизация комплексов металлов в матрице SiO2 может быть реализована с использованием одного из четырех основных методов (схема 4 ) [154].
1. Функционализированный алкоксисиланом лиганд сначала закрепляют на гидратированной поверхности SiO2. Затем модифицированный SiO2 реагирует с металлсодержащим прекурсором (обычно солью металла), приводя к закреплению комплекса на поверхности SiO2 [155–159].
2. Сначала синтезируют комплекс металла с алкоксисилансодержащим лигандом, после чего его смешивают с предварительно полученным SiO2, тем самым обеспечивая ковалентное связывание комплекса с поверхностными группами Si–OH [9, 160, 161].
3. Алкоксисилансодержащий лиганд согелируют с тетраалкоксисиланом Si(OR)4, в результате чего лиганд распределяется равномерно по всему объему материала; затем к нему добавляют металлсодержащий прекурсор с целью связывания последнего с поверхностными лигандами [160, 162].
4. Подход представляет собой видоизмененный аналог метода 2, где синтезированный предварительно комплекс металла с алкоксисилансодержащим лигандом согелируют с Si(OR)4 [131, 133, 134, 163–168]. При синтезе гибридных аэрогелей на основе SiO2 чаще всего используется именно этот подход. Заметим, что на практике возможны два способа реализации подхода 4. Первый – когда сначала выделяют комплекс [(R'O)3Si–X–A]yMLm, а затем согелируют его с Si(OR)4; второй – когда одновременно согелируют смесь лиганда (R'O)3Si–X–A, раствора Mn+ и Si(OR)4. Первый способ реализован в [166], второй – в [165, 167].
Четыре основных методов иммобилизации металлокомплексов в матрице SiO2 [154] представлены на схеме 4 .

Схема 4 .
Методы 1 и 2 являются классическими методами закрепления комплексов металлов на поверхности материалов, тогда как методы 3 и 4 представляют собой пример использования золь-гель технологии для получения материалов, содержащих комплексы металлов, равномерно распределенных по всему объему матрицы SiO2.
Выбор способа модификации оказывает значительное влияние на стабильность и функциональные свойства получаемых гибридных материалов. В частности, установлено, что использование метода 4 позволяет получить каталитические материалы (в частности, комплексы родия, иммобилизованные в матрице SiO2) с более высокой активностью, чем при использовании иных методов [169, 170]. Напротив, Чапка и соавт. [171] показали, что катализаторы, полученные закреплением родиевого комплекса в матрице SiO2 по методу 4, оказались менее активными по сравнению с полученными по методу 2. Наблюдаемый эффект, предположительно обусловлен недоступностью отдельных каталитически активных центров в материалах, полученных методом 4.
При иммобилизации металлокомплексов с применением органозамещенных алкоксисиланов наиболее сложными и трудоемкими этапами являются синтез и очистка алкоксисилансодержащего лиганда, а также соответствующего металлокомплекса. Следует отметить, что не всегда удается получить монокристаллы комплекса [(R'O)3Si–X–A]yMLm, чтобы охарактеризовать его структуру детально. Зачастую подобные комплексы вообще не кристаллизуются и выделяются в виде маслянистой жидкости [172]. Последующий золь-гель синтез с получением металлокомплекса, иммобилизованного в матрице SiO2, является относительно тривиальной задачей. Тем не менее выбор условий золь-гель синтеза должен быть тщательно продуман, так как формирующаяся пористая структура конечного материала, зависящая от параметров синтеза, определяет функциональные свойства модифицированного аэрогеля.
Золь-гель технологии обеспечивают гомогенизацию прекурсоров на молекулярном уровне. Однако, несмотря на то что в начале реакции реагенты действительно оказываются распределенными равномерно, если различия в скоростях поликонденсации прекурсоров достаточно велики, результатом синтеза может стать гетерогенный материал. Такое фазовое разделение, очевидно, будет существенно сказываться на конечных свойствах материала [152].
Среди материалов на основе SiO2, используемых в качестве матрицы для иммобилизации металлокомплексов, аэрогели занимают особое место. Высокие мезопористость и удельная площадь поверхности, характерные для аэрогелей SiO2, дают возможность получить материалы, в которых молекулы металлокомплекса стохастически распределены по объему матрицы и изолированы друг от друга в открытой пористой структуре, и, таким образом, легкодоступны [134, 166, 167].
Впервые аэрогели SiO2, содержащие химически закрепленные комплексы, были получены Хейнрикс и соавт. в 1997 г. [168]. В качестве образцов сравнения были также синтезированы соответствующие ксерогели. Схемы реакций, протекающих при иммобилизации комплекса Pd(II) в матрице аэрогеля SiO2: согелирование (а) и пост-модификация (б) [168] представлены на схеме 5 (см. также схему 4 , методы 2 и 4). Порошок Pd(Аcac)2 суспендировали в этаноле, после чего к нему добавляли 3-(2-аминоэтил)аминопропилтриметоксисилан. Суспензию перемешивали при комнатной температуре до образования прозрачного желтого раствора (цвет, характерный для комплекса палладия с аминами). Затем полученный раствор комплекса согелировали с Si(OC2H5)4 (схема 5 а). Лиогели были подвергнуты сушке в CК-этаноле при 327°С и 12 МПа.

Схема 5 .
Для проведения постмодификации сначала синтезировали аэрогель SiO2, после чего отжигали его при 400°С на воздухе. Далее отожженный аэрогель помещали в раствор комплекса (схема 5 б). После пропитки аэрогель снова высушивали при 300°С на воздухе. СК-сушка в этаноле приводила к восстановлению Pd2+ до Pd0 с размерами частиц ~2.2 нм. При этом в образцах, полученных согелированием, кристаллиты Pd при отжиге агрегируют в существенно меньшей степени. Отметим, что восстановление металлокомплекса в процесс СК-сушки в спиртах наблюдалось и в других работах [173].
N-донорные лиганды, такие как (3-аминопропил)триметоксисилан (APTMS) и 3-(2-аминоэтил)аминопропилтриметоксисилан (EDTMS), наиболее часто применяются при координировании переходных металлов, поскольку эти металлы образуют с ним прочные комплексы. Например, с использованием EDTMS в качестве лиганда в матрицу SiO2 были успешно иммобилизованы ионы Ni2+ и Pd2+ [174], Pt2+ [163], Fe3+ [131].
Аналогичный подход, основанный на согелировании Si(OCH3)4 и комплекса, полученного взаимодействием пивалата меди(II) с АРТМS, был использован при получении медьсодержащих аэрогелей [175]. При этом сушка в СК-CO2 позволяла синтезировать аэрогели, содержащие химически иммобилизованный комплекс меди(II), а сушка в сверхкритических спиртах (изопропанол, метанол) приводила к восстановлению двухвалентной меди до металлического состояния.
Модифицируя аминозамещенные алкоксисиланы, можно получать новые лиганды и комплексы на их основе с требуемыми характеристиками. Например, Мёрфи и соавт. [167] для закрепления в гелях комплексов Co(II) и Cu(II) с N,N'-этилен-бис(салицилиденимин)ами (Salen) – основаниями Шиффа – проводили реакцию между салициловым альдегидом и (3-аминопропил)триэтоксисиланом в растворе солей указанных металлов. Последующее согелирование с ТЭОС приводило к получению гибридного лиогеля. На схеме 6 представлены синтез и иммобилизация комплексов [M(Salen)] в матрице аэрогеля SiO2 (M = Co(II), Cu(II) (а) [167] и синтез нового лиганда (1), соответствующего рутениевого комплекса (2) и иммобилизация последнего в аэрогель SiO2 (б) [166].

Схема 6 .
Сечкин и соавт. [166] для получения аэрогеля с иммобилизованным металлокомплексом сначала получили 3-(4,5-дигидроимидазол-1-ил)пропилтриэтоксисилан из 3-(2-аминоэтил) аминопропилтриэтоксисилана (1 в схеме 6 б). Дальнейшая реакция этого лиганда 1 с комплексом рутения [RuCl2(p-Me2CHC6H4Me)] (см. схему 6 б) приводила к получению комплекса 2 (схема 6 б). В результате гидролиза и согелирования комплекса 2 с ТЭОС происходило закрепление комплекса в матрице аэрогеля (4 в схема 6 б).
Известны работы, в которых для введения в матрицу SiO2 комплекс металла предварительно модифицируют лигандами с концевыми силанольными группами, после чего согелируют его с ТЭОС [133, 134]. На схеме 7 представлены исходный фосфиновый комплекс рутения (1), комплекс, модифицированный органотриалкоксисиланом (2), и аэрогель с иммобилизованным комплексом (3a) [134].
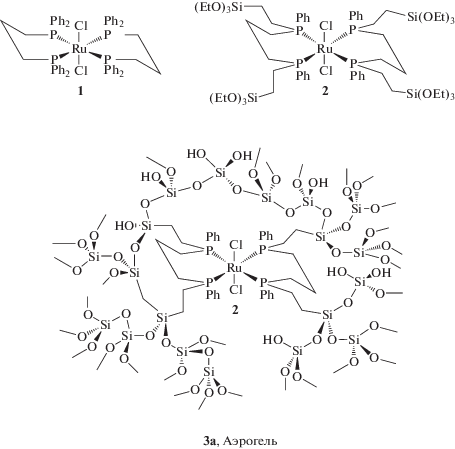
Схема 7 .
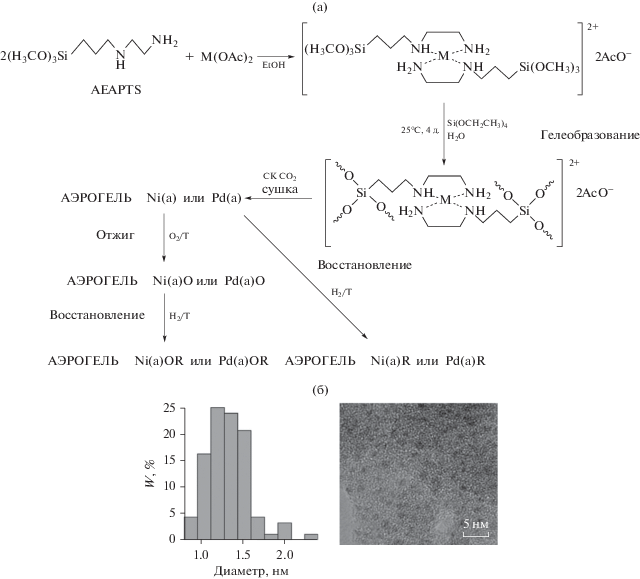
Постсинтетическая обработка иммобилизованных металлокомплексов позволяет получать оксидные или металлические наночастицы, однородно распределенные по поверхности аэрогеля (рис. 2) [174].
Рис. 2.
Схема иммобилизации комплексов Ni(II) или Pd(II) в матрице аэрогеля SiO2 и постсинтетической обработки полученных образцов (а), распределение наночастиц Pd по размерам согласно данным просвечивающей электронной микроскопии [174] (б).
Помимо N-донорных лигандов, для модификации кремнийоксидных аэрогелей широко применяются и O-донорные лиганды. Так, Шуберт для закрепления в матрице аэрогеля соединений переходных металлов (Ti и Zr) использовал прекурсоры на основе β-дикетонзамещенных алкоксисиланов [163]. Синтез таких прекурсоров для получения функционализированных аэрогелей на основе SiO2 представлен на схеме 8 .

Схема 8 .
Таким образом, варьируя лиганд L и металл M в координационных соединениях состава [(RO)3Si(CH2)nL]nMXm, можно иммобилизовать комплексы практически любых металлов в матрице SiO2, в том числе в виде аэрогеля. Сочетание возможностей золь-гель технологии и координационной химии позволяет в широких пределах варьировать синтетические параметры и тем самым получать как гибридные аэрогели на основе SiO2 с закрепленными металлокомплексами, так и смешанно-оксидные аэрогели с разнообразными функциональными свойствами.
Химическая модификация аэрогелей на основе Al2O3: основные синтетические подходы. Основными способами модификации аэрогелей Al2O3, описанными в литературе, являются согелирование гидроксосоединений алюминия с раствором/суспензией модификатора и пропитка лиогеля или аэрогеля гидратированного оксида алюминия либо раствором, либо парами модификатора. В табл. 4 представлены основные существующие синтетические подходы к модификации аэрогелей на основе Al2O3 [31, 146, 176–236 ].
Таблица 4.
Основные синтетические подходы, используемые для модификации аэрогелей Al2O3
| Подход | Исходные соединения алюминия и модификатора* | Литература |
|---|---|---|
| Модифицирование в ходе формирования лиогеля | Алкоксид алюминия и соль модификатора | [176–180, 186, 197, 208, 219, 230, 234–236] |
| Соль алюминия и соль модификатора | [181–185, 187–191] | |
| Алкоксид алюминия и алкоксид модификатора | [192–196, 198] | |
| Смешанный алкоксид алюминия и модификатора | [31, 199, 200] | |
| Золь гидратированного оксида алюминия и золь гидратированного оксида модификатора | [201–206] | |
| Золь гидратированного оксида алюминия и соль модификатора | [196, 198, 202, 207, 209–218] | |
| Пропитка лиогеля или аэрогеля гидратированного оксида алюминия | Лиогель гидратированного оксида алюминия и раствор соединения модификатора | [146, 194, 220–222] |
| Аэрогель гидратированного оксида алюминия и раствор соединения модификатора | [184, 186, 196, 198, 212, 214, 218, 223–226, 230, 235] | |
| Аэрогель гидратированного оксида алюминия и газообразное соединение модификатора | [227, 228] | |
| Введение хелатирующих лигандов в координационные соединения алюминия (в т.ч. алкоксиды) для контроля скоростей процессов гидролиза и поликонденсации | [31, 179, 186, 193, 197, 207, 221, 229, 231–233, 235] | |
| Модификация лиогеля гидратированного оксида алюминия в процессе сверхкритической сушки | [75, 228] | |
Для модификации аэрогелей на основе Al2O3 также широко применяются подходы координационной химии. В [191] получен аэрогель Al2O3 с использованием водного раствора AlCl3 и пропиленоксида в качестве инициатора гелеобразования. При синтезе также был использован поливиниловый спирт, химически модифицированный ацетоуксусным эфиром (Аcac-PVA) (рис. 3а). Последний, взаимодействуя с ионами Al3+ и поверхностью частиц золя нанодисперсного гидратированного оксида алюминия, выполняет функции темплата, диспергирующего и комплексообразующего агента (рис. 3б). Аэрогели Al2O3, полученные с использованием Аcac-PVA, характеризовались большой механической прочностью при высоком содержании мезопор.
Рис. 3.
(а) Pеакция этерификации поливинилового спирта и метилацетоацетата с образованием Acac-PVA, (б) иллюстрация механизма образования геля на основе Al2O3 в присутствии Acac-PVA [191].

Хелатирование алкоксидов алюминия для снижения их реакционной способности широко используется при синтезе аэрогелей на основе Al2O3. Использование хелатирующих агентов особенно актуально, когда необходимо получать аэрогели на основе смешанных оксидов (с целью выравнивания скоростей гидролиза–поликонденсации). В качестве исходного соединения при получении аэрогелей на основе Al2O3 обычно используют втор-бутоксид Al(OBusec)3 или изопропоксид A1(OPri)3 алюминия, а в качестве хелатирующих агентов – ацетилацетон или ацетоуксусный эфир.
Следует отметить, что концентрация хелатирующего агента может в значительной степени определять состав и структуру конечного продукта – аэрогеля Al2O3. Например, Бихан и соавт. [232] изучили влияние мольного соотношения (С) втор-бутоксида алюминия к хелатирующему агенту на закономерности золь-гель перехода и свойства получаемых аэрогелей Al2O3. В качестве хелатирующих агентов были выбраны бутандиол-1,3 и ацетилацетон. Использование ацетилацетона приводило к формированию кристаллического Al(Аcac)3 во всем исследованном диапазоне параметра C (1–3). В случае C = 2, 3 происходило образование осадка Al(Аcac)3 и формирования геля не наблюдалось, а при более низких значениях C присутствие кристаллического ацетилацетоната алюминия отмечено в конечных продуктах синтеза – аэрогелях. Напротив, при использовании бутандиола-1,3 удалось получить высокопористые аэрогели на основе Al2O3 при всех значениях параметра C.
Для иммобилизации комплексов в матрице аэрогелей Al2O3 также широко используются органозамещенные алкоксиды кремния. Ли и соавт. [177] для нанесения наночастиц золота на аэрогель Al2O3 применяли следующий подход. К раствору изопропроксида алюминия в безводном iPrOH добавляли раствор золотохлористоводородной кислоты в гептане. Затем к полученному раствору добавляли (3-аминопропил)триэтоксисилан (APTES). При этом формировался комплекс Au(III) с APTES. После перемешивания полученного раствора в течение 1 ч к нему добавляли раствор боргидрида натрия в изопропаноле для восстановления Au3+ до Au0. Механизм формирования аэрогеля Al2O3 с нанесенными наночастицами золота (золь-гель переход и восстановление золота (1); отжиг при 500°C в течение 5 ч (2)), представлен на схеме 9 [177].
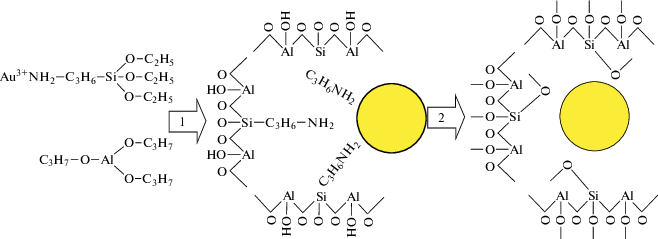
Схема 9 .
Наночастицы золота, распределенные в матрице аэрогеля Al2O3, продемонстрировали превосходную стабильность при отжиге (500°С, 5 ч). Полученные аэрогели Au/SiO2@Al2O3 обладают значительно лучшей каталитической активностью при аэробном окислении бензилового спирта по сравнению с эталонным катализатором, полученным традиционным способом (пропиткой).
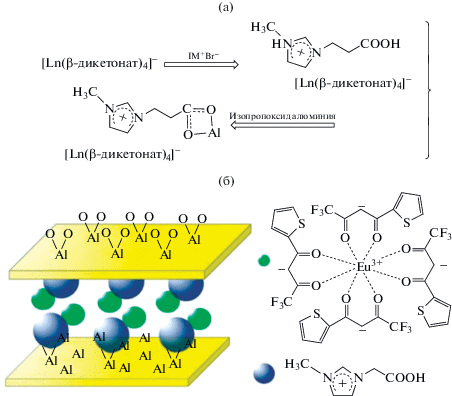
Для химической иммобилизации металлокомплексов в матрице Al2O3 можно использовать лиганды, содержащие карбоксильную группу [176, 202, 215, 236]. Ян и соавт. [176] для закрепления комплексов РЗЭ в матрице Al2O3 разработали следующую процедуру. Сначала получили комплекс европия с теноилтрифторацетоном (ТТА) состава Na[Eu(TTA)4]. Далее к этанольному раствору Na[Eu(TTA)4] добавляли бромид 3-(2-карбоксипропил)-1-метилимидазолия (IM+Br–). В результате был выделен комплекс [Eu(TTA)4]–IM+, который затем согелировали с изопропоксидом алюминия в мольном соотношении Eu : Al = 1 : 10 (рис. 4). Разработанный подход позволил получить гибридные пористые материалы, демонстрирующие интенсивную люминесценцию. Аналогичный подход был использован теми же авторами и в других работах [215, 236].
Рис. 4.
Схема синтеза гибридного ксерогеля Al2O3, модифицированного комплексом [Eu(L)4]–IM+ (а), схема структуры полученного материала [176] (б).
Как отмечалось выше, алкоксиды алюминия, как и многие другие алкоксиды металлов, чрезвычайно склонны к гидролизу, в связи с чем для получения аэрогелей на основе Al2O3 более предпочтительным является метод, основанный на эпоксид-индуцированном золь-гель переходе. Использование эпоксидов для гелирования растворов, содержащих координационные соединения алюминия, может стать основой и для получения аэрогелей Al2O3, содержащих химически иммобилизованные металлокомплексы. Так, с использованием данного подхода были получены аэрогели Al2O3, модифицированные люминесцентным 8-оксихинолинатом алюминия [237]. Отметим, что формирование гелей в системе, содержащей устойчивые комплексы алюминия и 8‑оксихинолина, происходило за существенно большее время, чем в отсутствие 8-оксихинолина (около 100 и 20 ч соответственно).
С целью получения сверхлегких люминесцентных материалов на основе Al2O3 был предложен метод химической модификации предварительно полученных высокопористых материалов на основе Al2O3 [238]. В качестве матрицы для иммобилизации люминесцентного 8-оксихинолината алюминия был выбран аэрогелеподобный материал, получаемый окислением металлического алюминия в условиях контролируемой влажности через слой амальгамы алюминия [239–243]. Химическую модификацию матрицы проводили 8-оксихинолином через газовую фазу. Результатом такой модификации стало приобретение материалом супергидрофобных свойств (контактный угол смачивания водой достигал 153°). Отметим, что существенным достоинством метода химической модификации аэрогелей и аэрогелеподобных материалов с использованием газообразных реагентов является сохранение структуры исходной матрицы, что крайне важно для получения высокопористых каталитических систем. В частности, модифицирование аэрогелей на основе оксида олова газообразным хлористым сульфурилом и другими сульфатирующими агентами позволило получить материалы, характеризующиеся высокой каталитической активностью в реакциях изомеризации и олигомеризации олефинов [49, 244].
Химическая модификация аэрогелеподобных материалов на основе Al2O3 путем введения в их структуру наночастиц металлов или оксидов металлов также приводит к получению эффективных катализаторов скелетной изомеризации гексанов и гексенов [245–249]. Нанесенные наночастицы могут служить катализаторами формирования углеродных нанотрубок методом химического осаждения из газовой фазы (CVD) [240].
ФУНКЦИОНАЛЬНЫЕ СВОЙСТВА АЭРОГЕЛЕЙ, МОДИФИЦИРОВАННЫХ С ИСПОЛЬЗОВАНИЕМ ПОДХОДОВ КООРДИНАЦИОННОЙ ХИМИИ
Комбинируя подходы золь-гель технологии и координационной химии, можно в значительной степени расширить возможности получения функциональных материалов на основе аэрогелей, обладающих каталитическими, фотокаталитическими и люминесцентными свойствами.
Каталитические свойства аэрогелей на основе SiO2 и Al2O3. Более 90% процессов в современной нефтехимической и пищевой промышленности являются каталитическими, при этом более 90% используемых катализаторов гетерогенны [150]. Гетерогенные катализаторы являются удобными в использовании, поскольку их можно легко отделить от реакционной смеси и регенерировать, однако по каталитической активности они обычно сильно уступают гомогенным. В свою очередь, основным недостатком гомогенных процессов является сложность отделения катализатора от реакционной смеси и его регенерации, а также предотвращение возможного загрязнения катализатором конечного продукта.
В 70-х годах XX в. в качестве каталитических материалов было предложено использовать твердофазные носители с химически иммобилизованными координационными соединениями металлов [137]. Следуя классификации Хартли [137], гетерогенные катализаторы являются катализаторами первого поколения, гомогенные – второго поколения, а каталитические системы, содержащие иммобилизованные металлокомплексы, можно отнести к катализаторам третьего поколения.
Реализация концепции иммобилизации металлокомплексов для каталитических применений предполагает решение ряда задач, связанных с конструированием металлокомплексов и носителей, обеспечивающих высокую концентрацию активных центров, а также возможность выделения катализатора из системы и его регенерации. Решение этих задач подразумевает переход от эмпирического к научно обоснованному подбору катализаторов, к предсказанию их каталитического действия [126].
Общая стратегия гетерогенизации гомогенного катализатора заключается в нанесении каталитически активных соединений на твердофазные (неорганические) носители с высокой удельной поверхностью, например на металлооксидные аэрогели или другие высокопористые оксидные материалы, получаемые золь-гель методом [150, 250]. Среди золь-гель материалов наиболее часто в качестве носителей выступают аэрогели на основе SiO2 и Al2O3, обладающие высокой пористостью и термической стабильностью.
Например, Шмидт и соавт. [134] выполнили синтез каталитически активных аэрогелей и ксерогелей SiO2 с закрепленным комплексом рутения RuCl2[Ph2P(CH2)3PPh2]2 (схема 7 ). Полученные гибридные материалы были использованы в качестве гетерогенного катализатора при синтезе N,N-диэтилформамида из CO2, H2 и диэтиламина. Было показано, что активность аэрогеля (TOF = = 18 400 ч–1) значительно выше активности ксерогеля (TOF = 2210 ч–1) при одних и тех же условиях эксперимента, что связано с улучшенными текстурными характеристиками аэрогеля, в частности c присутствием большого количества мезопор, что устраняет диффузионные ограничения на протекание процесса и обеспечивает высокую доступность активных центров. Более того, каталитическая активность аэрогеля оказалась даже выше собственной активности исходного комплекса при использовании его в качестве гомогенного катализатора. Такое поведение, по предположению авторов, связано с влиянием матрицы на электронное строение комплекса. Увеличение каталитической активности комплексов после их иммобилизации в матрице аэрогеля по сравнению с соответствующими гомогенными катализаторами наблюдалось в [131, 155].
При разработке гетерогенных катализаторов важным является вопрос о вымывании активной фазы с поверхности носителя в ходе каталитических реакций. Ковалентное закрепление металлокомплекса в матрице аэрогеля позволяет минимизировать данное явление. Не менее важным фактором является устойчивость металлокомплекса в условиях каталитической реакции. Так, Мёрфи и соавт. [167] показали, что выдерживание аэрогелей SiO2 с закрепленным комплексом Cu(Salen) или Co(Salen) (см. схему 6 а) в среде ДМСО–ДМФА или этилбензола при 110°С в течение 24 ч не приводит к разрушению комплекса, что указывает на высокую стабильность иммобилизованных комплексов в относительно жестких реакционных условиях. Полученные материалы проявили каталитическую активность в процессах окисления этилбензола, при этом были достигнуты высокая селективность по отношению к ацетофенону и количественная конверсия этилбензола [251].
Возможность прочного связывания каталитически активных металлокомплексов с поверхностью аэрогелей продемонстрирована также на ряде других примеров [131, 155]. В частности, Грау и соавт. [131] получили аэрогели с закрепленным комплексом Fe(III) с 3-(2-аминоэтиламино)пропил-триметоксисиланом (EDTMS, L1) и 2-(дифенилфосфино)этилтриэтоксисиланом (L2) (схема 10 ).

Схема 10 .
Полученные композиционные аэрогели были использованы в качестве гетерогенных катализаторов реакций аминирования аллильных спиртов в водной среде. Было показано, что активность гетерогенного катализатора превышает активность гомогенного катализатора FeCl3 ∙ 6H2O. Полученные катализаторы сохраняли высокую каталитическую активность в течение как минимум шести реакционных циклов.
Высокую стабильность при реализации реакции Мизороки–Хека продемонстрировали аэрогели SiO2, модифицированные металлическими или оксидными наночастицами Ni и Pd, полученными постсинтетической обработкой соответствующих ковалентно закрепленных металлокомплексов (рис. 2) [174].
Таким образом, оксидные аэрогели (в первую очередь, на основе SiO2) с иммобилизованными координационными соединениями представляют несомненный практический интерес с точки зрения создания высокоэффективных каталитических систем.
Люминесцентные свойства аэрогелей на основе SiO2. Люминесцентные материалы на основе аэрогелей чаще всего получают путем введения люминесцентных центров в матрицу аэрогеля. В качестве люминесцентных центров обычно выступают:
• органические люминофоры [129, 138, 252];
• оксиды [253–256], соли РЗЭ и переходных металлов [138, 257, 258] и комплексные соединения металлов [259];
• наночастицы [260–262], включая квантовые точки [263], фуллерены [264] и нанокристаллы кремния [265, 266].
Следует отметить, что индивидуальные аэрогели на основе SiO2 также демонстрируют люминесцентные свойства в синей области спектра при облучении УФ-светом (365 нм), которые обусловлены наличием в матрице SiO2 кислородных вакансий [267, 268].
Люминесцентные аэрогели можно использовать при конструировании сенсоров [129], при создании дисплеев [138], сцинтилляторов [139], лазеров [140] и т.д. Среди комплексов, используемых при получении люминесцентных аэрогелей, особое место занимают координационные соединения РЗЭ, содержащие органические лиганды, выполняющие роль “антенны” [127, 269, 270]. По сравнению с солями или оксидами металлов, металлоорганические комплексы РЗЭ обладают более высоким коэффициентом экстинкции, что приводит к более эффективной люминесценции ионов лантанидов.
Работы, описывающие иммобилизацию комплексов РЗЭ в матрице аэрогелей SiO2, достаточно фрагментарны. Гарфия-Мурило и соавт. [258] синтезировали люминесцентные аэрогели на основе индивидуального оксида Y2O3 и смешанных оксидов Y2O3–SiO2, допированных ионами Eu3+. Было показано, что интенсивность люминесценции увеличивается с ростом концентрации Eu3+ в аэрогеле. Интенсивность люминесценции аэрогелей на основе Y2O3 было в среднем на 50% выше, чем аэрогелей на основе смешанных оксидов Y2O3–SiO2. Данный результат предположительно обусловлен низкой прозрачностью смешанного аэрогеля и присутствием Y(OH)3 в его составе.
Стан и соавт. [270] получили люминесцентный аэрогель на основе SiO2, модифицированный комплексом тербия(III) с N-гидроксисукцинимидом. Было показано, что комплекс в составе матрицы аэрогеля характеризуется более высокой термической стабильностью, что указывает на взаимодействие комплекса с матрицей SiO2. Вместе с тем сопоставление спектров возбуждения и люминесценции свободного и иммобилизованного в матрицу аэрогеля комплекса показало, что матрица аэрогеля практически не влияет на его фотофизические характеристики, лишь снижая общую интенсивность фотолюминесценции. По-видимому, это обусловлено уменьшением глубины проникновения возбуждающего излучения в объем аэрогеля за счет поглощения и рассеяния света. Несмотря на это, введение комплекса в структуру аэрогеля SiO2 обеспечивает ряд преимуществ, таких как долговременное сохранение функциональных характеристик комплекса при контакте с окружающей средой (свободный комплекс чувствителен к влаге) [271].
Все описанные методы синтеза люминесцентных аэрогелей на основе комплесов РЗЭ, рассмотренные выше, основаны либо на совместном гелеобразовании модификатора и алкоксисилана/золя SiO2 в процессе золь-гель перехода, либо на пост-модификации предварительно полученного аэрогеля.
Синтетический подход, основанный на ковалентном закреплении комплексов РЗЭ в матрице аэрогелей SiO2 был впервые описан Ёровым с соавт. [272] (рис. 5). Необходимо подчеркнуть, что материалы на основе аэрогелей, как правило, характеризуются невысокими люминесцентными характеристиками, что связано с тушением люминесценции в результате взаимодействия люминесцентных центров с OH-группами, присутствующими в составе аэрогелей. Для решения данной проблемы было предложено иммобилизовать в матрице аэрогеля биметаллический люминесцентный комплекс, включающий в себя люминесцентный центр (ион европия) и линкерный нелюминесцентный ион (ион цинка), обеспечивающий химическое связывание комплекса с матрицей аэрогеля, экранируя ион европия от присутствующих на поверхности аэрогеля гидроксильных групп [272].
Рис. 5.
Схема синтеза (а) и внешний вид (б) люминесцентного аэрогеля SiO2 с иммобилизованным гетерометаллическим комплексом цинка и европия [272].
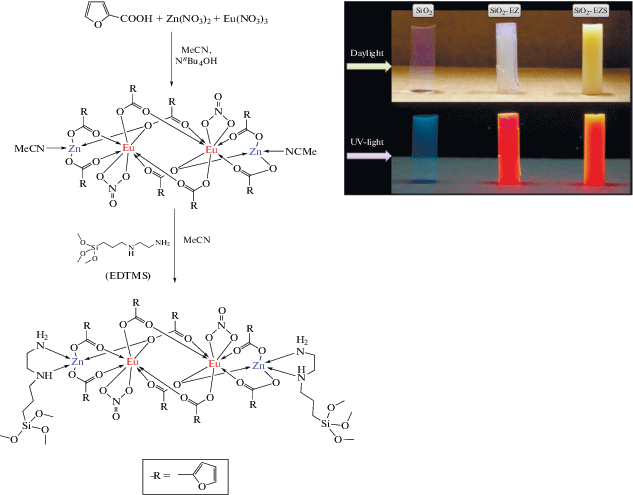
Полученные материалы характеризовались интенсивной люминесценцией при УФ-облучении, характерной для координационных соединений европия (см. рис. 5б), сохраняя присущую аэрогелям высокую удельную поверхность и пористость.
Сенсорные свойства аэрогелей, модифицированных координационными соединениями. Детектирование химических веществ в воздухе, воде и других средах имеет большое значение для широкого спектра применений, включая экологический мониторинг опасных химических веществ, медицину, криминалистику и др. Высокая удельная площадь поверхности, высокая пористость и низкая плотность делают аэрогели особенно привлекательными для применений в сенсорике [273].
До настоящего времени основная масса работ по созданию сенсорных систем на основе золь-гель материалов, в том числе модифицированных координационными соединениями металлов, приходилась на ксерогели [274]. В качестве матрицы для создания сенсоров на основе аэрогелей обычно используют SiO2. Это обусловлено его высокой прозрачностью в УФ- и видимом диапазоне, что важно при конструировании оптических сенсоров [154].
Левентис и соавт. [275] для получения сенсоров на кислород использовали аэрогель SiO2, модифицированный трис-(1,10-фенантролинат)ом рутения(II) и комплексами рутения 1 и 2 с электроноакцепторными лигандами (рис. 6а). Наилучшие сенсорные свойства продемонстрировал аэрогель с комплексом 1 (рис. 6б, 6в). Интересно, что люминесцентные характеристики комплексов в аэрогеле были аналогичны характеристикам замороженного раствора комплекса; при охлаждении интенсивность люминесценции комплекса увеличивалась, наблюдается гипсохромный сдвиг.
Рис. 6.
Структурные формулы комплексов рутения(II) с бис(1,10-фенантролин)моно(3,8-бис(N-метил-4-(4-этинилбензоил)пиридин)-1,10-фенантролин) тетра(тетрафторборат) моногидратом (1) и комплекса бис(1,10-фенантролин)моно[3,8-бис(N-(4-этинилбензил)-N-метил-4,4′-бипиридин)-1,10-фенантролин)]рутения(II) гекса(тетрафторбората) (2) (а), сенсорный отклик аэрогеля SiO2, модифицированного комплексом (1) при чередовании потоков N2 и O2 при комнатной температуре (б), демонстрация тушения люминесценции в атмосфере O2 для аэрогеля, модифицированного комплексом (1) [275] (в).
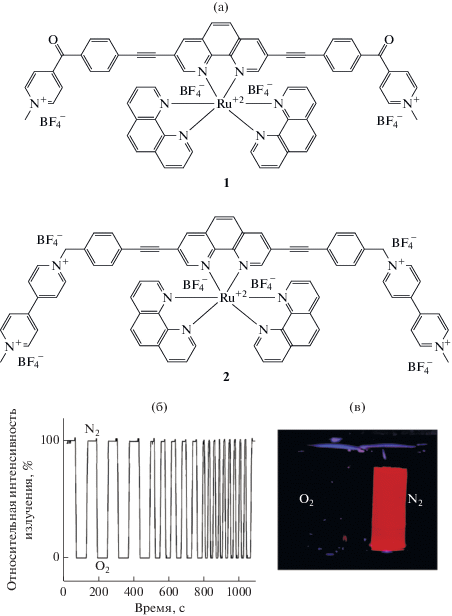
Плата и соавт. [276] провели сравнительный анализ сенсорных характеристик комплексов рутения(II) в аэрогелях и ксерогелях на основе SiO2. Было показано, что время отклика модифицированного аэрогеля в ~5 раз меньше времени отклика соответствующего ксерогеля, что, очевидно, связано с лучшей газопроницаемостью аэрогеля по сравнению с ксерогелем.
Аэрогели, модифицированные наночастицами благородных металлов, используются для детектирования органических молекул за счет усиления сигнала комбинационного рассеяния соответствующих молекул [277–279]. Для улучшения равномерности распределения наночастиц по поверхности матрицы аэрогелей широко применяются подходы координационной химии [151].
Фотокаталитические и фотопротекторные материалы на основе аэрогелей. В настоящее время создание высокоэффективных фотокаталитических систем является чрезвычайно актуальной задачей, поскольку такие системы широко востребованы для очистки воды и воздуха от органических и биогенных загрязнений, а также фотоэлектрохимического разложения воды [280–282].
Для получения фотокатализаторов на основе аэрогелей чаще всего используют аэрогели SiO2, модифицированные TiO2 [94, 103, 104, 110, 185, 283–289]. При этом матрица SiO2 обеспечивает высокую удельную поверхность и пористость, а TiO2 придает композитному аэрогелю фотокаталитическую активность (ФКА). При синтезе аэрогелей на основе SiO2–TiO2 в качестве фотокатализаторов важным является присутствие и равномерное распределение кристаллической фазы TiO2 (анатаза) в сочетании с высокой проницаемостью матрицы. Выбор метода CК-сушки бинарных аэрогелей оказывает существенное влияние на их ФКА. Аэрогели, полученные сушкой в органических растворителях, проявляют заметную ФКА, тогда как аэрогели, полученные с использованием СК-СО2, не обладают фотокаталитической активностью [285, 289]. Это связано с формированием нанокристаллического анатаза в первом случае и аморфного диоксида титана во втором.
В ряде случаев зависимость ФКА от содержания TiO2 в составе бинарных аэрогелей имеет немонотонный характер. Так, Ахмед и соавт. [110] показали, что в диапазоне концентраций TiO2 от 20 до 50 мол. % при увеличении содержания диоксида титана наблюдается уменьшение ФКА, связанное с уменьшением пористости и прозрачности композитного материала для УФ-излечения.
Для улучшения ФКА аэрогелей SiO2–TiO2 зачастую проводят их отжиг с целью кристаллизации диоксида титана и формирования нанокристаллического анатаза. Низкотемпературный отжиг (<500°C), в результате которого удаляются органические соединения, способствует увеличению удельной поверхности и пористости аэрогелей, а также приводит к формированию наночастиц анатаза и увеличению ФКА [284, 289, 290]. Отжиг при более высоких температурах (650°С и выше), как правило, ведет к уменьшению пористости и к уменьшению ФКА [290].
К настоящему времени практически отсутствуют работы, посвященные систематическому анализу влияния содержания хелатирующего агента, регулирующего относительную скорость гидролиза алкоксидов титана и кремния, на структуру и фотокаталитические свойства аэрогелей SiO2–TiO2. Малоизученным остается влияние типа используемого сверхкритического флюида на физико-химические свойства аэрогелей на основе SiO2–TiO2.
В этой связи Ёровым с соавт. было впервые обсуждено влияние условий СК-сушки (типа СК-растворителя) на физико-химические характеристики бинарных аэрогелей SiO2–TiO2 с содержанием диоксида титана до 50 мол. % [291, 292]. Было показано, что в различных сверхкритических средах возможно получение как аморфных аэрогелей SiO2– TiO2 с гомогенным распределением компонентов (при сушке в СО2), так и композитных аэрогелей, содержащих нанокристаллический анатаз (при сушке в изопропаноле и метил-трет-бутиловом эфире) и характеризующихся высокой удельной поверхностью (около 1000 м2/г). Важной характеристикой полученных материалов явилась их высокая термическая стабильность – отжиг при температурах до 600°С не приводил к значимому разрушению пористой структуры аэрогелей, и они сохраняли высокие значения удельной поверхности (>400 м2/г).
Аэрогели на основе SiO2, и прежде всего SiO2–TiO2, перспективны в качестве компонентов косметических препаратов. В 2018 г. было опубликовано сообщение об успешном использовании гидрофобного аэрогеля на основе SiO2 в качестве матирующего компонента крема для маскирования жирного блеска кожи [293]. Было показано, что концентрация аэрогеля в креме на уровне 1–2 мас. % эффективно предотвращает появление жирного блеска в течение не менее 6 ч. Аэрогель оказался намного более эффективен, чем другие минеральные добавки, которые традиционно применяются в матирующей косметике: перлит и тальк.
Аэрогели на основе SiO2–TiO2 могут быть перспективными и для применения в качестве компонентов солнцезащитной косметики. Известно, что традиционным компонентом солнцезащитных средств является диоксид титана [294, 295], который, однако, в силу выраженных фотокаталитических свойств вызывает повреждение кожи активными формами кислорода, образующимися при УФ-облучении [296]. В композитных аэрогелях инертная матрица SiO2 снижает фотоцитотоксичность наночастиц TiO2, блокируя их поверхность. Матрица SiO2 также служит физическим барьером между наночастицами диоксида титана и компонентами крема и/или себума (кожное сало), предотвращая повреждение кожи активными формами кислорода. В то же время диоксид титана в композите сохраняет свою функцию УФ-фильтра.
Впервые фотопротекторные свойства аэрогелей SiO2–TiO2, синтез которых был проведен с использованием алкоголятов титана, хелатированных ацетилацетоном, были проанализированы Ёровым и соавт. [297] с использованием стандартного метода ISO 24443. Было показано, что фазовый состав аэрогелей в существенной степени определяет их фотопротекторные характеристики, при этом он может быть направленным образом задан путем выбора растворителя, используемого на стадии СК-сушки материала (CO2, изопропанол, метил-трет-бутиловый эфир, гексафторизопропанол). Сверхкритическая сушка при температурах менее 200°С (в CO2 или гексафторизопропаноле) приводила к получению рентгеноаморфных материалов, тогда как при более высоких температурах 240–250°C (в изопропаноле или метил-трет-бутиловом эфире) происходило формирование нанокристаллического анатаза. Полученные аэрогели обладали низкой ФКА, хорошими матирующими свойствами и достаточно высокими фотопротекторными характеристиками в UVA-диапазоне. Отметим, что в данной работе было также показано, что традиционно используемые в солнцезащитной косметике материалы на основе TiO2 могут обладать ФКА, превышающей коммерческие фотокатализаторы.
В заключение отметим, что использование подходов координационной химии в золь-гель технологии в значительной степени расширяет спектр функциональных свойств и областей применения как самих комплексных соединений, так и аэрогелей. В частности, модификация аэрогелей координационными соединениями позволяет получать новые полифункциональные материалы, сочетающие превосходные каталитические, фотокаталитические, оптические и другие свойства с высокой пористостью и удельной площадью поверхности.
Наиболее распространенным способом получения гибридных аэрогелей, модифицированных координационными соединениями, является физическое связывание комплекса с матрицей геля в результате захвата молекул комплекса в процессе формирования геля, либо в результате внедрения молекул комплекса в уже сформированный гель/ аэрогель. Несмотря на простоту реализации, данный подход имеет ряд существенных недостатков, включая неравномерность распределения комплекса в матрице, блокировку пор, частичное разрушение пористой структуры матрицы за счет действия капиллярных сил и др. Перечисленные проблемы могут быть в значительной степени решены с применением подходов, основанных на химической иммобилизации координационных соединений с матрицей аэрогеля.
Известные подходы к химической иммобилизации координационных соединений на поверхности матрицы металлооксидных аэрогелей можно разделить на две группы.
Первая группа подходов основана на непосредственном связывании комплекса состава (RO)xMLy, где RO – алкоксидная группа, M – металл и L – лиганд, с металлооксидной матрицей в результате его взаимодействия с поверхностными OH-группами. Данный подход наиболее часто используется при получении смешанно-оксидных аэрогелей (в частности аэрогелей на основе SiO2–TiO2). В качестве комплекса выступает предварительно хелатированный алкоксид металла, реакционная способность которого значительно ниже, чем у исходного алкоксида, что важно для получения аэрогелей с однородным распределением компонентов в матрице.
Вторая группа подходов основана на закреплении комплексных соединений через химически инертный (углеводородный) фрагмент с использованием предварительно полученного гибридного соединения состава [(RO)3Si(CH2)nL]nMXm, где L – донорная группа, например –NH2, –SH и т.п., X – противоион или лиганд) за счет конденсации фрагментов Si–OR с поверхностными OH-группами металлооксидной матрицы. Иммобилизацию полученного комплекса осуществляют на разных стадиях синтеза аэрогелей. Наиболее эффективным является согелирование комплекса с золем гидратированного оксида металла или алкоксидом матрицеобразующего элемента. При реализации данного подхода наиболее сложными и трудоемкими этапами являются синтез и очистка алкоксисилансодержащего лиганда, а также соответствующего металлокомплекса. Кроме того, в большинстве случаев выделить монокристаллы силансодержащего комплекса не удается, что затрудняет однозначную идентификацию полученных комплексов как в индивидуальном состоянии, так и в составе аэрогельной матрицы. Вторая группа подходов в основном используется при иммобилизации комплексов переходных металлов. Отметим, что работы, описывающие иммобилизацию комплексов РЗЭ в матрице аэрогелей SiO2, достаточно фрагментарны. Существующие подходы к синтезу аэрогелей, модифицированных комплексами РЗЭ, основаны либо на совместном гелеобразовании модификатора и алкоксисилана/золя SiO2 в процессе золь-гель перехода, либо на постмодификации предварительно полученного аэрогеля.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Pierre A.C., Pajonk G.M.G.M. // Chem. Rev. 2002. V. 102. № 11. P. 4243. https://doi.org/10.1021/cr0101306
Kistler S.S. // J. Phys. Chem. 1931. V. 36. № 1. P. 52. https://doi.org/10.1021/j150331a003
Hüsing N., Schubert U. // Angew. Chemie Int. Ed. 1998. V. 37. № 1/2. P. 22. https://doi.org/10.1002/1521-3773(19980202)37:1/2<22:: AID-ANIE22>3.3.CO;2-9
Du A., Zhou B., Zhang Z. et al. // Materials (Basel). 2013. V. 6. № 3. P. 941. https://doi.org/10.3390/ma6030941
Qin G., Guo S. // Carbon. 2001. V. 39. № 12. P. 1935. https://doi.org/10.1016/S0008-6223(01)00162-2
Lermontov S.A., Malkova A.N., Sipyagina N.A. et al. // J. Fluor. Chem. 2021. V. 244. P. 109742. https://doi.org/10.1016/j.jfluchem.2021.109742
Lermontov S.A., Malkova A.N., Sipyagina N.A. et al. // Mendeleev Commun. 2018. V. 28. № 1. P. 102. https://doi.org/10.1016/j.mencom.2018.01.035
Shimizu T., Kanamori K., Maeno A. et al. // Chem. Mater. 2016. V. 28. № 19. P. 6860. https://doi.org/10.1021/acs.chemmater.6b01936
Corma A., Garcia H. // Adv. Synth. Catal. 2006. V. 348. № 12–13. P. 1391. https://doi.org/10.1002/adsc.200606192
Kistler S.S. // Nature. 1931. V. 127. № 3211. P. 741. https://doi.org/10.1038/127741a0
Jain A., Panchal S., Sharma S. et al. // Green Chemistry. Fundamentals and Applications. N.J. Apple Acad. Press, 2013. P. 138. https://doi.org/10.1201/b15500
Teichner S.J., Nicolaon G.A., Vicarini M.A. et al. // Adv. Colloid Interface Sci. 1976. V. 5. № 3. P. 245. https://doi.org/10.1016/0001-8686(76)80004-8
Gash A.E., Tillotson T.M., Satcher Jr. J.H. et al. // J. Non. Cryst. Solids. 2001. V. 285. № 1–3. P. 22. https://doi.org/10.1016/S0022-3093(01)00427-6
Danks A.E., Hall S.R., Schnepp Z. // Mater. Horizons. 2016. V. 3. № 2. P. 91. https://doi.org/10.1039/c5mh00260e
Maleki H., Durães L., García-González C.A. et al. // Adv. Colloid Interface Sci. 2016. V. 236. P. 1. https://doi.org/10.1016/j.cis.2016.05.011
Iler R.K. The Chemistry of Silica. Chichester: John Wiley and Sons, 1979. V. 24. P. 886.
Nakanishi K., Minakuchi H., Soga N. et al. // J. Sol-Gel Sci. Technol. 1998. V. 13. № 1–3. P. 163. https://doi.org/10.1023/A:1008644514849
Wagh P.B.B., Begag R., Pajonk G.M.M. et al. // Mater. Chem. Phys. 1999. V. 57. № 3. P. 214. https://doi.org/10.1016/S0254-0584(98)00217-X
Deng Z., Wang J., Wei J. et al. // J. Sol-Gel Sci. Technol. 2000. V. 19. № 1–3. P. 677. https://doi.org/10.1023/A:1008754504788
Venkateswara Rao A., Bhagat S.D., Hirashima H. et al. // J. Colloid Interface Sci. 2006. V. 300. № 1. P. 279. https://doi.org/10.1016/j.jcis.2006.03.044
Kostenko M.O., Ustinovich K.B., Pokrovskii O.I. // Russ. J. Inorg. Chem. 2020. V. 65. № 10. P. 1577. https://doi.org/10.1134/S0036023620100125
Lermontov S.A., Sipyagina N.A., Malkova A.N. et al. // Russ. J. Inorg. Chem. 2015. V. 60. № 4. P. 488. https://doi.org/10.1134/S0036023615040117
Livage J., Henry M., Sanchez C. // Prog. Solid State Chem. 1988. V. 18. № 4. P. 259. https://doi.org/10.1016/0079-6786(88)90005-2
Livage J., Sanchez C. // J. Non. Cryst. Solids. 1992. V. 145. № C. P. 11. https://doi.org/10.1016/S0022-3093(05)80422-3
Zherebtsov D.A., Kulikovskikh S.A., Viktorov V. V. et al. // Russ. J. Inorg. Chem. 2019. V. 64. № 2. P. 165. https://doi.org/10.1134/S0036023619020220
Babonneau F., Coury L., Livage J. // J. Non. Cryst. Solids. 1990. V. 121. № 1–3. P. 153. https://doi.org/10.1016/0022-3093(90)90122-3
Wang C.T., Willey R.J. // Catal. Today. 1999. V. 52. № 1. P. 83. https://doi.org/10.1016/S0920-5861(99)00065-6
Mishakov I.V., Ilyina E.V., Bedilo A.F. et al. // React. Kinet. Catal. Lett. 2009. V. 97. № 2. P. 355. https://doi.org/10.1007/s11144-009-0041-1
Abouarnadasse S., Pajonk G.M., Teichner S.J. // Stud. Surf. Sci. Catal. 1988. V. 41. P. 371. https://doi.org/10.1016/S0167-2991(09)60835-1
Tleimat-Manzalji R., Manzalji T., Pajonk G.M. // J. Non. Cryst. Solids. 1992. V. 147–148. P. 744. https://doi.org/10.1016/S0022-3093(05)80709-4
Mizushima Y., Hori M. // J. Mater. Res. 1994. V. 9. № 9. P. 2272. https://doi.org/10.1557/JMR.1994.2272
Löbmann P., Glaubitt W., Gross J. et al. // J. Non. Cryst. Solids. 1995. V. 186. P. 59. https://doi.org/10.1016/0022-3093(95)00081-X
Li S., Li B., Qi X.B. et al. // J. Non. Cryst. Solids. 2010. V. 356. № 43. P. 2263. https://doi.org/10.1016/j.jnoncrysol.2010.08.017
Einarsrud M.-A., Pedersen S., Larsen E. et al. // J. Eur. Ceram. Soc. 1999. V. 19. № 3. P. 389. https://doi.org/10.1016/S0955-2219(98)00199-X
Dagan G., Tomkiewicz M. // J. Phys. Chem. 1993. V. 97. № 49. P. 12651. https://doi.org/10.1021/j100151a001
Schneider M., Baiker A. // J. Mater. Chem. 1992. V. 2. № 6. P. 587. https://doi.org/10.1039/jm9920200587
Lecomte A., Blanchard F., Dauger A. et al. // J. Non. Cryst. Solids. 1998. V. 225. № 1–3. P. 120. https://doi.org/10.1016/S0022-3093(98)00107-0
Tang P.E., Sakamoto J.S., Baudrin E. et al. // J. Non. Cryst. Solids. 2004. V. 350. P. 67. https://doi.org/10.1016/j.jnoncrysol.2004.07.072
Maurer S.M., Ng D., Ko E.I. // Catal. Today. 1993. V. 16. № 3–4. P. 319. https://doi.org/10.1016/0920-5861(93)80071-8
Yao N., Cao S., Yeung K.L. // Microporous Mesoporous Mater. 2009. V. 117. № 3. P. 570. https://doi.org/10.1016/j.micromeso.2008.08.020
Himmel B., Gerber T., Bürger H. et al. // J. Non. Cryst. Solids. 1995. V. 186. P. 149. https://doi.org/10.1016/0022-3093(95)00045-3
Ilyina E. V., Mishakov I. V., Vedyagin A.A. // Inorg. Mater. 2009. V. 45. № 11. P. 1267. https://doi.org/10.1134/s0020168509110144
Pierre A.C. Aerogels Handbook. New York: Springer, 2011. P. 3. https://doi.org/10.1007/978-1-4419-7589-8_1
Itoh H., Tabata T., Kokitsu M. et al. // J. Ceram. Soc. Jpn. 1993. V. 101. № 1177. P. 1081. https://doi.org/10.2109/jcersj.101.1081
Dobinson B., Hofmann W., Stark B.P. et al. The Determination of Epoxide Groups: Monographs in Organic Functional Group Analysis. Elsevier Science, 1969. P. 18.
Gash A.E., Tillotson T.M., Satcher J.H. et al. // Chem. Mater. 2001. V. 13. № 3. P. 999. https://doi.org/10.1021/cm0007611
Baumann T.F., Gash A.E., Satcher J.H. Aerogels Handbook. N.Y.: Springer, 2011. P. 155. https://doi.org/10.1007/978-1-4419-7589-8_8
Gash A.E., Satcher J.H., Simpson R.L. et al. // Chem. Mater. 2003. V. 15. № 17. P. 3268. https://doi.org/10.1021/cm034211p
Lermontov S.A., Yurkova L.L., Straumal E.A. et al. // Russ. J. Inorg. Chem. 2018. V. 63. № 3. P. 303. https://doi.org/10.1134/S0036023618030142
Straumal E.A., Ivanov V.K., Malkova A.N. et al. // J. Sol-Gel Sci. Technol. 2017. V. 84. № 3. P. 377. https://doi.org/10.1007/s10971-017-4429-5
Plantier K.B., Pantoya M.L., Gash A.E. // Combust. Flame. 2005. V. 140. № 4. P. 299. https://doi.org/10.1016/j.combustflame.2004.10.009
Prentice D., Pantoya M.L., Gash A.E. // Energy and Fuels. 2006. V. 20. № 6. P. 2370. https://doi.org/10.1021/ef060210i
Baumann T.F., Gash A.E., Chinn S.C. et al. // Chem. Mater. 2005. V. 17. № 2. P. 395. https://doi.org/10.1021/cm048800m
Wei T.Y., Chen C.H., Chang K.H. et al. // Chem. Mater. 2009. V. 21. № 14. P. 3228. https://doi.org/10.1021/cm9007365
Laberty-Robert C., Long J.W., Lucas E.M. et al. // Chem. Mater. 2006. https://doi.org/10.1021/cm051385t
Zhang H.D., Li B., Zheng Q.X. et al. // J. Non. Cryst. Solids. 2008. V. 354. № 34. P. 4089. https://doi.org/10.1016/j.jnoncrysol.2008.05.044
Reibold R.A., Poco J.F., Baumann T.F. et al. // J. Non. Cryst. Solids. 2003. V. 319. № 3. P. 241. https://doi.org/10.1016/S0022-3093(03)00012-7
Shobe A.M., Gill S.K., Hope-Weeks L.J. // J. Non. Cryst. Solids. 2010. V. 356. P. 1337. https://doi.org/10.1016/j.jnoncrysol.2010.03.002
Chervin C.N., Clapsaddle B.J., Chiu H.W. et al. // Chem. Mater. 2005. V. 17. № 13. P. 3345. https://doi.org/10.1021/cm0503679
Brown P., Hope-Weeks L.J. // J. Sol-Gel Sci. Technol. 2009. V. 51. № 2. P. 238. https://doi.org/10.1007/s10971-009-1985-3
Guo Y., Meyer-Zaika W., Muhler M. et al. // Eur. J. Inorg. Chem. 2006. № 23. P. 4774. https://doi.org/10.1002/ejic.200600550
Baumann T.F., Kucheyev S.O., Gash A.E. et al. // Adv. Mater. 2005. V. 17. № 12. P. 1546. https://doi.org/10.1002/adma.200500074
Long J.W., Logan M.S., Rhodes C.P. et al. // J. Am. Chem. Soc. 2004. V. 126. № 51. P. 16879. https://doi.org/10.1021/ja046044f
Suh D.J., Park T.-J., Kim W.-I. et al. // J. Power Sources. 2003. V. 117. № 1–2. P. 1. https://doi.org/10.1016/S0378-7753(02)00617-1
Chen L., Zhu J., Liu Y.M. et al. // J. Mol. Catal. A. 2006. V. 255. P. 260. https://doi.org/10.1016/j.molcata.2006.04.043
Zhong L., Chen X., Song H. et al. // RSC Adv. 2014. V. 4. № 60. P. 31666. https://doi.org/10.1039/c4ra04601c
Sisk C.N., Hope-Weeks L.J. // J. Mater. Chem. 2008. V. 18. № 22. P. 2607. https://doi.org/10.1039/b802174k
Gao Y.P., Sisk C.N., Hope-Weeks L.J. // Chem. Mater. 2007. V. 19. № 24. P. 6007. https://doi.org/10.1021/cm0718419
Gash A.E., Satcher J.H., Simpson R.L. // J. Non. Cryst. Solids. 2004. V. 350. P. 145. https://doi.org/10.1016/j.jnoncrysol.2004.06.030
Bisson A., Rigacci A., Lecomte D. et al. // Dry. Technol. 2003. V. 21. № 4. P. 593. https://doi.org/10.1081/drt-120019055
Pons A., Casas L., Estop E. et al. // J. Non. Cryst. Solids. 2012. V. 358. № 3. P. 461. https://doi.org/10.1016/j.jnoncrysol.2011.10.031
Matson D.W., Smith R.D. // J. Am. Ceram. Soc. 1989. V. 72. № 6. P. 871. https://doi.org/10.1111/j.1151-2916.1989.tb06237.x
Kirkbir F., Murata H., Meyers D. et al. // J. Non. Cryst. Solids. 1998. V. 225. P. 14. https://doi.org/10.1016/S0022-3093(98)00003-9
Yoda S., Ohshima S. // J. Non. Cryst. Solids. 1999. V. 248. № 2–3. P. 224. https://doi.org/10.1016/S0022-3093(99)00250-1
Lermontov S.A., Straumal E.A., Mazilkin A.A. et al. // J. Phys. Chem. C. 2016. V. 120. № 6. P. 3319. https://doi.org/10.1021/acs.jpcc.5b10461
Lermontov S.A., Sipyagina N.A., Malkova A.N. et al. // RSC Adv. 2014. V. 4. № 94. P. 52423. https://doi.org/10.1039/c4ra06974a
Lermontov S.A., Malkova A.N., Yurkova L.L. et al. // Mater. Lett. 2014. V. 116. P. 116. https://doi.org/10.1016/j.matlet.2013.10.080
Lermontov S., Malkova A., Yurkova L. et al. // J. Supercrit. Fluids. 2014. V. 89. P. 28. https://doi.org/10.1016/j.supflu.2014.02.011
Petukhov D.I., Baranchikov A.E., Sipyagina N.A. et al. // Russ. J. Inorg. Chem. 2015. V. 60. № 4. P. 488. https://doi.org/10.1134/s0036023615040117
Lermontov S.A., Malkova A.N., Sipyagina N.A. et al. // Russ. J. Inorg. Chem. 2015. V. 60. № 10. P. 1169. https://doi.org/10.1134/S0036023615100125
Sipyagina N.A., Ivanov V.K., Malkova A.N. et al. // Russ. J. Inorg. Chem. 2014. V. 59. № 12. P. 1392. https://doi.org/10.1134/s0036023614120134
Sipyagina N.A., Ivanov V.K., Petukhov D.I. et al. // Russ. J. Inorg. Chem. 2015. V. 60. № 5. P. 541. https://doi.org/10.1134/s0036023615050137
Lermontov S.A., Baranchikov A.E., Sipyagina N.A. et al. // Russ. J. Inorg. Chem. 2020. V. 65. № 2. P. 255. https://doi.org/10.1134/S0036023620020084
Lermontov S.A., Malkova A.N., Sipyagina N.A. et al. // Inorg. Mater. 2017. V. 53. № 12. P. 1270. https://doi.org/10.1134/S002016851712007X
Beghi M., Chiurlo P., Costa L. et al. // J. Non. Cryst. Solids. 1992. V. 145. № C. P. 175. https://doi.org/10.1016/S0022-3093(05)80451-X
Ingemar Odenbrand C.U., Lars S., Andersson T. et al. // J. Catal. 1990. V. 125. № 2. P. 541. https://doi.org/10.1016/0021-9517(90)90325-E
Baranchikov A.E., Kopitsa G.P., Yorov Kh.E. et al. // Russ. J. Inorg. Chem. 2021. V. 66. P. 874. https://doi.org/10.1134/S003602362106005X
Schneider M., Baiker A. // Catal. Today. 1997. V. 35. № 3. P. 339. https://doi.org/10.1016/S0920-5861(96)00164-2
Miller J.B., Johnston S.T., Ko E.I. // J. Catal. 1994. V. 150. P. 311. https://doi.org/10.1006/jcat.1994.1349
Dutoit D.C.M., Schneider M., Baiker A. // J. Catal. 1995. V. 153. P. 165. https://doi.org/10.1006/jcat.1995.1118
Dutoit D.C.M.C.M., Schneider M., Hutter R. et al. // J. Catal. 1996. V. 161. P. 651. https://doi.org/10.1006/jcat.1996.0227
Calvino J.J., Cauqui M.A., Cifredo G. et al. // J. Sol-Gel Sci. Technol. 1994. V. 2. № 1–3. P. 831. https://doi.org/10.1007/BF00486360
Calvino J.J., Cauqui M.A., Cifredo G. et al. // J. Mater. Sci. 1993. V. 28. № 8. P. 2191. https://doi.org/10.1007/BF00367583
Ismail A.A., Ibrahim I.A. // Appl. Catal. A. 2008. V. 346. № 1–2. P. 200. https://doi.org/10.1016/j.apcata.2008.05.031
Ingale S. V., Wagh P.B., Tripathi A.K. et al. // J. Sol-Gel Sci. Technol. 2011. V. 58. № 3. P. 682. https://doi.org/10.1007/s10971-011-2445-4
Shao G.N., Hilonga A., Jeon S.J. et al. // Powder Technol. 2013. V. 233. P. 123. https://doi.org/10.1016/j.powtec.2012.08.025
Ingale S. V., Sastry P.U., Wagh P.B. et al. // Mater. Chem. Phys. 2012. V. 135. № 2–3. P. 497. https://doi.org/10.1016/j.matchemphys.2012.05.014
Kim W.-I.I., Hong L.K., Hong I.-K. // J. Ind. Eng. Chem. 2003. V. 9. № 6. P. 728.
Aravind P.R., Shajesh P., Mukundan P. et al. // J. Sol-Gel Sci. Technol. 2009. V. 52. № 3. P. 328. https://doi.org/10.1007/s10971-009-2042-y
Wang X., Liu J., Shi F. et al. // J. Porous Mater. 2014. V. 21. № 3. P. 293. https://doi.org/10.1007/s10934-013-9774-3
Liu J.-X.X., Shi F., Bai L.-N.N. et al. // J. Sol-Gel Sci. Technol. 2014. V. 69. P. 93. https://doi.org/10.1007/s10971-013-3190-7
Yoda S., Tasaka Y., Uchida K. et al. // J. Non. Cryst. Solids. 1998. V. 225. P. 105. https://doi.org/10.1016/S0022-3093(98)00013-1
Cheng S., Liu X., Yun S. et al. // Ceram. Int. 2014. V. 40. P. 13781. https://doi.org/10.1016/j.ceramint.2014.05.093
Kim Y.N., Shao G.N., Jeon S.J. et al. // Chem. Eng. J. 2013. V. 231. P. 502. https://doi.org/10.1016/j.cej.2013.07.072
Yoldas B.E. // J. Non. Cryst. Solids. 1980. V. 38–39. P. 81. https://doi.org/10.1016/0022-3093(80)90398-1
Shishmakov A.B., Mikushina Y.V., Koryakova O.V. et al. // Russ. J. Inorg. Chem. 2019. V. 64. P. 864. https://doi.org/10.1134/S0036023619070155
Re N. // J. Non. Cryst. Solids. 1992. V. 142. P. 1. https://doi.org/10.1016/S0022-3093(05)80002-X
Bernal S., Calvino J.J., Cauqui M.A. et al. // Stud. Surf. Sci. Catal. 1995. V. 91. P. 461. https://doi.org/10.1016/S0167-2991(06)81783-0
Drobot D.V., Nikishina E.E. // Russ. J. Inorg. Chem. 2020. V. 65. P. 981. https://doi.org/10.1134/S0036023620070062
Ahmed M.S., Attia Y.A. // J. Non. Cryst. Solids. 1995. V. 186. P. 402. https://doi.org/10.1016/0022-3093(95)00085-2
Lee J.H., Choi S.Y., Kim C.E. et al. // J. Mater. Sci. 1997. V. 32. P. 3577. https://doi.org/10.1023/A:1018665910396
Brinker C.J., Scherer G.W. Sol-Gel Sci. Boston: Elsevier, 1990. P. 96. https://doi.org/10.1016/B978-0-08-057103-4.50008-8
Smith G.D., Caughlan C.N., Campbell J.A. // Inorg. Chem. 1972. V. 11. P. 2989. https://doi.org/10.1021/ic50118a024
Leaustic A., Babonneau F., Livage J. // Chem. Mater. 1989. V. 1. P. 240. https://doi.org/10.1007/BF01544593
Bradley D.C., Holloway C.E. // J. Chem. Soc. A. 1969. P. 282. https://doi.org/10.1039/j19690000282
Errington R.J., Ridland J., Clegg W. et al. // Polyhedron 1998. V. 17. P. 659. https://doi.org/10.1016/S0277-5387(97)00418-X
Jung M. // Int. J. Inorg. Mater. 2001. V. 3. P. 471. https://doi.org/10.1016/S1466-6049(01)00050-2
Nakagawa K., Wang F., Murata Y. et al. // Chem. Lett. 2005. V. 34. P. 736. https://doi.org/10.1246/cl.2005.736
Sadeghzadeh Attar A., Sasani Ghamsari M., Hajiesmaeilbaigi F. et al. // J. Phys. D. 2008. V. 41. 155318. https://doi.org/10.1088/0022-3727/41/15/155318
Shih W.-H., Lu Q. // Ferroelectrics. 1994. V. 154. P. 241. https://doi.org/10.1080/00150199408017293
Lermontov S.A., Straumal E.A., Mazilkin A.A. et al. // Mater. Lett. 2018. V. 215. P.19. https://doi.org/10.1016/j.matlet.2017.12.031
Fattakhova Z.A., Zakharova G.S., Andreikov E.I. et al. // Russ. J. Inorg. Chem. 2019. V. 64. P. 857. https://doi.org/10.1134/S0036023619070076
Zakharova G.S., Fattakhova Z.A., Andreikov E.I. et al. // Russ. J. Inorg. Chem. 2019. V. 64. P. 283. https://doi.org/10.1134/S0036023619030227
Miller J.B., Mathers L.J., Ko E.I. // J. Mater. Chem. 1995. V. 5. P. 1759. https://doi.org/10.1039/JM9950501759
Rupp W., Hüsing N., Schubert U. // J. Mater. Chem. 2002. V. 12. P. 2594. https://doi.org/10.1039/b204956b
Yermakov Y.I., Kuznetsov B.N., Zakharov V.A. Catalysis by Supported Complexes. Amsterdam: Elsevier, 1981. 542 p.
Binnemans K. // Chem. Rev. 2009. V. 109. P. 4283. https://doi.org/10.1021/cr8003983
Badini G.E., Grattan K.T.V., Tseung A.C.C. // Rev. Sci. Instrum. 1995. V. 66. P. 4034. https://doi.org/10.1063/1.1145413
Leventis N., Elder I.A., Rolison D.R. et al. // Chem. Mater. 1999. V. 11. P. 2837. https://doi.org/10.1021/cm9901966
Dang T.T., Shan S.P., Ramalingam B. et al. // RSC Adv. 2015. V. 5. P. 42399. https://doi.org/10.1039/C5RA07225E
Grau A., Baeza A., Serrano E. et al. // ChemCatChem. 2015. V. 7. P. 87. https://doi.org/10.1002/cctc.201402599
Garon S., Lau E.K.C., Chew S.L. et al. // J. Soc. Inf. Disp. 2005. V. 13. P. 405. https://doi.org/10.1889/1.1927732
Rohr M., Günther M., Jutz F. et al. // Appl. Catal. A. 2005. V. 296. P. 238. https://doi.org/10.1016/j.apcata.2005.08.025
Schmid L., Rohr M., Baiker A. // Chem. Commun. 1999. P. 2303. https://doi.org/10.1039/a906956i
Kröcher O., Köppel R.A., Fröba M. et al. // J. Catal. 1998. V. 178. P. 284. https://doi.org/10.1006/jcat.1998.2151
Kröcher O., Köppel R.A., Baiker A. // Chem. Commun. 1996. P. 1497. https://doi.org/10.1039/CC9960001497
Hartley F.R., Vezey P.N. // Adv. Organomet. Chem. 1977. V. 15. P. 189. https://doi.org/10.1016/S0065-3055(08)60129-X
Glauser S.A.C., Lee H.W.H. // MRS Online Proc. L-ibr. 1997. V. 471. P. 331. https://doi.org/10.1557/proc-471-331
Golovkin S.V., Vasil’chenko V.G., Solovev A.S. IHEP Preprint 97-11. 1997. 4 p. http://web.ihep.su/library/pubs/prep1997/ps/97-11.pdf
Reisfeld R. // Opt. Mater. 2001. V. 16. P. 1. https://doi.org/10.1016/S0925-3467(00)00052-5
Bockhorst M., Heinloth K., Pajonk G.M. et al. // J. Non. Cryst. Solids 1995. V. 186. P. 388. https://doi.org/10.1016/0022-3093(95)00064-X
Gupta K.C., Kumar Sutar A., Lin C.C. // Coord. Chem. Rev. 2009. V. 253. P. 1926. https://doi.org/10.1016/j.ccr.2009.03.019
Annis D.A., Jacobsen E.N. // J. Am. Chem. Soc. 1999. V. 121. P. 4147. https://doi.org/10.1021/ja984410n
Çetinkaya B., Seçkin T., Özdemir I. et al. // J. Mater. Chem. 1998. V. 8. P. 1835. https://doi.org/10.1039/a802576b
Pajonk G.M.M. // Catal. Today 1997. V. 35. P. 319. https://doi.org/10.1016/S0920-5861(96)00163-0
Bang Y., Park S., Han S.J. et al. // Appl. Catal. B. 2016. V. 180. P. 179. https://doi.org/10.1016/j.apcatb.2015.06.023
Debecker D.P., Mutin P.H. // Chem. Soc. Rev. 2021. V. 41. P. 3624. https://doi.org/10.1039/C2CS15330K
Fadieiev Y.M., Smola S.S., Rusakova M.Y. et al. // J. Lumin. 2018. V. 194. P. 631. https://doi.org/10.1016/j.jlumin.2017.09.025
Tagaya M., Ogawa M. // Chem. Lett. 2005. V. 35. P. 108. https://doi.org/10.1246/cl.2006.108
Serrano E., Linares N., Garcia-Martinez J. et al. // ChemCatChem. 2013. V. 5. P. 844. https://doi.org/10.1002/cctc.201200938
Amonette J.E., Matyáš J. // Microporous Mesoporous Mater. 2017. V. 250. P. 100. https://doi.org/10.1016/j.micromeso.2017.04.055
Hüsing N., Schubert U., Misof K. et al. // Chem. Mater. 1998. V. 10. P. 3024. https://doi.org/10.1021/cm980706g
Schubert U., Tewinkel S., Lamber R. // Chem. Mater. 1996. V. 8. P. 2047. https://doi.org/10.1021/cm960162n
Watton S.P., Taylor C.M., Kloster G.M. et al. Progress in Inorganic Chemistry. N.Y.: John Wiley & Sons, 2002. V. 51. P. 333. https://doi.org/10.1002/0471267287
Al-Oweini R., Aghyarian S., El-Rassy H. // J. Sol-Gel Sci. Technol. 2012. V. 61. P. 541. https://doi.org/10.1007/s10971-011-2657-7
Faghihian H., Nourmoradi H., Shokouhi M. // Polish J. Chem. Technol. 2012. V. 14. P. 50. https://doi.org/10.2478/v10026-012-0059-4
Sharma R.K., Gulati S., Pandey A. // J. Porous Mater. 2013. V. 20. P. 937. https://doi.org/10.1007/s10934-012-9671-1
Sharma R.K., Sharma S., Gaba G. // RSC Adv. 2014. V. 4. P. 49198. https://doi.org/10.1039/C4RA10384J
Faghihian H., Nourmoradi H., Shokouhi M. // Desalin. Water Treat. 2014. V. 52. P. 305. https://doi.org/10.1080/19443994.2013.785367
Collis A.E.C., Horváth I.T. // Catal. Sci. Technol. 2011. V. 1. P. 912. https://doi.org/10.1039/c1cy00174d
Allum K.G., Hancock R.D., Howell I. V. et al. // J. Organomet. Chem. 1975. V. 87. P. 203. https://doi.org/10.1016/S0022-328X(00)91286-5
Štandeker S., Veronovski A., Novak Z. et al. // Desalination. 2011. V. 269. P. 223. https://doi.org/10.1016/j.desal.2010.10.064
Schubert U. // Adv. Eng. Mater. 2004. V. 6. P. 173. https://doi.org/10.1002/adem.200300548
Rupp W., Hüsing N., Schubert U. // J. Mater. Chem. 2002. V. 12. P. 2594. https://doi.org/10.1039/B204956B
Trimmel G., Lembacher C., Kickelbick G. et al. // New J. Chem. 2002. V. 26. P. 759. https://doi.org/10.1039/b110612k
Seçkin T., Çetinkaya B., Özdemir I. // Polym. Bull. 2000. V. 44. P. 47. https://doi.org/10.1007/s002890050572
Murphy E.F., Schmid L., Bürgi T. et al. // Chem. Mater. 2001. V. 13. P. 1296. https://doi.org/10.1021/cm001187w
Heinrichs B., Noville F., Pirard J.P. // J. Catal. 1997. V. 170. P. 366. https://doi.org/10.1006/jcat.1997.1772
Čapka M., Czakoová M., Schubert U. // Appl. Organomet. Chem. 1993. V. 7. P. 369. https://doi.org/10.1002/aoc.590070603
Schubert U., Hiising N., Lorenz A. // Chem. Mater. 1995. V. 7. P. 2010. https://doi.org/10.1021/cm00059a007
Čapka M., Schubert U., Heinrich B. et al. // Collect. Czechoslov. Chem. Commun. 1992. V. 57. P. 2615. https://doi.org/10.1135/cccc19922615
Yan B., Kong L.L., Zhou B. // J. Non. Cryst. Solids. 2009. V. 355. P. 1281. https://doi.org/10.1016/j.jnoncrysol.2009.05.006
Lermontov S.A., Sipyagina N.A., Malkova A.N. et al. // Russ. J. Inorg. Chem. 2015. V. 60. P. 1459. https://doi.org/10.1134/S0036023615120177
Martínez S., Moreno-Mañas M., Vallribera A. et al. // New J. Chem. 2006. V. 30. P. 1093. https://doi.org/10.1039/b604544h
Sipyagina N.A., Minin V. V., Efimov N.N. et al. // Russ. J. Inorg. Chem. 2015. V. 60. P. 1459. https://doi.org/10.1134/s0036023615120177
Yan Z.Y., Yan B. // New J. Chem. 2014. V. 38. P. 2604. https://doi.org/10.1039/c3nj01639k
Li Z., Ji Y., Cadigan C. et al. // Catal. Sci. Technol. 2014. V. 4. P. 2520. https://doi.org/10.1039/c3cy01064c
Pérez L.L., Zarubina V., Mayoral A. et al. // Catal. Today. 2015. V. 250. P. 115. https://doi.org/10.1016/j.cattod.2014.05.017
Pakharukova V.P., Shalygin A.S., Gerasimov E.Y. et al. // J. Solid State Chem. 2016. V. 233. P. 294. https://doi.org/10.1016/j.jssc.2015.11.007
Yang J., Wang Q., Wang T. et al. // RSC Adv. 2016. V. 6. P. 26271. https://doi.org/10.1039/c5ra28053b
Fujita K., Tokudome Y., Nakanishi K. et al. // J. Non. Cryst. Solids. 2008. V. 354. P. 659. https://doi.org/10.1016/j.jnoncrysol.2007.06.091
Gill S.K., Brown P., Ogundiya M.T. et al. // J. Sol-Gel Sci. Technol. 2010. V. 53. P. 635. https://doi.org/10.1007/s10971-009-2142-8
Wang J., Ma Y., Zeng M. et al. // Appl. Catal. A. 2011. V. 396. P. 123. https://doi.org/10.1016/j.apcata.2011.02.001
Bang Y., Han S.J., Yoo J. et al. // Appl. Catal. B. 2014. V. 148–149. P. 269. https://doi.org/10.1016/j.apcatb.2013.11.012
Zu G., Shen J., Wang W. et al. // ACS Appl. Mater. Interfaces. 2015. V. 7. P. 5400. https://doi.org/10.1021/am5089132
Ponthieu E., Grimblot J., Elaloui E. et al. // J. Mater. Chem. 1993. V. 3. P. 287. https://doi.org/10.1039/jm9930300287
Cao F., Ren L., Li X. // RSC Adv. 2015. V. 5. P. 18025. https://doi.org/10.1039/c5ra00424a
Ren L., Li X., Cui S. // J. Nanomater. 2016. V. 2016. P. 1. https://doi.org/10.1155/2016/8135043
Zhang R., Jiang N., Duan X.J. et al. // New Carbon Mater. 2017. V. 32. P. 258. https://doi.org/10.1016/S1872-5805(17)60122-7
Xie M., Wu X., Liu J. et al. // Ceram. Int. 2017. V. 43. P. 9896. https://doi.org/10.1016/j.ceramint.2017.04.175
Zhang X., Zhang R., Jin S. et al. // J. Sol-Gel Sci. Technol. 2018. V. 87. P. 486. https://doi.org/10.1007/s10971-018-4679-x
Miller J.B., Ko E.I. // Catal. Today 1998. V. 43. P. 51. https://doi.org/10.1016/S0920-5861(98)00134-5
Krompiec S., Mrowiec-Białoń J., Skutil K. et al. // J. Non. Cryst. Solids. 2003. V. 315. P. 297. https://doi.org/10.1016/S0022-3093(02)01609-5
Younes M.K., Ghorbel A., Rives A. et al. // J. Sol-Gel Sci. Technol. 2003. V. 26. P. 677. https://doi.org/10.1023/A:1020769331891
Domínguez G., Phillips M.L.F., Jones S.M. et al. // J. Non. Cryst. Solids. 2004. V. 350. P. 385. https://doi.org/10.1016/j.jnoncrysol.2004.06.027
Courthéoux L., Popa F., Gautron E. et al. // J. Non. Cryst. Solids. 2004. V. 350. P. 113. https://doi.org/10.1016/j.jnoncrysol.2004.06.051
Kobayashi H., Tadanaga K., Minami T. // J. Mater. Chem. 1998. V. 8. P. 1241. https://doi.org/10.1039/a800778k
Popa A.F., Courthéoux L., Gautron E. et al. // Eur. J. Inorg. Chem. 2005. V. 2005. P. 543. https://doi.org/10.1002/ejic.200400657
Yu Z.Q., Wang C.X., Li C. et al. // J. Cryst. Growth. 2003. V. 256. P. 210. https://doi.org/10.1016/S0022-0248(03)01313-7
Peterlik H., Rennhofer H., Torma V. et al. // J. Non. Cryst. Solids. 2007. V. 353. P. 1635. https://doi.org/10.1016/j.jnoncrysol.2007.01.016
Horiuchi T., Chen L., Osaki T. et al. // Catal. Lett. 1999. V. 58. P. 89. https://doi.org/10.1023/A:1019069631852
Nava R., Halachev T., Rodríguez R. et al. // Microporous Mesoporous Mater. 2005. V. 78. P. 91. https://doi.org/10.1016/j.micromeso. 2004.09.021
Osaki T., Nagashima K., Watari K. et al. // J. Non. Cryst. Solids. 2007. V. 353. P. 2436. https://doi.org/10.1016/j.jnoncrysol.2007.04.016
Li X., Qin G., Wang Y. et al. // J. Porous Mater. 2014. V. 21. P. 611. https://doi.org/10.1007/s10934-014-9807-6
Yu Y., Zhu M., Fang J. // RSC Adv. 2017. V. 7. P. 1540. https://doi.org/10.1039/c6ra26601k
Yang J., Wang Q., Wang T. et al. // J. Porous Mater. 2017. V. 24. P. 889. https://doi.org/10.1007/s10934-016-0328-3
Mizushima Y., Hori M. // J. Non. Cryst. Solids. 1994. V. 170. № 3. P. 215. https://doi.org/10.1016/0022-3093(94)90050-7
Kantam M.L., Jaya V.S., Sreedhar B. et al. // J. Mol. Catal. A. 2006. V. 256. P. 273. https://doi.org/10.1016/j.molcata.2006.04.054
Osaki T., Horiuchi T., Sugiyama T. et al. // J. Non. Cryst. Solids. 1998. V. 225. P. 111. https://doi.org/10.1016/S0022-3093(98)00015-5
Kudrawiec R., Podhorodecki A., Mirowska N. et al. // Mater. Sci. Eng. B. 2003. V. 105. P. 53. https://doi.org/10.1016/j.mseb.2003.08.055
Corrias A., Casula M.F., Falqui A. et al. // Chem. Mater. 2004. V. 16. P. 3130. https://doi.org/10.1021/cm049796h
Al-Yassir N., Le Van Mao R. // Can. J. Chem. 2008. V. 86. P. 146. https://doi.org/10.1139/v07-138
Novakovic T., Rozic L., Vukovic Z. et al. // Hem. India. 2011. V. 65. P. 355. https://doi.org/10.2298/HEMIND110406031N
Osaki T., Yamada K., Watari K. et al. // Catal. Lett. 2012. V. 142. P. 95. https://doi.org/10.1007/s10562-011-0692-1
Xu X.Y., Yan B. // RSC Adv. 2014. V. 4. P. 38761. https://doi.org/10.1039/c4ra05704j
Wang W., Zhang Z., Zu G. et al. // RSC Adv. 2014. V. 4. P. 54864. https://doi.org/10.1039/c4ra08832h
Fu T., Tang J., Chen K. et al. // Appl. Opt. 2016. V. 55. P. 705. https://doi.org/10.1364/AO.55.000705
Zou W., Wang X., Wu Y. et al. // J. Sol-Gel Sci. Technol. 2017. V. 84. P. 507. https://doi.org/10.1007/s10971-017-4380-5
Al-Yassir N., Le Van Mao R. // Appl. Catal. A Gen. 2007. V. 317. P. 275. https://doi.org/10.1016/j.apcata.2006.10.030
Bao W., Guo F., Zou H. et al. // Powder Technol. 2013. V. 249. P. 220. https://doi.org/10.1016/j.powtec.2013.08.001
Jung I.K., Ha T.J., Park H.H. et al. // Surf. Coatings Technol. 2013. V. 231. P. 185. https://doi.org/10.1016/j.surfcoat.2012.05.049
Li X., He H., Ren L. // J. Porous Mater. 2017. V. 24. P. 679. https://doi.org/10.1007/s10934-016-0304-y
Wang X., Guo Y., Lu G. et al. // Catal. Today. 2007. V. 126. P. 369. https://doi.org/10.1016/j.cattod.2007.06.011
Al-Yassir N., Mao R.L. Van // Appl. Catal. A. 2007. V. 332. P. 273. https://doi.org/10.1016/j.apcata.2007.08.023
Xiaohong W., Guanzhong L.U., Yun G. et al. // Chinese J. Catal. 2008. V. 29. P. 1043. https://doi.org/10.1016/S1872-2067(08)60081-9
Soloviev S.O., Kapran A.Y., Orlyk S.N. et al. // J. Nat. Gas Chem. 2011. V. 20. P. 184. https://doi.org/10.1016/S1003-9953(10)60149-1
Fanelli A.J., Burlew J. V., Marsh G.B. // J. Catal. 1989. V. 116. P. 318. https://doi.org/10.1016/0021-9517(89)90099-7
Zu G., Shen J., Zou L. et al. // Chem. Mater. 2013. V. 25. P. 4757. https://doi.org/10.1021/cm402900y
Mizushima Y., Hori M., Sasaki M. // J. Mater. Res. 1993. V. 8. P. 2109. https://doi.org/10.1557/JMR.1993.2109
Osaki T., Mori T. // J. Non. Cryst. Solids. 2009. V. 355. P. 1590. https://doi.org/10.1016/j.jnoncrysol.2009.06.006
Pierre A., Begag R., Pajonk G. // J. Mater. Sci. 1999. V. 34. P. 4937. https://doi.org/10.1023/A:1004703504103
Le Bihan L., Dumeignil F., Payen E. et al. // J. Sol-Gel Sci. Technol. 2002. V. 24. P. 113. https://doi.org/10.1023/A:1015235623713
Sinkó K., Kubuki S., Peterlik H. // Inorg. Chem. 2013. V. 52. P. 13238. https://doi.org/10.1021/ic402189c
Cutrufello M.G., Rombi E., Ferino I. et al. // J. Sol-Gel Sci. Technol. 2011. V. 60. P. 324. https://doi.org/10.1007/s10971-011-2460-5
Triki M., Ksibi Z., Ghorbel A. et al. // J. Sol-Gel Sci. Technol. 2011. V. 59. P. 1. https://doi.org/10.1007/s10971-011-2452-5
Cuan J., Yan B. // RSC Adv. 2014. V. 4. P. 1735. https://doi.org/10.1039/c3ra45281f
Yorov K.E., Yapryntsev A.D., Baranchikov A.E. et al. // J. Sol-Gel Sci. Technol. 2018. V. 86. P. 400. https://doi.org/10.1007/s10971-018-4647-5
Yorov K.E., Khodan A.N., Baranchikov A.E. et al. // Microporous Mesoporous Mater. 2020. V. 293. 109804. https://doi.org/10.1016/j.micromeso.2019.109804
Wislicenus H. // Z. Chem. Ind. Kolloide. 1908. V. 2. P. XI. https://doi.org/10.1007/BF01526935
Bai J.B., Vignes J.L., Fournier T. et al. // Adv. Eng. Mater. 2002. V. 4. P. 701. https://doi.org/10.1002/1527-2648(20020916)4:9<701:: AIDADEM701>3.0.CO;2-T
Vignes J.L., Mazerolles L., Michel D. // Key Eng. Mater. 2009. V. 132–136. P. 432. https://doi.org/10.4028/www.scientific.net/kem.132-136.432
Vignes J.-L.L., Frappart C., Di Costanzo T. et al. // J. Mater. Sci. 2008. V. 43. P. 1234. https://doi.org/10.1007/s10853-007-2260-z
Khodan A.N., Kopitsa G.P., Yorov K.E. et al. // J. Surf. Invest. 2018. V. 12. P. 296. https://doi.org/10.1134/S102745101802026X
Straumal E.A., Yurkova L.L., Baranchikov A.E. et al. // Russ. J. Inorg. Chem. 2021. V. 66. P. 288. https://doi.org/10.1134/S0036023621020194
Huang Z.R., Jiang D., Michel D. et al. // J. Mater. Res. 2002. V. 17. P. 3177. https://doi.org/10.1557/JMR.2002.0459
Mazerolles L., Michel D., Di Costanzo T., Vignes J.-L. // Ceram. Trans. 2002. V. 135. P. 227. https://doi.org/10.1002/9781118380826.ch20
Mazerolles L., Michel D., Di Costanzo T. et al. // Ceram. Eng. Sci. Proc. 2003. V. 24. P. 105. https://doi.org/10.1002/9780470294802.ch16
Logie V., Maire G., Michel D. et al. // J. Catal. 1999. V. 188. P. 90. https://doi.org/10.1006/jcat.1999.2600
Di-Grégorio F., Keller V., Di-Costanzo T. et al. // Appl. Catal. A. 2001. V. 218. P. 13. https://doi.org/10.1016/S0926-860X(01)00587-7
Wegener S.L., Marks T.J., Stair P.C. // Acc. Chem. Res. 2012. V. 45. P. 206. https://doi.org/10.1021/ar2001342
Chisem I.C., Rafelt J., Shieh M.T. et al. // Chem. Commun. 1998. P. 1949. https://doi.org/10.1039/a805420g
Jesenák K., Gáplovsky A., Lácová M. et al. // Solid State Phenom. 2003. V. 90–91. P. 487. https://doi.org/10.4028/www.scientific.net/SSP.90-91.487
Eid J., Pierre A.C., Baret G. // J. Non. Cryst. Solids. 2005. V. 351. P. 218. https://doi.org/10.1016/j.jnoncrysol.2004.11.020
El Mir L., El Ghoul J., Alaya S. et al. // Phys. B. 2008. V. 403. P. 1770. https://doi.org/10.1016/j.physb.2007.10.069
El Mir L., Amlouk A., Barthou C. et al. // Mater. Sci. Eng. C. 2008. V. 28. P. 771. https://doi.org/10.1016/j.msec.2007.10.019
Allison S.W., Baker E.S., Lynch K.J. et al. // Radiat. Phys. Chem. 2017. V. 135. P. 88. https://doi.org/10.1016/j.radphyschem.2017.01.045
Cheng W., Rechberger F., Niederberger M. // ACS Nano. 2016. V. 10. P. 2467. https://doi.org/10.1021/acsnano.5b07301
García-Murillo A., Carrillo-Romo F.J., Oliva-Uc J. et al. // Ceram. Int. 2017. V. 43. P. 12196. https://doi.org/10.1016/j.ceramint.2017.06.079
Gutzov S., Danchova N., Kirilova R. et al. // J. Lumin. 2017. V. 183. P. 108. https://doi.org/10.1016/j.jlumin.2016.11.029
El Mir L., Amlouk A., Barthou C. et al. // Physica. B. 2007. V. 388. P. 412. https://doi.org/10.1016/j.physb.2006.06.151
Amlouk A., El Mir L., Kraiem S. et al. // Mater. Sci. Eng. B. 2008. V. 146. P. 74. https://doi.org/10.1016/j.mseb.2007.07.056
Alattar A.M., Drexler M., Twej W.A.A. et al. // Photonics Nanostruct. 2018. V. 30. P. 65. https://doi.org/10.1016/j.photonics. 2018.04.004
Sorensen L., Strouse G.F., Stiegman A.E. // Adv. Mater. 2006. V. 18. P. 1965. https://doi.org/10.1002/adma.200600791
Zhu L., Li Y., Wang J. et al. // J. Appl. Phys. 1995. V. 77. P. 2801. https://doi.org/10.1063/1.358685
Aghajamali M., Iqbal M., Purkait T.K. et al. // Chem. Mater. 2016. V. 28. P. 3877. https://doi.org/10.1021/acs.chemmater.6b01114
Cao W., Hunt A.J. // Appl. Phys. Lett. 1994. V. 64. P. 2376. https://doi.org/10.1063/1.111619
Yan B., Zhang H.-J., Wang S.-B. et al. // J. Photochem. Photobiol. A. 1998. V. 112. P. 231. https://doi.org/10.1016/S1010-6030(97)00303-1
Ayers M. // J. Non. Cryst. Solids 1997. V. 217. P. 229. https://doi.org/10.1016/S0022-3093(97)00126-9
Utochnikova V.V., Kuzmina N.P. // Russ. J. Coord. Chem. 2016. V. 42. P. 679. https://doi.org/10.1134/S1070328416090074
Stan C.S., Marcotte N., Secula M.S. et al. // J. Sol-Gel Sci. Technol. 2014. V. 69. P. 207. https://doi.org/10.1007/s10971-013-3205-4
Stan C.S., Rosca I., Sutiman D. et al. // J. Rare Earths. 2012. V. 30. P. 401. https://doi.org/10.1016/S1002-0721(12)60061-1
Yorov K.E., Kottsov S.Y., Baranchikov A.E. et al. // J. Sol-Gel Sci. Technol. 2019. V. 92. P. 304. https://doi.org/10.1007/s10971-019-04958-9
Carroll M.K., Anderson A.M. // Aerogels Handbook. New York: Springer, 2011. P. 637. https://doi.org/10.1007/978-1-4419-7589-8_27
Lukowiak A., Strek W. // J. Sol-Gel Sci. Technol. 2009. V. 50. P. 201. https://doi.org/10.1007/s10971-009-1952-z
Leventis N., Rawashdeh A.M.M., Elder I.A. et al. // Chem. Mater. 2004. V. 16. P. 1493. https://doi.org/10.1021/cm034999b
Plata D.L., Briones Y.J., Wolfe R.L. et al. // J. Non. Cryst. Solids. 2004. V. 350. P. 326. https://doi.org/10.1016/j.jnoncrysol.2004.06.046
Atkinson E.J.O., Gilbert B.D., Carlson E.R. // J. Non. Cryst. Solids. 2014. V. 405. P. 16. https://doi.org/10.1016/j.jnoncrysol.2014.08.033
Anderson M.L., Rolison D.R., Merzbacher C.I. // Proc. Vol. 3790. Engineered Nanostructural Films and Materials. 1999. P. 38. https://doi.org/10.1117/12.351260
Tai Y., Watanabe M., Kaneko K. et al. // Adv. Mater. 2001. V. 13. P. 1611. https://doi.org/10.1002/1521-4095(200111)13:21<1611:: AID-ADMA1611>3.0.CO;2-V
Kudo A., Miseki Y. // Chem. Soc. Rev. 2009. V. 38. P. 253. https://doi.org/10.1039/B800489G
Fujishima A., Zhang X., Tryk D. // Surf. Sci. Rep. 2008. V. 63. P. 515. https://doi.org/10.1016/j.surfrep.2008.10.001
Hoffmann M.R., Martin S.T., Choi W. et al. // Chem. Rev. 1995. V. 95. P. 69. https://doi.org/10.1021/cr00033a004
Ingale S. V., Wagh P.B., Tripathi A.K. et al. // J. Sol-Gel Sci. Technol. 2011. V. 58. P. 682. https://doi.org/10.1007/s10971-011-2445-4
Wang H.-L., Liang W.-Z., Jiang W.-F. // Mater. Chem. Phys. 2011. V. 130. P. 1372. https://doi.org/10.1016/J.MATCHEMPHYS.2011.09.033
Yoda S., Ohtake K., Takebayashi Y. et al. // J. Mater. Chem. 2000. V. 10. P. 2151. https://doi.org/10.1039/b002019m
Malinowska B., Walendziewski J., Robert D. et al. // Appl. Catal. B. 2003. V. 46. P. 441. https://doi.org/10.1016/S0926-3373(03)00273-X
Ismail A., Ibrahim I., Ahmed M. et al. // J. Photochem. Photobiol. A. 2004. V. 163. P. 445. https://doi.org/10.1016/J.JPHOTOCHEM.2004.01.017
Wang J., Uma S., Klabunde K.. J. // Appl. Catal. B. 2004. V. 48. P. 151. https://doi.org/10.1016/j.apcatb. 2003.10.006
Cao S., Yeung K.L., Yue P.L. // Appl. Catal. B. 2006. V. 68. P. 99. https://doi.org/10.1016/j.apcatb.2006.07.022
Wang X., Liu J., Shi F. et al. // J. Porous Mater. 2014. V. 21. P. 293. https://doi.org/10.1007/s10934-013-9774-3
Yorov K.E., Sipyagina N.A., Malkova A.N. et al. // Inorg. Mater. 2016. V. 52. P. 163. https://doi.org/10.1134/S0020168516020035
Yorov K.E., Sipyagina N.A., Baranchikov A.E. et al. // Russ. J. Inorg. Chem. 2016. V. 61. P. 1339. https://doi.org/10.1134/S0036023616110048
Cassin G., Diridollou S., Flament F. et al. // Int. J. Cosmet. Sci. 2018. V. 40. P. 58. https://doi.org/10.1111/ics.12433
Han K., Yu M. // J. Appl. Polym. Sci. 2006. V. 100. P. 1588. https://doi.org/10.1002/app.23312
KRONOS Titanium Dioxide. https://kronostio2. com/en/data-sheets-and-literature/brochures/send/ 2-brochures/81-grades-and-applications
Kolesnik I. V., Shcherbakov A.B., Kozlova T.O. et al. // Russ. J. Inorg. Chem. 2020. V. 65. P. 960. https://doi.org/10.1134/S0036023620070128
Yorov K.E., Kolesnik I.V., Romanova I.P. et al. // J. Supercrit. Fluids. 2020. V. 169. P. 105099. https://doi.org/10.1016/j.supflu.2020.105099
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия