

Координационная химия, 2022, T. 48, № 3, стр. 185-192
Синтез и строение трис(пиразолил)боратных комплексов рения c о-донорными лигандами
И. В. Скабицкий 1, *, С. С. Шаповалов 1
1 Институт общей и неорганический химии им. Н.С. Курнакова РАН
Москва, Россия
* E-mail: skabitskiy@gmail.com
Поступила в редакцию 06.05.2021
После доработки 06.08.2021
Принята к публикации 07.08.2021
- EDN: PIIGOT
- DOI: 10.31857/S0132344X22030045
Аннотация
Реакцией TpReOCl2 (Tp = трис(пиразолил)боратный анион) c метанолом в присутствии триэтиламина получен новый алкоголятный комплекс рения TpReO(OMe)2 (I). В отличие от исходного комплекса, устойчивого к гидролизу, I реагирует с водой в толуоле с образованием димерного комплекса [TpReO(μ-O)]2 (II). Действие большего количества воды в ацетоне приводит также к разложению части Tp-лигандов и возникновению катионного комплекса [(TpReO)2(μ-O)(μ-C3H3N2)]ReO4 (III) с пиразолатным мостиковым лигандом и перренат анионом на внешней сфере. Действием уксусного ангидрида на комплекс I был получен трис(пиразолил)боратный комплекс рения, содержащий терминальные ацетатные заместители TpReO(OAc)2 (IV). Полученные комплексы охарактеризованы ИК- и ЯМР-спектроскопией, их строение установлено методом РСА (CIF files CCDC № 2081834 (I), 2081835 (II), 2081836 (III), 2081837 (IV)).
Трис(пиразолил)боратные анионы (Tp– = = ${\text{HB(}}{{{\text{C}}}_{{\text{3}}}}{{{\text{H}}}_{{\text{3}}}}{{{\text{N}}}_{2}})_{3}^{ - }$), являющиеся тридентатными лигандами и образующие стабильные комплексы с переходными металлами, нашли широкое применение в координационной химии [1]. При этом трис(пиразолил)боратные анионы являются лигандами более слабого поля, хотя их часто рассматривают как аналог циплопентадиенильных анионов. Соответствующие комплексы, содержащие Tp–, в отличие от диамагнитных ферроценов, часто оказываются высокоспиновыми [2]. В случае рения известны комплексы с трис-пиразоллилборатным лигандом для степеней окисления с I по VIII, причем наиболее изученными типами таких комплексов являются LReOX2 (L = HBPz3 или BPz4, Pz = пиразол, X = моноанионный лиганд) и производные TpReI(CO)3, образующиеся при замещении одной и более групп CO на различные лиганды [3]. Среди оксокомплексов Re(V) первого типа заметную долю занимают алкоголятные комплексы (X = OR) в основном с хелатными алкоголятными заместителями.
Алкоголятные комплексы (BPz4)ReO(OR)2 (R = = Me, iPp, Ph), и (BPz4)ReO(OXO) (X = CH2CH2, o-C6H4) были получены действием соответствующих спиртов на оксомостиковый [(BPz4)ReO(μ-O)]2 [4]. Реакция бис-метилатного комплекса с диолами также использовалась для синтеза комплексов с бидентантными алкоголятами с размером хелатного цикла от 5 до 7 (X = CHMeCH2, CHMeCHMe, CMe2CMe2, CH2CH2CH2, C6H10, CH2CH2CH2CH2), которые также могут быть получены одностадийным синтезом из KBPz4- (PPh3)2ReCl3O и соответствующих диолов [5]. Аналогичный комплекс TpReO(OCHMeCH2O) был получен действием HOCHMeCH2OH на TpReOCl2 в присутствии триэтиламина в ацетонитриле и выделен в энантиомерно чистой форме как потенциальный объект для наблюдения нарушения пространственной четности спектроскопическими методами [6]. В похожих условиях, но с использованием метанола в качестве растворителя были получены диолатные комплексы производные ангидроэритрита и метил-β-D-галактопиранозы [7]. Также наблюдалось образование фенолятных комплексов TpReOCl(OPh) и TpReO(O2C6H4) в реакциях окисления фенильной группы на атоме рения в TpReOPh(OTf) под действием оксида пиридина [8].
В отличие от алкоголятных комплексов, карбоксилатные производные трис(пиразолил)бората рения ограничены лишь несколькими примерами. Оксалатный комплекс TpReO(C2O4) (схема 1 ) был получен действием щавелевой кислоты на TpReO(OCH2CH2O) в ацетонитриле [9]. В той же работе были исследованы его фотохимические превращения, связанные с возникновением интермедиата TpReO при фотохимическом отщеплении оксалатного лиганда. Вторым примером являются комплексы с карбоксилаталкоголятным лигандом LReO(Ph2C(O)COO) (схема 1 ), возникающие в результате присоединения дифенилкетена к LReO3 (L = Tp, PhBPz3) [10].

Схема 1 .
В данной работе мы синтезировали трис(пиразолил)боратные оксокомплексы Re(V), содержащие метилатные и ацетатные заместители, а также охарактеризовали продукты их гидролиза.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Все операции в ходе синтеза и выделения продуктов проводили в абсолютных растворителях в атмосфере чистого аргона. ИК-спектры регистрировали при помощи спектрометра Bruker Alpha с приставкой НПВО Bruker ATR Diamond. Спектры ЯМР 1H и 13C регистрировали на приборе Bruker AV 300; химические сдвиги приведены относительно тетраметилсилана. Коммерческие триэтиламин и уксусный ангидрид использовали без дополнительной очистки. Синтез TpReOCl2 [11] проводили согласно литературной методике.
Синтез TpReO(OMe)2 (I). К суспензии 408 мг (0.84 ммоль) TpReOCl2 в 60 мл метанола добавляли 235 мкл триэтиламина (1.7 ммоль). Реакционную смесь перемешивали при 50°C в течение ночи, полученый прозрачный синий раствор концентрировали до 1/4 объема. Выпавшие при выдерживании при –25°C синие кристаллы I · MeOH отделяли декантацией, промывали охлажденным до –80°C метанолом и высушивали в вакууме. Выход 374 мг (87%).
ИК (ν, см–1): 3289 ш.о.сл, 3120 сл, 3106 о.сл, 2916 ш.о.сл, 2819 о.сл, 2804 о.сл, 2518 о.сл, 1504 сл, 1435 о.сл, 1404 ср, 1390 о.сл, 1312 с, 1207 с, 1161 о.сл, 1118 сл, 1103 о.сл, 1072 о.сл, 1049 сл, 1036 о.сл, 1017 с, 991 о.сл, 961 о.с, 858 о.сл, 802 сл, 789 о.сл, 768 с, 728 о.сл, 710 с, 672 о.сл, 653 ср, 620 ср, 560 сл, 535 с.
ЯМР 1H (CDCl3; 298 K; δ, м.д.): 1.18 (к., 3JH–H = = 5.5 Гц, 1H, MeOH), 3.44 (д., 3JH–H = 5.5 Гц, 3H, MeOH), 4.66 (c. 6H, MeO), 6.07 (т., 3JH–H = 2.2 Гц, 1H), 6.41 (т., 3JH–H = 2.2 Гц, 2H), 7.42 (д.д., 3JH–H = = 2.1 Гц, 4JH–H ≈ 0.6 Гц, 1H), 7.60 (д., 3JH–H = 2.1 Гц, 1H), 7.75 (д.д., 3JH–H = 2.5 Гц, 4JH–H ~ 0.6 Гц, 2H), 7.89 (д., 3JH–H = 2.1 Гц, 2H).
ЯМР 13С{1H} (CDCl3; 298 K; δ, м.д.): 50.93 (MeOH), 73.45 (OMe), 106.18, 107.82, 134.48, 137.97, 143.11, 147.34.
Образец I, не содержащий сольватного метанола, получали нагреванием I · MeOH в вакууме при 80°C в течение 20 мин.
| Найдено, %: | C 28.83; | H 3.43; | N 17.30. |
| Для C11H16BN6O3Re (M = 477) | |||
| вычислено, %: | C 27.68; | H 3.38; | N 17.60. |
ЯМР 1H (d8-толуол; 298 K; δ, м.д.): 4.83 (c. 6H, MeO), 5.51 (т., 3JH–H = 2.2 Гц, 1H), 5.78 (т., 3JH–H = = 2.3 Гц, 2H), 6.86 (д., 3JH–H = 2.5 Гц, 1H), 7.16 (д., 3JH–H = 2.4 Гц, 2H), 7.46 (д., 3JH–H = 2.1 Гц, 1H), 7.74 (д., 3JH–H = 2.2 Гц, 2H).
Синтез [TpReO(μ-O)]2 (II). Суспензию 50 мг (0.11 ммоль) I и 200 мкл воды в 30 мл толуола выдерживали в ультразвуковой бане в течение 20 мин до растворения исходного. Выпадавший при выдерживании в течение 2 сут желто-коричневый осадок отделяли декантацией, промывали 2 мл толуола, высушивали в вакууме. Выход 31 мг (65%).
| Найдено, %: | C 25.55; | H 2.73; | N 19.50. |
| Для C18H20B2N12O4Re2 (M = 853) | |||
| Вычислено, %: | C 25.07; | H 2.34; | N 19.49. |
ИК (ν, см–1): 3108 ш.сл, 2532 о.сл, 1707 ср, 1698 о.сл, 1505 ср, 1430 о.сл, 1405 о.с, 1392 о.сл, 1365 ср, 1310 о.с, 1282 сл, 1208 о.с, 1183 ср, 1122 ср, 1108 о.сл, 1077 о.сл, 1050 о.с, 1025 ср, 996 о.сл, 975 с, 896 ш.о.с, 814 о.сл, 763 ср, 724 о.сл, 710 ш.о.с, 672 о.сл, 650 сл, 612 ср, 530 сл, 498 сл, 459 о.сл, 441 о.сл, 422 о.сл, 409 о.сл.
Пригодные для РСА кристаллы II · CDCl3 получали выдерживанием раствора I в CDCl3, содержащем следы воды, в течение недели.
Синтез [(TpReO)2(µ-O)(µ-C3H3N2)]ReO4 · · 2Me2CO (III). Раствор 100 мг (0.22 ммоль) I в смеси 2 мл воды и 30 мл ацетона выдерживали при комнатной температуре в течение недели. Образующиеся крупные черные кристаллы отделяли декантацией, промывали 10 мл ацетона и высушивали в вакууме. Выход 37 мг (40%).
| Найдено, %: | C 24.45; | H 2.68; | N 15.80. |
| Для C27H35B2N14O9Re3 (M = 1280) | |||
| вычислено, %: | C 25.34; | H 2.76; | N 15.32. |
ЯМР 1H (d6-ДМСО; 298 K; δ, м.д.): 2.08 (Me2CO), 4.63 (ш.с. 2H, HB), 6.16 (т. 3JH–H = 2.2 Гц, 2H), 6.50 (д. 3JH–H = 2.3 Гц, 2H), 6.81, (т. 3JH–H = 2.4 Гц, 2H), 6.83 (т. 3JH–H = 2.3 Гц, 2H), 7.30 (т. 3JH–H = 2.5 Гц, 1H), 7.94 (д. 3JH–H = 2.2 Гц, 2H), 8.19 (д. 3JH–H = = 2.3 Гц, 2H), 8.41 (д. 3JH–H = 2.4 Гц, 2H), 8.60 (д. 3JH–H = 2.3 Гц, 2H), 8.88 (д. 3JH–H = 2.1 Гц, 2H), 8.92 (д. 3JH–H = 2.6 Гц, 2H).
ЯМР 13С{1H} (d6-ДМСО; 298 K; δ, м.д.): 30.67 (Me2CO), 106.82, 109.23, 109.86, 116.39, 136.80, 139.79, 141.50, 141.61, 145.61, 148.40, 151.28, 206.48 (Me2CO).
ИК (ν, см–1): 3855 о.сл, 3736 о.сл, 3650 о.сл, 3146 о.сл, 3121 о.сл, 3086 о.сл, 2531 о.сл, ш, 2502 о.сл, 1504 ср, 1404 сл, 1388 с, 1307 с, 1210 о.с, 1187 о.сл, 1124 о.сл, 1116 ср, 1098 о.сл, 1072 о.сл, 1047 о.с, 986 сл, 948 о.с, 918 о.сл, 859 о.сл, 817 сл, 793 о.сл, 779 о.сл, 759 о.с, 730 о.сл, 713 ср, 678 о.сл, 655 сл, 631 о.сл, 617 о.с, 554 сл, 452 о.сл, 440 о.сл.
Синтез TpReO(OAc)2 (IV). Раствор 50 мг (0.11 ммоль) I и 0.5 мл (5.29 ммоль) Ac2O в 20 мл хлористого метилена перемешивали в течение суток. Сине-зеленую реакционную смесь концентрировали в вакууме до 1 мл и выдерживали при –25°C в течение часа. Выпадавшие синие кристаллы IV промывали диэтиловым эфиром и высушивали в вакууме. Выход 23 мг.
Дополнительно получили 8 мг кристаллов IV, пригодных для РСА, диффузией паров диэтилового эфира в маточный раствор в течение 3 сут. Суммарный выход 31 мг (53%).
| Найдено, %: | C 29.23; | H 2.79; | N 15.86. |
| Для C13H16BN6O5Re (M = 533). | |||
| вычислено, %: | C 29.28; | H 3.02; | N 15.76. |
ЯМР 1H (CDCl3; 298 K; δ, м.д.): 2.35 (c. 6H, MeCOO), 6.08 (т., 3JH–H = 2.2 Гц, 1H), 6.47 (т., 3JH–H = = 2.4 Гц, 2H), 7.41 (д.д., 3JH–H = 2.2 Гц, 4JH–H ≈ 0.6 Гц, 1H), 7.85 (д.д., 3JH–H = 2.5 Гц, 4JH–H ≈ 0.6 Гц, 2H), 8.00 (д., 3JH–H = 2.3 Гц, 2H), 8.15 (д., 3JH–H = 2.1 Гц, 1H).
ЯМР 13С{1H} (CDCl3, 298K, δ м.д.): 23.46 (OOCMe), 105.99, 108.53, 134.83, 139.13, 144.94, 148.46, 182.78 (OOCMe).
ИК (ν, см−1): 3141 о.сл, 3121 о.сл, 3101 о.сл, 2539 ш.о.сл, 1686 ср, 1661 ш.ср, 1504 сл, 1441 о.сл, 1404 ср, 1361 ср, 1312 сл, 1283 с, ш, 1243 сл, 1210 ср, 1186 о.сл, 1126 о.сл, 1116 сл, 1084 о.сл, 1052 о.с, 986 ср, 913 сл, 811 о.сл, 772 о.с, 711 ср, 680 о.сл, 670 о.сл, 647 с, 620 сл, 602 о.сл, 565 о.сл, 543 сл, 532 о.сл.
РСА выполнен на дифрактометре Bruker APEX II CCD для комплексов I, III и на дифрактометре Bruker Venture D8 для комплексов II, IV. Поглощение учтено методом множественного измерения эквивалентных отражений по программе SADABS [12]. Структуры I–IV определены прямым методом и уточнены методом наименьших квадратов относительно F 2 в анизотропном приближении неводородных атомов с использованием пакетов программ SHELX-2014 [13] и OLEX2 [14]. Положения атомов Н рассчитаны геометрически. Кристаллографические данные и параметры уточнения структуры I–IV приведены в табл. 1. Основные длины связей и валентные углы в I–IV приведены в подписях к рис. 1–4.
Таблица 1.
Кристаллографические данные и параметры уточнения структуры комплексов I–IV
| Параметр | I | II | III | IV |
|---|---|---|---|---|
| Брутто-формула | C12H20BN6O4Re | C20H22B2N12O4Cl6Re2 | C27H35B2N14O9Re3 | C13H16BN6O5Re |
| M | 509.35 | 1101.21 | 1279.91 | 533.33 |
| Излучение (λ , Å) | MoKα (λ = 0.71073) | |||
| Температура съемки, K | 296 | 100 | 150 | 100 |
| Сингония | Моноклинная | Моноклинная | Орторомбическая | Моноклинная |
| Пр. гр. | P21/n | P21/c | Pbcn | P21/n |
| а, Å | 13.838(6) | 7.7589(2) | 28.7444(6) | 7.5114(4) |
| b, Å | 8.610(4) | 20.2699(6) | 15.6392(4) | 16.3728(9) |
| c, Å | 15.160(5) | 10.4552(3) | 16.9346(4) | 13.6934(8) |
| α, град | 90 | 90 | 90 | 90 |
| β, град | 104.580(13) | 97.1230(10) | 90 | 90.739(2) |
| γ, град | 90 | 90 | 90 | 90 |
| V, Å3 | 1748.1(12) | 1631.62(8) | 7612.8(3) | 1683.91(16) |
| Z | 4 | 2 | 8 | 4 |
| ρ(выч.), г/cм–3 | 1.935 | 2.241 | 2.233 | 2.104 |
| μ, мм–1 | 6.980 | 7.954 | 9.585 | 7.256 |
| F(000) | 984.0 | 1040.0 | 4816.0 | 1024.0 |
| Интервал сканирования, град | 4.606–59.84 | 5.62–60.016 | 4.538–54.378 | 3.878–57.346 |
| Тип сканирования | ω | |||
| Независимых отражений (N1) | 4625 (Rint = 0.0739) | 4752 (Rint = 0.0581) | 8470 (Rint = 0.0727) | 4330 (Rint = 0.0404) |
| Отражений с I > 2σ(I) (N2) | 3475 | 4316 | 7706 | 3922 |
| Число уточняемых параметров | 225 | 209 | 501 | 240 |
| GOОF (F 2) | 1.067 | 1.085 | 1.251 | 1.090 |
| R1 для N2 | 0.0509 | 0.0180 | 0.0418 | 0.0187 |
| wR2 для N1 | 0.0942 | 0.0375 | 0.0675 | 0.0443 |
| Δρmax/Δρmin, e Å–3 | 1.20/–1.83 | 0.85/–0.84 | 1.06/–2.34 | 0.93/–0.72 |
Рис. 1.
Молекулярная структура I (сольватная молекула метанола не показана). Некоторые длины связей: Re(1)–O(1) 1.684(5), Re(1)–O(2) 1.933(5), Re(1)–O(3) 1.969(5), Re(1)–N(1) 2.102(6), Re(1)–N(3) 2.145(5), Re(1)–N(5) 2.241(6) Å и валентные углы: O(1)Re(1)O(2) 108.8(2)° O(1)Re(1)O(3) 103.7(2)°, O(2)Re(1)O(3) 85.6(2)°, C(10)O(2)Re(1) 124.9(4)°, C(11)O(3)Re(1) 124.9(5)°.

Рис. 2.
Катионная часть молекулярной структуры II. Некоторые длины связей: Re(1)–Re(1)i 3.1049(2), Re(1)–O(1) 1.689(2), Re(1)–O(2)i 1.948(2), Re(1)–O(2) 1.974(2), Re(1)–N(1) 2.299(2), Re(1)–N(3) 2.100(2), Re(1)–N(5) 2.134(2) Å и валентные углы: O(1)Re(1)O(2) 104.18(8)°, O(1)Re(1)O(2)i 107.52(8)°, Re(1)iO(2)Re(1) 104.70(8)°.

Рис. 3.
Молекулярная структура III. Некоторые длины связей: Re(1)–O(1) 1.938(4), Re(1)–O(2) 1.676(4), Re(1)–N(1) 2.046(5), Re(1)–N(3) 2.248(5), Re(1)–N(5) 2.133(5), Re(1)–N(7) 2.071(5), Re(2)–O(1) 1.924(4), Re(2)–O(3) 1.688(4), Re(2)–N(2) 2.055(5), Re(2)–N(9) 2.237(5), Re(2)–N(11) 2.159(5), Re(2)–N(13) 2.067(5) Å и валентные углы: Re(2)O(1)Re(1) 130.5(2)°, O(2)Re(1)O(1) 105.3(2)°, O(2)Re(1)N(1) 103.8(2)°, O(3)Re(2)O(1) 105.7(2)°, O(3)Re(2)N(2) 100.2(2)°.
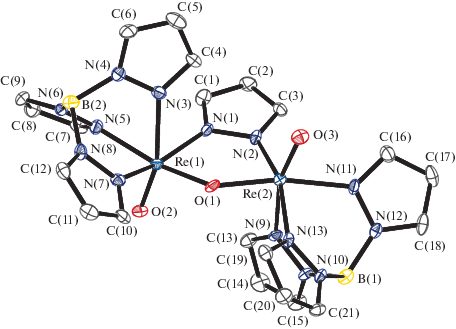
Рис. 4.
Молекулярная структура IV. Некоторые длины связей: Re(1)–O(1) 1.669(2), Re(1)–O(2) 2.005(2), Re(1)–O(4) 1.976(2), Re(1)–N(1) 2.069(2), Re(1)–N(3) 2.098(2), Re(1)–N(5) 2.255(2), O(2)–C(1) 1.273(4), O(3)–C(1) 1.236(5), O(4)–C(3) 1.331(4), O(5)–C(3) 1.216(4) Å и валентные углы: O(1)Re(1)O(2) 111.0(1)°, O(1)Re(1)O(4) 105.9(1)°, O(4)Re(1)O(2) 82.9(1)°, C(1)O(2)Re(1) 130.8(3)°, C(3)O(4)Re(1) 124.2(2)°.

Координаты атомов и другие параметры структур I–IV депонированы в Кембриджском банке структурных данных (CCDC № 2081834–2081837 соответственно; http://www.ccdc.cam.ac.uk/ data_request/cif).
Квантово-химические расчеты проводили в рамках теории функционала плотности с помощью программного комплекса ORCA 4.21 [15]. Оптимизацию геометрии комплексов и переходных состояний проводили с использованием функционала PBE [16, 17] c дважды расщепленным полноэлектронным базисом def2-SVP [18] и использованием эмпирической поправки к дисперсионным взаимодействиям D3BJ [19, 20]. Энергию активации рассчитывали с использованием гибридного функционала PBE0 [21] с трижды расщепленным полноэлектронным базисом def2-TZVP [18].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Действием триэтиламина в кипящем метаноле на TpReOCl2 был получен новый метоксидный комплекс TpReO(OMe)2 (I). В отличие от устойчивого к гидролизу хлоридного исходного, I медленно реагирует с водой: при действии воды в толуоле образуется новый оксомостиковый димерный комплекс [TpReO(μ-O)]2 (II). Действие избытка воды в ацетоне приводит также к расщеплению части трис(пиразолил)боратных лигандов с образованием катионного димера [(TpReO)2(µ-O)(µ-C3H3N2)]ReO4 (III), в котором один из кислородных мостиковых лигандов заменен на пиразолатный.
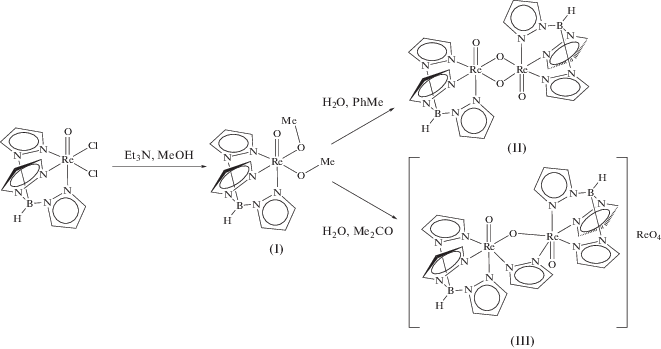
Схема 2 .
Строение комплексов было определено РСА, в частности, молекулярная структура I представлена на рис. 1. Связи ReO с метилатными групами (1.969(5), 1.933(5) Å) заметно более длинные, чем кратная связь с оксолигандом (1.684(5) Å), но при этом заметно короче суммы ковалентных радиусов (2.17 Å) [22]. Этот факт, как и увеличение углов ReOС (124.9(5)°) по сравнению с тетраэдрическим, свидетельствует о заметном дополнительном связывании Re-метилат за счет подачи неподеленной пары кислорода на разрыхляющую π-орбиталь кратной связи Re–O с оксолигандом. Расстояния Re–N для атомов азота в транс-положении к метоксидным (2.102(6), 2.145(5) Å) и кислородному (2.242(6) Å) лигандам заметно неэквивалентны, по видимому вследствие более сильного транс-влияния оксолиганда. При этом расстояния Re–N в транс-положении к метоксидным группам в среднем оказываются меньше, чем расстояния Re–N (2.147 и 2.175 Å) в транс-положении к тиолатным заместителям в пропилтиолатном аналоге TpReO(SC3H7)2 [23].
В оксомостиковом димере II строение ТpRe фрагмента аналогично его строению в I (рис. 2). Расстояния Re–N в транс-положении к концевому лиганду оказываются еще более удлиненными (2.299(5) Å) по сравнению с двумя другими (2.100(2), 2.134(2) Å ). Расстояния Re–O с мостиковыми атомами кислорода (1.948(2), 1.974(2) Å) значительно больше, чем с концевыми (1.689(2) Å) и практически эквивалентны расстояниям Re–OMe в исходном комплексе I. Расстояние Re…Re (3.1049(2) Å) оказывается лишь незначительно большим, чем связывающее расстояние Re–Re в карбониле рения (3.042 Å). Такой короткий контакт Re…Re, скорее всего, является следствием геометрии мостиковых лигандов, однако наличие дополнительного взаимодействия неподеленной пары Re(V) с разрыхляющими π-орбиталями кратной связи Re–O на соседнем атоме рения не может быть исключено.
В катионном димере III (рис. 3) замещение одного из кислородных мостиковых лигандов на пиразолат приводит к увеличению расстояния Re…Re до 3.51434 Å и угла ReORe до 130.5(3)°, однако расстояния Re–O c кислородным мостиком даже несколько укорачиваются (1.924(6), 1.935(6) Å). Расстояния Re–N в транс-положении к пиразолатному мостику (2.046(5), 2.067(5) Å) заметно короче, чем в транс-положении к кислородному (2.133(5), 2.159(5) Å).
В спектрах ЯМР 1H и 13С I наблюдается один сигнал метильных групп и два набора сигналов пиразолатов в соотношении 1 : 2, что свидетельствует об исчезновении в растворе неэквивалентности метильных групп, наблюдаемой в РСА. Для аналогичных тиолатных комплексов TpReO(SR)2 [23] наблюдалось заторможенное вращение тиолатных групп с барьерами 13 и 12.6 ккал/моль (для R = СMe3, н-C3H7 соответственно). В связи с этим было интересно исследовать температурную зависимость 1H-спектра I, демонстрирующего особенности строения, схожие с тиолатными комплексами. Однако до 253 К спектр I не претерпевает существенных изменений, что свидетельствует о более низком барьере вращения OMe по сравнению с тиолатными заместителями.
Теоретический расчет барьера вращения в I методом функционала электронной плотности дает меньшее значение (8.2 ккал/моль), чем для тиолатных комплексов (14.1 и 12.3 ккал/моль). В отличие от тиолатных комплексов, в переходном состоянии не происходит удлинения связи Re–O с вращающейся группой OMe, но угол ReOСMe увеличивается до 140°. Этот факт позволяет предположить, что снижение барьера вращения при переходе от серы к кислороду в основном связано с возможностью дополнительного донирования двух неподеленных пар на кислороде на разрыхляющие π-орбитали кратной связи Re–O с терминальным кислородом вне зависимости от диэдрального угла OReOСMe, тогда как в случае серы пара электронов, имеющая s-характер, практически не принимает участия в дополнительном связывании Re–S.
ЯМР спектры II не удалось зарегистрировать из-за крайне низкой растворимости в обычных дейтерированных растворителях. При нагревании II до 80°C в d6-ДМСО образуется бесцветный раствор, содержащий уширенный синглет BH-фрагмента (3.37 м.д.) и один набор сигналов пиразолатного кольца (6.53, 8.18, 8.37 м.д.). По-видимому, комплекс окисляется под действием ДМСО до TpReO3 [24], аналогично образованию (Pz4B)ReO3 в реакции [(Pz4B)ReO2]2 с ДМСО [4].
Реакция комплекса I с карбоновыми кислотами (AcOH, PhCOOH) приводит только к образованию комплекса II с низким выходом, по-видимому, из-за реакции со следами воды. Реакция I с избытком уксусного ангидрида приводит к замещению метилатных заместителей на ацетатные с образованием комплекса IV.
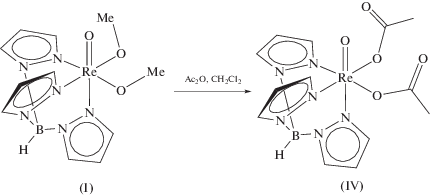
Схема 3 .
ИК-спектр IV содержит две полосы ассиметричных колебаний карбоксилатных групп при 1686 и 1661 см–1, отсутствующие в исходном комплексе. Полосы симметричных колебаний OCO, по-видимому, наблюдаются при 1284 и 1244 см–1, но их отнесение менее очевидно, поскольку в исходном комплексе также присутствуют интенсивные полосы в этой области. Полоса колебаний BH смещается от 2518 до 2539 см–1 при переходе от I к IV, скорее всего, в связи с различиями в кристаллической упаковке: в кристалле I связь BH направлена в сторону π-системы одного из пиразолатных колец соседней молекулы, тогда как в IV связь BH направлена между кислородами ацетатных групп (HB…O 2.89, 3.12 Å). Как и для I, спектры ЯМР 1H и 13С комплекса IV содержат один сигнал метильных групп и два набора сигналов пиразолатов в соотношении 1 : 2, показывая эквивалентность ацетатных заместителей в растворе.
По данным РСА, в комплексе IV (рис. 4) ацетатные группы координированы только одним атомом кислорода с заметной асимметрией расстояний C–O для связанных с рением (1.273(4), 1.331(4) Å) и не связанных (1.236(5), 1.216(4) Å) атомов кислорода. Связи Re–O с ацетатными группами (2.005(2), 1.976(2) Å) длиннее, чем ReO с метилатными группами (1.969(5), 1.933(5) Å) в I, а расстояния Re–N для атомов азота в транс-положении к ацетатным группам (2.069(2), 2.098(2) Å) и расстояние ReO с оксолигандом (1.669(2) Å) несколько укорочены по сравнению с аналогичными расстояниями в I. Такие особенности геометрии говорят о более слабом связывании рения с ацетатами по сравнению с метилатами в таких комплексах, что согласуется с меньшей стабильностью IV по отношению к гидролизу и невозможностью его получения обменной реакцией I с уксусной кислотой.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Trofimenko S. // Chem. Rev. 1993. V. 93. P. 943.
Calogero S., Lobbia G.G., Cecchi P. et al. // Polyhedron. 1994. V. 13. P. 87
Lail M., Pittard K.A., Gunnoe T.B. // Adv. Organomet. Chem. 2008. V. 56. P. 95.
Paulo A., Domingos A., Marcalo J. et al. // Inorg. Chem. 1995. V. 34. P. 2113.
Nunes D., Domingos A., Paulo A. et al. // Inorg. Chim. Acta. 1998. V. 271. P. 65.
Lassen P.R., Guy L., Karame I. et al. // Inorg. Chem. 2006. V. 45. P. 10230.
Klüfers P., Krotz O, Oßberger M. // Eur. J. Inorg. Chem. 2002. P. 1919.
Brown S.N., Mayer J.M. // J. Am. Chem. Soc. 1996. V. 118. 12119.
Brown S.N., Mayer J.M. // Inorg. Chem. 1992. V. 31. P. 4091.
Middleditch M., Anderson J.C., Blake A.J., Wilson C. // Inorg. Chem. 2007. V. 46. P. 2797.
Abrams M.J., Davison A., Jones A.G. // Inorg. Chim. Acta. 1984. V. 82. P. 125.
SADABS. Version 2008/1. Madison (WI, USA): Bruker AXS Inc., 2008.
Sheldrick G.M. // Acta Crystallogr. A. 2008. V. 64. P. 112.
Dolomanov O.V., Bourhis L.J., Gildea R.J. et al. // J. A-ppl. Cryst. 2009. V. 42. P. 339.
Neese F. // WIREs Comput. Mol. Sci. 2012. V. 2. P. 73.
Perdew J.P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1996. V. 77. P. 3865.
Perdew J.P., Burke K., Ernzerhof M. // Phys. Rev. Lett. 1997. V. 78. P. 1396 (Erratum).
Weigend F., Ahlrichs R. // Phys. Chem. Chem. Phys. 2005. V. 7. P. 3297.
Grimme S., Ehrlich S., Goerigk L. // J. Comput. Chem. 2011. V. 32. P. 1456.
Grimme S., Antony J., Ehrlich S., Krieg H. // J. Chem. Phys. 2010. V. 132. P. 154104.
Adamo C., Barone V. // J. Chem. Phys. 1999. V. 110. P. 6158.
Cordero B., Gómez V., Platero-Prats A.E. et al. // Dalton Trans. 2008. V. 21. P. 2832.
Скабицкий И.В., Сахаров С.Г., Пасынский А.А., Эшмаков Р.С. // Коорд. химия. 2019. T. 45. C. 539.
Degnan I.A., Herrmann W.A., Herdtweck E. // Chem. Ber. 1990. V. 123. P 1347.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия