

Координационная химия, 2022, T. 48, № 4, стр. 206-214
Исследование экстракционных и координационных свойств 2,3-бис(дифенилфосфинил)бута-1,3-диена и 3,4-бис(дифенилфосфинил)-2,5-диметилгекса-2,4-диена
В. К. Брель 1, *, О. И. Артюшин 1, В. П. Моргалюк 1, А. В. Вологжанина 1, А. Н. Туранов 2, В. К. Карандашев 3
1 Институт элементоорганических соединений им. А.Н. Несмеянова РАН
Москва, Россия
2 Институт физики твердого тела им. Ю.А. Осипьяна РАН
Черноголовка, Россия
3 Институт проблем технологии микроэлектроники и особо чистых материалов РАН
Черноголовка, Россия
* E-mail: v_brel@mail.ru
Поступила в редакцию 08.09.2021
После доработки 07.10.2021
Принята к публикации 08.10.2021
- EDN: VWEMTH
- DOI: 10.31857/S0132344X22040016
Аннотация
Изучены экстракционные свойства 2,3-бис(дифенилфосфинил)бута-1,3-диена (L1) и 3,4-бис(дифенилфосфинил)-2,5-диметилгекса-2,4-диена (L2) по отношению к f-элементам на примере извлечения микроколичеств U(VI) и Th(IV) из растворов HNO3 растворами экстрагентов в 1,2-дихлорэтане. Установлено, что введение метильных групп в 1,3-бутадиеновый углеродный скелет бисфосфиноксида приводит к увеличению эффективности экстракции U(VI). Координационные свойства би-сфосфиноксидов L1 и L2 изучены на примере комплексов с нитратом уранила. Кристаллическое строение бисфосфиноксида L1 · THF (CIF file CCDC № 2107403), его комплекса с нитратом уранила I (CIF file CCDC № 2107404) и комплекса II (CIF file CCDC № 2107405) подтверждено рентгенодифракционными данными.
Несмотря на достаточно длительную историю исследования оксидов фосфора, в последние десятилетия наблюдается возросший интерес к созданию эффективных и селективных фосфорорганических лигандов к ионам различных металлов. Бионеорганическая химия [1], катализ [2], создание новых материалов [3], процессы разделения и извлечения редких и рассеянных элементов [4], гидрометаллургия и очистка отходов ядерных электростанций от радиоактивных элементов [5] – вот краткое перечисление областей, где активно используют моно-, ди- и полидентатные фосфиноксиды. Наибольший интерес вызывают комплексы металлов с бисфосфиноксидами, демонстрирующие более богатую структурную топологию, чем монофосфиноксиды. Так, в литературе описаны координационные полимеры [6–9], макроциклические соединения [10] и комплексы клеточного строения [11] на основе бисфосфиноксидов.
Структура получаемых комплексов во многом зависит от строения используемого бисфосфиноксида. Например, метилен бисфосфиноксид, как правило, хелатирует ионы металлов с образованием шестичленных комплексов [12, 13]. В случае удлинения мостика связывающего две фосфиноксидные группы Ph2P(O)(CH2)nP(O)Ph2 (n = 2, 4 или 6), как правило, происходит образование комплексов димерного или полимерного строения [14–16].
Продолжая изучение координационных свойств алкиленбисфосфиноксида [17] и его аналогов [18], нами выполнены исследования, связанные с попыткой выяснения влияния структурных изменений углеродного скелета бисфосфиноксидов на их координационные и экстракционные свойства. В качестве объектов исследования, выбраны бисфосфиноксидные лиганды на 1,3-алкадиеновой платформе: 2,3-бис(дифенилфосфинил)бута-1,3-диен (L1) [19] и 3,4-бис(дифенилфосфинил)-2,5-диметилгекса-2,4-диен (L2) [20].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали СНСl3, СН2Сl2 и ацетонитрил марки “х. ч.”, которые перегоняли над P2O5 для обезвоживания и хранили над CaH2; ТГФ перегоняли и хранили над CaH2, отгоняя перед применением. Коммерческие 1,2-дихлорэтан и этанол, использовали без предварительной очистки.
Спектры ЯМР 1Н, 13С и 31Р регистрировали в CDCl3 на спектрометре Bruker Avance 400 (400.13, 100.61 и 161.98 МГц соответственно). Спектры ЯМР 1Н, 31Р комплекса зарегистрированы в ДМФА-d7. ИК-спектры регистрировали на VER-TEX 70v Fourier-transform ИК спектрометре (Германия). Масс-спектры высокого разрешения регистрировали на приборе Bruker micrOTOF II методом электрораспыления (ESI). Элементный анализ (C, H, N) выполнен на автоматическом анализаторе CarloErba 1106.
Синтез 3,4-бис(дифенилфосфинил)-2,5-диметилгекса-2,4-диена (L2) выполняли в инертной атмосфере. К суспезии 0.31 г (12.8 ммоль) NaH в 6 мл ТГФ при интенсивном перемешивании добавляли 0.9 г (6.4 ммоль) 2,5-диметил-3-гексин-2,5-диола и через 5 мин по каплям добавляли 3.12 г (14.1 ммоль) дифенилхлорфосфина, поддерживая температуру реакционной среды 20°С. После прекращения газовыделения перемешивали 4 ч при 20°С. Осадок отфильтровавыли и промывали 2 × 3 мл ТГФ. К фильтрату добавляли 0.2 г Cu(OTf)2 (0.7 ммоль, 10 мол. %) и после образования желто-оранжевого раствора оставляли на 48 ч. Добавляли 20 мл CH2Cl2, промывали 2 × 3 мл 25%-ного раствора NH3, 2 × 5 мл водой и сушили над Na2SO4. Осушитель отфильтровывали, промывали 2 × 5 мл CH2Cl2, фильтрат упаривали в вакууме. После хроматографирования на силикагеле в системе CHCl3–бензол–метанол 30 : 30 : 1 получили: 2.39 г (72%) 3,4-бис(дифенилфосфинил)-2,5-диметилгекса-2,4-диена (L2), 0.21 г (9%) (5-хлор-2,5-диметилгекс-3-ин-2-ил)дифенилфосфиноксида (L3) и 0.14 г (7%) (2,5-диметилгекс-5-ен-3-ин-2-ил)дифенилфосфиноксида (L4).
3,4-Бис(дифенилфосфинил)-2,5-диметилгекса-2,4-диен (L2). Бесцветные кристаллы. Тпл = = 186–188°C [20]. ЯМР 1H (δ, м.д.): 8.00–7.96 м. (4H, o-H, Ph), 7.48–7.44 м. (6H, o,п-H, Ph), 7.38–7.35 м. (2H, п-H, Ph), 7.20–7.16 м. (4H, м-H, Ph), 7.14–7.08 м. (4H, м-H, Ph), 1.76 с. (6H, 2CH3), 1.69 с. (6H, 2CH3). ЯМР 13C (δС, м.д.): 153.88 д.д. ((CH3)2C=C), 2JP,C = 8.02 Гц, 134.75 д. (ipso-C, 1JP,C = 102.13 Гц), 134.20 д. (ipso-C, 1JP,C = = 99.21 Гц), 132.15 д.д. (п-C, Ph, 4JP,C = 11.68 Гц, 4JP,C = 10.21 Гц), 131.20 уш.д. (o-C, Ph, 2JP,C = = 31.36 Гц), 129.02 д. ((CH3)2C=C, 1JP,C = 8.02 Гц), 128.38 д. (м-C, Ph, 3JP,C = 10.95 Гц), 128.38 д. (м-C, Ph, 3JP,C = 11.68 Гц), 25.30–25.23 м. ((CH3)2C=C), 25.12–25.09 м. ((CH3)2C=C). ЯМР 31P (δP, м.д.): 27.60 с. ИК (ν, см–1): 3051 ν(CH, Ph); 2910, 1436 ν(CH3); 1608, 1589 ν(Ph); 1628 ν(C=C); 1162 ν(P=O). Масс-спектр, m/z: 511.1950 [M + H]+ (вычислено для C32H33O2P2: 511.1961).
(5-Хлор-2,5-диметилгекс-3-ин-2-ил)дифенилфосфиноксида (L3). Бесцветное масло. ЯМР 1H (δ, м.д.): 7.72–7.68 м. (4H, o-H, Ph), 7.52–7.50 м. (2H, п-H, Ph), 7.48–7.43 м. (4H, м-H, Ph), 1.46 д. (6H, 2CH3, 3JP,H = 6.0 Гц), 1.44 с. (6H, 2CH3). ЯМР 13C (δС, м.д.): 133.29 д. (ipso-C, Ph, 1JP,C = 104.32 Гц), 131.74 д. (п-C, Ph, 4JP,C = 2.91 Гц), 131.36 д. (o-C, Ph, 2JP,C = 9.48 Гц), 128.25 д. (м-C, Ph, 3JP,C = 11.67 Гц), 102.95 уш.с. (C≡C), 101.93 уш.с. (C≡C), 99.74 д. (C‒P, 1JP,C = 13.86 Гц), 73.57 д. (C–Cl, 4JP,C = = 5.11 Гц), 30.74 д. (2CH3, 5JP,C = 2.91 Гц), 19.00 д. (2CH3, 2JP,C = 5.84 Гц). ЯМР 31P (δP, м.д.): 34.37 с. Масс-спектр, m/z: 345.1170 [M + H]+ (вычислено для C20H23ClOP: 345.1181).
(2,5-Диметилгекс-5-ен-3-ин-2-ил)-дифенилфосфиноксид (L4). Бесцветные кристаллы. Тпл = = 152–154°C. ЯМР 1H (δ, м.д.): 7.70–7.65 м. (4H, o‑H, Ph), 7.47–7.39 м. (8H, Ph), 5.46 с. (1H, =C(H)H), 5.02 с. (1H, =C(H)H), 1.84 с. (3H, CH3(=CH2)), 1.36 д. (6H, 2CH3, 3JP,H = 6.1 Гц). ЯМР 13C (δС, м.д.): 136.17 д. (CH2=C–CH3, 5JP,C = = 7.29 Гц), 133.21 д. (ipso-C, Ph, 1JP,C = 105.04 Гц), 131.18 с. (o-C, Ph, 2JP,C = 9.48 Гц), 131.41 с11, Ph, 128.14 д. (Ph, 3JP,C = 12.41 Гц), 117.12 д. (CH2=C–CH3, 4JP,C = = 3.65 Гц), 101.09 уш.с. (C≡C), 100.08 уш.с. (C≡C), 99.45 д. (C–P, 1JP,C = 13.14 Гц), 23.00 д. (CH2=C–CH3, 5JP,C = 5.84 Гц), 19.10 д. (2CH3, 2JP,C = 5.83 Гц). ЯМР 31P (δP, м.д.): 31.57 с. Масс-спектр, m/z: 309.1403 [M + H]+ (вычислено для C20H22OP: 309.1414).
Синтез 2,3-бис(дифенилфосфинил)-бут-1,3-диендинитратуранила (I). Раствор UO2(NO3)2 ⋅ 6H2O (104 мг, 0.2 ммоль) в 5 мл ацетонитрила медленно добавили к раствору фосфиноксида L1 (90.8 мг, 0.2 ммоль) в 5 мл хлороформа и перемешивали 1 ч. Выпавшие желтые кристаллы отфильтровывали, промывали 2 × 5 мл ацетонитрила, перекристаллизовывали из смеси ДМФА–диэтиловый эфир (1 : 1) и высушивали в вакууме 0.1 мм рт. ст. до постоянного веса. Выход комплекса I 160 мг (80%), Тразл = 255–257°С. ИК (КBr; ν, см–1): 539, 582, 692, 730, 929 (UO2), 1122, 1145, 1161, 1273 (UO2), 1292, 1385, 1487 (NO3), 1516. ЯМР 1Н (400 МГц; ДМФА-d7; δ, м.д.): 8.4–7.6 м. (20Н, 4С6Н5), 6.87 д. (2Н, 2JHH = 42 Гц, СН2=), 6.18 уш.с. (2Н, СН2=). ЯМР 13С (100 МГц; ДМФА-d7; δ, м.д.): 138.94 д.д. (1JPC = = 95.0 Гц, 2JPC = 8.0, Р–С=), 136.38 уш.с. (Н2С=), 134.04 уш.с. (С6Н5), 132.57 д. (3JPC = 9.7 Гц, С6Н5), 129.68 д. (2JPC = 12.0 Гц, С6Н5). ЯМР 31Р (162 МГц; ДМФА-d7; δ, м.д.): 46.44 уш.с.
Синтез 2,3-бис(дифенилфосфинил)-1,1,4,4-тетраметилбут-1,3-диендинитратуранила (II). Раствор UO2(NO3)2 ⋅ 6H2O (104 мг, 0.2 ммоль) в 5 мл этанола осторожно добавляли на поверхность раствора фосфиноксида L2 (102 мг, 0.2 ммоль) в 5 мл ДМФА. На следующий день выпавшие светло-зеленые кристаллы отфильтровывали, промывали эфиром и сушили в вакууме 0.1 мм рт. ст. до постоянного веса. Выход комплекса II 150 мг (73%), Тразл = 320–322°С. ИК (КBr; ν, см–1): 545, 575, 699, 748, 932 (UO2), 1097, 1121, 1144, 1278 (UO2), 1296, 1385, 1439, 1484 (NO3), 1512, 1529.
Изучение экстракционных свойств для соединений L1, L2, L5. В качестве органического растворителя использовали 1,2-дихлорэтан марки “х. ч.” Растворы экстрагентов готовили по точной навеске. Исходные водные растворы U(VI) и Th(IV) готовили растворением соответствующих нитратов в воде с последующим добавлением HNO3 до требуемой концентрации. Исходная концентрация ионов металлов составляла 2 × 10–6 моль/л. Равные объемы органической и водной фаз перемешивали при комнатной температуре на роторном аппарате для перемешивания со скоростью 60 об./мин в течение 1 ч, что достаточно для установления постоянных значений коэффициентов распределения.
Концентрацию U(VI) и Th(IV) в исходных и равновесных водных растворах определяли методом масс-спектрометрии с ионизацией пробы в индуктивно связанной плазме (ICP-MS) с использованием масс-спектрометра XSeriesII (Thermo Scientific, США) при следующих параметрах работы: выходная мощность генератора 1300 Вт; комплект стандартных никелевых конусов; концентрический распылитель PolyCon; кварцевая коническая распылительная камера, охлаждаемая до 3°С; расход плазмообразующего потока аргона 13 л/мин; расход вспомогательного потока аргона 0.9 л/мин; расход потока аргона в распылителе 0.95 л/мин; расход анализируемого образца 0.8 мл/мин. Градуировку спектрометра проводили с использованием одно- и многоэлементных стандартов производства High-Purity Standards (США). Концентрацию U(VI) и Th(IV) в равновесной органической фазе определяли как разницу между концентрациями в исходных и равновесных водных растворах. Коэффициенты распределения элементов (D) рассчитывали как отношение их концентраций в равновесных органической и водной фазах. Погрешность определения коэффициентов распределения не превышала 5%. Концентрацию HNO3 в равновесной водной фазе определяли потенциометрическим титрованием стандартным раствором NaOH.
РСА. Монокристаллы соединения L1 · THF получали перекристаллизацией из ТГФ, а комплексы I и II брали из реакционной смеси. Интенсивности отражений измерены с помощью дифрактометров Bruker Apex II (L1 · THF и I при 120 K) и Bruker Quest (II, 100 K) с использованием MoKα-излучения (λ = 0.71073 Å). Структуры решены с помощью алгоритма SHELXT [21] и уточнены в полноматричном приближении по F 2 (hkl). Неводородные атомы уточнены анизотропно. Положения атомов водорода найдены из геометрических соображений и уточнены в изотропном приближении в модели “наездника” с Uiso(H) = 1.2Ueq(X). Монокристаллы L1 · THF являются двойниками, которые удалось разделить с помощью программы PLATON [22] и уточнить с применением инструкций BASF/HKLF 5. Все расчеты проведены с использованием программ SHELXL2014 [23] и OLEX2 [24]. Параметры кристаллов и результаты уточнения приведены в табл. 1.
Таблица 1.
Кристаллографические данные и параметры эксперимента для соединений L1 · THF, I и II
| Параметр | Значение | ||
|---|---|---|---|
| L1 · THF | I | II | |
| Формула | C32H32O3P2 | C28H24N2O10P2U | C32H32N2O10P2U |
| Mw | 526.51 | 848.46 | 904.56 |
| Сингония | Моноклинная | Моноклинная | Орторомбическая |
| Пр. гр. | P21 | P21/n | P212121 |
| Z | 2 | 4 | 4 |
| a, Å | 8.3734(8) | 12.1672(13) | 9.5958(2) |
| b, Å | 16.2032(15) | 19.666(2) | 16.4917(3) |
| c, Å | 11.1046(10) | 12.8628(14) | 20.7474(4) |
| β, град | 112.1243(18) | 105.899(4) | 90 |
| V, Å3 | 1395.7(2) | 2960.1(6) | 3283.30(11) |
| ρ(выч.), г см–3 | 1.253 | 1.904 | 1.830 |
| μ, см–1 | 0.187 | 5.651 | 5.101 |
| F(000) | 556 | 1632 | 1760 |
| Измеренных отражений | 12 645 | 39 731 | 106 772 |
| Независимых отражений (Rint) | 12 645 (0.080) | 9080 (0.048) | 10 047 (0.045) |
| Отражений с I > 2σ(I) | 11 567 | 7023 | 9671 |
| Число параметров | 333 | 388 | 429 |
| R1 | 0.0442 | 0.0273 | 0.0173 |
| wR2 | 0.0907 | 0.0544 | 0.0385 |
| GOОF | 0.996 | 1.020 | 1.005 |
| Δρmax/Δρmin, e Å–3 | 0.54/–0.33 | 0.55/–1.39 | 1.29/–0.42 |
| Параметр Флека | –0.01(3) | 0.040(3) | |
Дополнительная кристаллографическая информация для L1 · THF, I, II представлена в Кембриджском банке структурных данных (CCDC № 2107403 (L1 · THF), 2107404 (I), 2107405 (II); http:// www.ccdc.cam.ac.uk/structures).
Поверхности полиэдров Вороного построены и проанализированы в комплексе структурно-топологических программ ToposPro [25].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Соединение I получено по ранее описанной методике [19]. Для синтеза 1,3-алкадиена II за основу была взята методика [20], которая после модификации была применена в данном исследовании. 2,5-Диметил-3-гексин-2,5-диол был введен в реакцию с дифенилхлорфосфином в присутствии NaH и 10 мол. % Cu(OTf)2 в ТГФ при 20°С (схема 1 ).
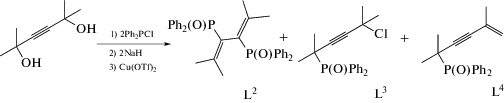
Схема 1 .
Целевой продукт, алкадиен L2, получен и выделен с помощью колоночной хроматографии с выходом 72%. В качестве побочных продуктов образуются (5-хлор-2,5-диметилгекс-3-ин-2-ил)дифенилфосфиноксид (L3) и (2,5-диметилгекс-5-ен-3-ин-2-ил)-дифенилфосфиноксид (L4) с выходами 9 и 7% соответственно. Строение соединений L2–L4 установлено методами ЯМР 1Н, 13С и 31P, ИК-спектроскопии, а состав – с помощью элементного анализа и масс-спектрометрии.
Экстракционные свойства соединений L1, L2 по отношению к f-элементам изучены на примере извлечения микроколичеств U(VI) и Th(IV) из растворов HNO3 растворами экстрагентов в 1,2-дихлорэтане. В качестве соединения сравнения был использован 1,2-бис(дифенилфосфинил)этан Ph2P(O)CH2CH2P(O)Ph2 (L5).
Было рассмотрено влияние концентрации HNO3 в равновесной водной фазе на изменение коэффициентов распределения U(VI) (рис. 1) и Th(IV) (рис. 2) при экстракции растворами 0.001 моль/л соединений L1, L2, L5 в дихлорэтане. Увеличение до 1–2 М HNO3 сопровождается ростом DU и DTh, что связано с высаливающим действием ионов ${\text{NO}}_{3}^{ - }$ и соответствует экстракции этих ионов в виде координационно-сольватированных нитратов [26]. Дальнейший рост концентрации HNO3 приводит к некоторому снижению DU и DTh, что вызвано соэкстракцией азотной кислоты, приводящей к снижению концентрации свободного экстрагента в органической фазе.
Рис. 1.
Зависимость коэффициентов распределения U(VI) от равновесной концентрации HNO3 в равновесной водной фазе при экстракции растворами 0.001 моль/л соединений L1, L2, L5 в дихлорэтане.

Рис. 2.
Зависимость коэффициентов распределения Th(IV) от равновесной концентрации HNO3 в равновесной водной фазе при экстракции растворами 0.001 моль/л соединений L1, L2, L5 в дихлорэтане.

Стехиометрическое соотношение металл : экстрагент в экстрагируемых комплексах определено методом сдвига равновесия. Полученные данные (рис. 3, 4) показали, что соединения L1, L2, L5 в дихлорэтане экстрагируют Th(IV) из азотнокислых растворов в форме дисольватов, а U(VI) преимущественно в форме моносольватов.
Рис. 3.
Зависимость коэффициентов распределения U(VI) от концентрации соединений L1, L2, L5 в дихлорэтане при экстракции из раствора 3 моль/л HNO3.

Рис. 4.
Зависимость коэффициентов распределения Th(IV) от концентрации соединений L1, L2, L5 в дихлорэтане при экстракции из раствора 3 M HNO3.

Для сравнения экстракционной способности соединений L1, L2, L5 по отношению к U(VI) и Th(IV), а также фактора разделения тория и урана (βTh/U = DTh/DU) в табл. 2 представлены данные по экстракции U(VI) и Th(IV) из азотнокислых растворов в сопоставимых условиях. Можно видеть, что введение аллильных и особенно метильных заместителей в алкиленовый мостик молекулы Ph2P(O)CH2CH2P(O)Ph2 (L5) приводит к увеличению эффективности экстракции U(VI) и некоторому снижению экстракционной способности соединений L1 и L2 по отношению к Th(IV).
Таблица 2.
Коэффициенты распределения U(VI) и Th(IV) при их экстракции из раствора 3 М HNO3 0.001 М растворами L1, L2, L5 в дихлорэтане
| Экстрагент | lg DTh | lg DU | βTh/U |
|---|---|---|---|
| L1 | 1.64 | 0.78 | 7.2 |
| L2 | 1.42 | 2.62 | 0.06 |
| L5 | 1.75 | –0.04 | 61.6 |
Селективность экстракции Th(IV) снижается в ряду соединений L5 > L1 > L2 и соединение L2 экстрагирует U(VI) значительно более эффективно, чем Th(IV). Для этого соединения величина фактора разделения урана и тория (βU/Th) уменьшается от 200 до 16 с ростом концентрации HNO3 в равновесной водной фазе от 0.3 до 3 моль/л. Различие в стехиометрии экстрагируемых комплексов U(VI) и Th(IV) приводит к снижению βU/Th с ростом концентрации соединения L2 в органической фазе.
С целью установления причин, влияющих на экстракционные свойства бисфосфиноксидов L1 и L2, были проведены их структурные исследования. Структура и состав лигандов L1и L2 были установлены с помощью рентгеновской дифракции. Бисфосфиноксид L2 кристаллизуется в виде моногидрата аналогично ранее представленным данным [20], в то время как соединение L1 кристаллизуется с молекулой ТГФ. Данные рентгеновской дифракции показали, что молекула L1 в кристалле L1 · THF реализует плоскую s-транс-конформацию (с углом С=С–С=С, равным 162.6(3)°) (рис. 5), в то время как L2 в L2 · H2O [20] имеет скошенную s‑цис-конформацию с углом поворота между непредельными фрагментами –С=С(СН3)2 86.5(2)°.

Нарушение компланарности алкадиенового фрагмента для соединения L2, вероятно, вызвано стерическими взаимодействиями между терминальными метильными группами с бисфосфиноксидными фрагментами. Компланарность фрагмента 1,3-алкадиена в L1 способствует делокализации плотности заряда вдоль этой цепи, что приводит к некоторому удлинению связей C=C (табл. 2). Кроме того, в L1 по сравнению с L2 удлинены связи фосфорильной группы P=O и связи фосфора с 1,3-алкадиеновым скелетом P–C.
Для установления конформационного состава соединений L1 и L2 в жидкой фазе, была привлечена УФ-спектроскопия. Известно, что для определения конформационного состава гибких молекул типа 1,3-алкадиенов широко используется электронная спектроскопия [27]. В частности, было установлено, что при нарушении сопряжения между винильными фрагментами углеродного скелета и реализации скошенной s-цис-конформации, происходит заметное снижение интенсивности полосы поглощение в УФ-спектрах. Данный эффект был ранее продемонстрирован при исследовании фосфорорганических 1,3-алкадиелов [28]. Используя данный прием, нам удалось показать, что УФ-спектры соединений L1 и L2, зарегистрированные в растворе, сильно отличаются. Интенсивность полосы в области 220 нм для соединения L1 почти в полтора больше, чем для соединения L2 (рис. 6). Таким образом, с большой долей вероятности можно утверждать, что конформационный состав бисфосфиноксидов L1 и L2 в твердой фазе и в растворе идентичен.

Координационные свойства бисфосфиноксидов L1 и L2 исследованы на примере их реакций с UO2(NO3)2 · 6H2O (схема 2 ). Уранильные комплексы I и II получены с использованием в качестве растворителя СН3СN для лиганда L1 и ДМФА для лиганда L2.

Схема 2 .
С помощью рентгеновской дифракции монокристаллов комплексов I и II установлено, что лиганды L1 и L2 образуют уранильные комплексы в соотношении UO2 : L = 1 : 1. Молекулярный вид комплексов представлен на рис. 7. Координационные полиэдры U(VI)O8 имеют формы гексагональных бипирамид с атомами кислорода уранила в осевых положениях и шестью атомами кислорода в экваториальных. Уранильные группы в I и II практически линейны (177.2(1)° и 178.2(1)°; табл. 3).

Таблица 3.
Некоторые межатомные расстояния (Å) в структурах L1, L2, I и II
| Параметр | L1 | L2 [20] | I | II | [Mn(L1)3][MBr4] [29] | [Mn(L1)3][M(SCN)4] [30] |
|---|---|---|---|---|---|---|
| U(VI) | Mn(II) | |||||
| U(VI)=O | 1.750(2)–1.768(2) | 1.768(2)–1.769(2) | ||||
| M−OP=O | 2.356(2)–2.371(2) | 2.334(2)–2.367(2) | 2.140(3)–2.154(3) | 2.131(2)–2.168(2) | ||
| ${\text{M}}{\kern 1pt} - {\kern 1pt} {{{\text{O}}}_{{{\text{N}}{{{\text{O}}}_{3}}}}}$ | 2.515(2)–2.540(2) | 2.513(2)–2.546(2) | ||||
| P=O | 1.489(2)–1.494(2) | 1.475(1)–1.479(1) | 1.503(2)–1.505(2) | 1.509(2)–1.517(2) | 1.486(3)–1.493(3) | 1.483(2)–1.488(2) |
| P–Cдиен | 1.828(2)–1.834(2) | 1.803(2)–1.806(2) | 1.810(3)– 1.812(3) | 1.812(3)–1.814(3) | 1.804(3)–1.811(4) | 1.801(3)–1.818(4) |
| P–CPh | 1.801(3)–1.812(2) | 1.788(2)–1.796(2) | 1.787(3)–1.796(3) | 1.792(3)–1.808(3) | 1.794(4)–1.811(5) | 1.785(3)–1.803(3) |
| C=C | 1.341(3)–1.350(3) | 1.333(2)–1.338(2) | 1.333(4)–1.336(4) | 1.344(4)–1.347(4) | 1.307(5)–1.326(6) | 1.309(5)–1.323(5) |
| Cдиен–Cдиен | 1.489(3) | 1.494(3) | 1.490(4) | 1.515(4) | 1.499(6)–1.511(6) | 1.496(5)–1.500(6) |
Связи U(VI)−OP=O короче, чем связи U(VI)−ON. Длины связей в бисфосфиноксидах L3 и L4 близки к связям в исходном лигаде L2 · H2O [20]. Сходство конформаций лигандов для L2, I и II по сравнению с L1 представлено на рис. 8.
Рис. 8.
Сравнение конформаций лигандов в кристаллах L1 · THF (оранжевый), L2 · H2O [20] (зеленый), I (синий) и II (красный). Наложены атомы групп P=O. Атомы водорода не изображены.

Как следует из рис. 8, конфигурации лиганда в комплексах I и II очень близки друг к другу из-за жесткости семичленного металлоцикла, который допускает вращение только вдоль связей P–CPh. Конформации фосфиноксида L2 · H2O [20] и его в комплексе II в целом также близки друг к другу за исключением поворота одного фрагмента Ph2P=O вдоль связи P–Cдиен на угол около 120°. В то же время конфигурация молекулы L1 в L1 · THF значительно отличается от конфигурации в L3 из-за необычных торсионных углов C=C–P=O. Таким образом, s-транс-конформация лиганда L1, может быть причиной сниженной его экстракционной способности к f-элементам по сравнению с его алкилзамещенными аналогами, в частности лиганда L2.
Другая возможность различного поведения лигандов L1и L2 при экстракции может быть связана с различным составом атомов, формирующих молекулярную поверхность и, как следствие, влияющих на растворимость вещества в гидрофобных растворителях. Удобным приближением, позволяющим оценить вклад различных атомов в площадь молекулярной поверхности и их участие в различных межмолекулярных контактах, являются поверхности молекулярных полиэдров Вороного, образованных всеми областями кристалла, которые ближе к данной молекулe, чем молекулам (или ионам) окружения [31]. Данное представление молекул в кристалле позволяет успешно анализировать особенности упаковки и межмолекулярных взаимодействий в полиморфах [31–33], конформационно-гибких молекулах [33, 34] и гомологических рядах соединений [35]. В частности, ранее было установлено, что рост вклада гидрофобных контактов H…H и контактов C…H в общую площадь молекулярной поверхности монокарбоновых кислот сопровождается увеличением их склонности образовывать с двойными солями урана(VI) полиядерные комплексы [35]. В случае L1 · THF и L2 · H2O площадь поверхности молекулы Вороного, которая переходит в гидрофобные взаимодействия C…C, C–H…C и C–H…H–C, равна 460 и 506 Å2 (или 87.5 и 91.7% от общей площади молекулы). В комплексах I и II частичный вклад гидрофобных взаимодействий в площадь молекулы равен 52.9 и 59.6% (327 и 387 Å2). Таким образом, как и следовало ожидать, алкилзамещенные диены и их комплексы должны иметь большую молекулярную поверхность, способную образовывать гидрофобные взаимодействия.
В результате выполненного исследования были изучены экстракционные и координационные свойства 2,3-бис(дифенилфосфинил)бута-1,3-диена (L1) и 3,4-бис(дифенилфосфинил)-2,5-диметилгекса-2,4-диена (L2). С использованием рен-тгеноструктурного анализа и электронной спектроскопии установлено, что соединение L1 имеет преимущественно плоскую s-цис-конформацию. Введение в 1,3-бутадиеновый фрагмент дополнительных заместителей в виде метильных групп (соединение L2) приводит к реализации скошенной s-цис-конформации с углом поворота между непредельными фрагментами –С=С(СН3)2 86.5(2)°. Схожая конформация лигандов была определена и для их уранильных комплексов I и II. При исследовании экстракционных свойств бисфосфиноксидов L1 и L2 была выявлена значительная эффективность экстрагента L2 по сравнению с соединением L1 при извлечении микроколичеств U(VI) из растворов HNO3. Вероятно, это можно объяснить повышенной гидрофобностью бисфосфиноксида L2 и уранильного комплекса II, а также более выгодной конформацией лиганда L2 для формирования комплекса с U(VI).
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Demkowicz S., Rachon J., Das'ko M., Kozak W. // RSC Adv. 2016. V. 6. P. 7101.
Ni H., Chan W.-L., Lu Y. // Chem. Rev. 2018. V. 118. P. 9344.
Baumgartner T., Réau R. // Chem. Rev. 2006. V. 106. P. 4681.
Rozen A.M., Krupnov B.V. // Russ. Chem. Rev. 1996. V. 65. P. 973.
Myasoedov B.F., Kalmykov S.N. // Mendeleev Commun. 2015. V. 25. № 5. P. 319.
Mathew M., Palenik G.J. // Inorg. Chim. Acta. 1971. V. 5. P. 573.
Harrison P.G., Sharpe N.W., Pelizzi C. et al. // Dalton Trans. 1983. P. 921.
Pettinari C., Marchetti F., Cingolani A. et al. // Inorg. Chim. Acta. 1991. V. 312. P. 125.
Spichal Z., Necas M., Pinkas J. // Inorg. Chem. 2005. V. 44. P. 2074.
Al-Resayes S.I., Hitchcock P.B., Nixon J.F. // Chem. Commun. 1991. P. 78.
Beagley B., Dyer G., McAuliffe C.A. et al. // Chem. Commun. 1991. P. 965.
Jin Q.-H., Wu J.-Q., Zhang Y.-Y. et al. // NCS. 2009. V. 224. P. 428.
Lees A.M.J., Platt A.W.G. // Inorg. Chem. 2003. V. 42. P. 4673.
Spichal Z., Necas M., Pinkas J. // Inorg. Chem. 2005. V. 44. P. 2070.
Spichal Z., Petricek V., Pinkas J., Necas M. // Polyhedron. 2008. V. 27. P. 283.
Spichal Z., Necas M., Pinkas J., Zdrahal Z. // Polyhedron. 2006. V. 25. P. 2006.
Berezin A.S., Davydova M.P., Bagryanskaya I.Yu. et al. // Inorg. Chem. Commun. 2019. V. 107. 107473.
Artem’ev A.V., Davydova M.P. et al. // Dalton Trans. 2019. V. 48. № 43. P. 16448.
Pollok T., Schmidbaur H. // Tetrahedron Lett. 1987. V. 28. P. 1085.
Chen F., Xia Y., Lin R. et al. // Org. Lett. 2019. V. 21. P. 579.
Sheldrick G.M. // Acta Crystrallog. A. 2015. V. 71. P. 3.
Spek A.L. // Acta Crystrallog. C. 2015. V. 71. P. 9.
Sheldrick G.M. // Acta Crystrallog. C. 2015. V. 71. P. 3.
Dolomanov O.V., Bourhis L.J., Gildea R.J. et al. // J. A-ppl. Cryst. 2009. V. 42. P. 339.
Blatov V.A., Shevchenko A.P., Proserpio D.M. // Cryst. Growth Des. 2014. V. 14. P. 3576.
Turanov A.N., Karandashev V.K., Kharitonov A.V. et al. // Solvent Extr. Ion Exch. 2000. V. 18. P. 1109.
Штерн Э., Тиммонс Е. Электронная абсорбционная спектроскопия в органической химии. М.: Мир, 1974.
Ратовский Г.В., Сергиенко Л.М., Брель В.К. et al. // Журн. общ. химии. 1983. T. 53. С. 1186.
Berezin A.S., Samsonenko D.G., Brel V., Artem’ev A.V. // Dalton Trans. 2018. V. 47. P. 7306.
Davydova, M.P., Bauer I.A., Brel V.K. et al. // Eur. J. Inorg. Chem. 2020. P. 695.
Serezhkin V.N., Serezhkina L.B., Vologzhanina A.V. // Acta Crystallogr. B. 2012. V. 68. P. 305.
Zerezhkin A.N., Savchenkov A.V. // Cryst. Growth Des. 2015. V. 15. P. 2878.
Zerezhkin V.N., Savchenkov A.V. // Cryst. Growth Des. 2020. V. 20. P. 1997.
Vologzhanin A.V., Ushakov I.E., Korlyukov A.A. // Int. J. Mol. Sci. 2021. V. 21. P. 8970.
Savchenkov A.V., Klepov V.V., Vologzhanina A.B. et al. // CrystEngComm. 2015. V. 17. P. 740.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия