

Координационная химия, 2022, T. 48, № 5, стр. 259-268
Синтез, строение и свойства бис-хелатных комплексов Zn(II), Cu(II), Co(II) и Ni(II) на основе 2,4,9,11-тетра-трет-бутилбензо[5,6][1,4]оксазино[2,3-b]феноксазин-1-ола
М. Г. Чегерев 1, *, О. П. Демидов 2, П. А. Князев 1, Н. И. Макарова 1, А. Г. Стариков 1, Е. П. Ивахненко 1, В. И. Минкин 1
1 Научно-исследовательский институт физической и органической химии Южного федерального университета
Ростов-на-Дону, Россия
2 Северо-Кавказский федеральный университет
Ставрополь, Россия
* E-mail: mchegerev@sfedu.ru
Поступила в редакцию 12.10.2021
После доработки 21.10.2021
Принята к публикации 24.10.2021
- EDN: PVTXRX
- DOI: 10.31857/S0132344X22050036
Аннотация
Синтезирована серия бис-хелатных комплексов цинка, меди, кобальта и никеля (I–IV) с 2,4,9,11-тетра-трет-бутилбензо[5,6][1,4]оксазино[2,3-b]феноксазин-1-олом (LН) общей формулы ML2. Пространственное и электронное строение полученных координационных соединений исследовано методом теории функционала плотности DFT B3LYP/6-311++g(d,p). Молекулярное строение аддукта комплекса меди с пиридином (II · Py) установлено методом РСА (CIF file CCDC № 2113818). Определены спектральные характеристики лиганда и синтезированных на его основе комплексов.
Поиск новых эффективных люминофоров и изучение процессов трансформации энергии в области органических светодиодов (OLED) являются актуальными задачами современной химии и материаловедения [1–5]. Широкое применение в качестве фото- и электролюминесцентных материалов имеют комплексы металлов главных групп с лигандами оксихинолинового типа [6–8]. Среди наиболее часто используемых комплексов для получения органических электронных устройств можно выделить хелатные трис(8-оксихинолин)алюминий и бис(8-оксихинолин)цинк, способные выполнять роль эмиссионных и электрон-транспортных материалов [9–13].
Структурными аналогами оксихинолинов являются производные 4-гидроксиакридина и 1-гидроксифеназина, включающие хелатный фрагмент гидроксихинолина и π-расширенную ароматическую систему. В литературе имеется несколько работ, описывающих строение и люминесцентные свойства металлокомплексов на основе подобных лигандов [14–19]. Модификация рассмотренных выше лигандных систем путем введения в углеродный скелет дополнительных гетероатомов (O или N) приводит к классу редокс-активных ф-еноксазинонов. К настоящему времени опубликован ряд работ, посвященных изучению строения и свойств комплексов металлов (как переходных, так и непереходных) на основе феноксазинонов в различных редокс-состояниях (нейтральных и ион-радикальных) [20–31]. В исследованных системах обнаружены явления обратимого переноса электрона между металлом и лигандом, а также обменные взаимодействия между парамагнитными центрами различной природы.
В последние годы значительное внимание химиков уделено замещенным трифенодиоксазинам (TPDO) [32, 33], являющихся перспективными материалами для использования в полевых транзисторах [34–36], люминесцентных материалах [37] и в качестве красителей для создания эффективных солнечных батарей [38, 39]. Подобные соединения являются структурными аналогами пентаценов, однако, в отличие от последних, содержат гетероатомы (O, N, S) в углеродном скелете, что позволяет получать лигандные системы с тонко настраиваемыми электронными и структурными свойствами [40].
Настоящая работа посвящена синтезу новых комплексов Zn(II), Cu(II), Co(II) и Ni(II) на основе пространственно-затрудненного 2,4,9,11-тетра-трет-бутилбензо-[5,6][1,4]оксазино[2,3-b]фено-ксазин-1-ола (LH) и исследованию физико-химических свойств полученных соединений. Лиганд LH является гетеропентаценом, содержащим O,N-оксихинолиновый фрагмент, за счет которого возможно образование хелатных металлокомплексов. Изучение подобного класса соединений позволит расширить представления о пространственном и электронном строении комплексов, содержащих хелатные гетеропентаценовые лиганды.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Использованные в работе перхлоратные соли металлов являются коммерчески доступными. Лиганд LH получали согласно методике [33]. Использованные в работе растворители очищали и обезвоживали по стандартным методикам.
Элементный анализ выполняли на анализаторе Elementar Vario El cube. Спектры ЭПР регистрировали на спектрометре Bruker EMX Plus. В качестве стандарта при определении g-фактора использовали дифенилпикрилгидразил (gi = 2.0037). Симуляцию спектров ЭПР проводили при помощи программного пакета Easyspin для Matlab [41]. Электронные спектры поглощения (ЭСП) записывали для 2 × 10–5 М растворов на спектрофотометре Agilent 8453, снабженном термостатируемой кюветой. Все спектры записывали в стандартной 1 см кварцевой кювете при комнатной температуре. Флуоресцентные измерения проводили на спектрофлуориметре Cary Eclipse (Varian). Для приготовления растворов использовали дихлорметан (Acros Organics) спектральной степени чистоты. ИК-спектры снимали на спектрометре Varian Excalibur 3100 FTIR.
Синтез ZnL2 (I). К суспензии лиганда (0.05 г, 0.095 ммоль) в ТГФ (10 мл) добавляли небольшое количество Et3N (0.1 мл). К полученной смеси добавляли 0.017 г (0.047 ммоль) Zn(ClO4)2 ⋅ 6H2O. Полученную реакционную смесь нагревали на водяной бане (70°C) в течение 1 ч. Окраска реакционной смеси изменялась с красной на интенсивную сине-фиолетовую. После добавления к реакционной смеси этанола (10 мл) выпадал мелкодисперсный порошок. Осадок отфильтровывали на воздухе и промывали небольшими порциями этанола и дистиллированной воды. Выход комплекса I в виде темно-фиолетового порошка 81%.
Спектр ЯМР 1Н (ТГФ-d8; 20°С; δ, м.д.; J, Гц): 7.33 (д., 1H, 2.3, Hарил); 7.29 (д. 1H, 2.3, Hарил); 7.17 (с. 1H, Hарил); 6.89 (с., 1H, Hарил); 6.59 (с. 1H, Hарил); 1.34 (c., 9H, t-Bu); 1.40 (c., 9H, t-Bu); 1.49 (c., 9H, t-Bu); 1.46 (c., 9H, t-Bu).
ИК-спектр (ν, см–1): 637 с, 725 ср, 755 сл, 781 сл, 808 сл, 851 ср, 879 сл, 915 ср, 994 сл, 1008 сл, 1033 сл, 1081 с, 1164 с, 1193 ср, 1217 ср, 1234 ср, 1255 ср, 1281 ср, 1359 ср, 1380 ср, 1401 ср, 1445 сл, 1467 ср, 1539 с, 1557 ср, 1598 ср, 1634 сл, 2867 ср, 2904 ср, 2950 ср.
Синтез CuL2 (II) осуществляли по аналогичной методике с использованием Cu(ClO4)2 · 6H2O (0.017 г, 0.047 ммоль) в качестве источника ионов меди(II). Выход 79%.
ИК-спектр (ν, см–1): 638 с, 725 ср, 756 сл, 780 сл, 808 сл, 851 ср, 878 сл, 915 ср, 995 сл, 1008 сл, 1031 сл, 1081 с, 1163 с, 1193 ср, 1219 ср, 1234 ср, 1257 ср, 1280 ср, 1357 ср, 1379 ср, 1402 ср, 1444 сл, 1466 ср, 1538 с, 1559 ср, 1598 ср, 1633 сл, 2867 ср, 2906 ср, 2950 ср.
Синтез CoL2 (III) осуществлялся по аналогичной методике с использованием Co(ClO4)2 ⋅ 6H2O (0.017 г, 0.047 ммоль) в качестве источника ионов кобальта(II). Выход 75%.
ИК-спектр (ν, см–1): 636 с, 724 ср, 754 сл, 780 сл, 809 сл, 851 ср, 878 сл, 916 ср, 995 сл, 1009 сл, 1031 сл, 1080 с, 1162 с, 1193 ср, 1218 ср, 1235 ср, 1258 ср, 1280 ср, 1357 ср, 1377 ср, 1403 ср, 1442 сл, 1465 ср, 1538 с, 1559 ср, 1599 ср, 1633 сл, 2866 ср, 2905 ср, 2951 ср.
Синтез NiL2 (IV) осуществляли по аналогичной методике с использованием Ni(ClO4)2 ⋅ 6H2O (0.017 г, 0.047 ммоль) в качестве источника ионов никеля(II). Выход 84%.
ИК-спектр (ν, см–1): 634 с, 723 ср, 754 сл, 780 сл, 810 сл, 851 ср, 878 сл, 912 ср, 998 сл, 1009 сл, 1034 сл, 1082 с, 1162 с, 1193 ср, 1215 ср, 1235 ср, 1259 ср, 1280 ср, 1356 ср, 1377 ср, 1402 ср, 1442 сл, 1464 ср, 1538 с, 1558 ср, 1597 ср, 1633 сл, 2867 ср, 2902 ср, 2950 ср.
Квантово-химические расчеты выполняли с использованием программного пакета Gaussian 16 [42] методом теории функционала плотности (DFT) с применением функционала B3LYP [43] и базисного набора 6-311++g(d,p) для всех атомов. Эффекты растворителя (CH2Cl2) учитывали с использованием модели CPCM [44].
РСА аддукта II · Py проведен при 293 K на дифрактометре Agilent SuperNova при использовании микрофокусного источника рентгеновского излучения с анодом из Cu и координатным CCD-детектором Atlas S2. Сбор отражений, определение и уточнение параметров элементарной ячейки проведены с использованием специализированного программного пакета CrysAlisPro 171.41.93a [45]. Структуры расшифрованы с помощью программы ShelXT [46], уточнены по программе ShelXL [47].
Соединение II · Py кристаллизуется в пространственной группе I2/a (a = 23.1152(3), b = = 9.63830(10), с = 31.4930(4) Å, α = 90°, β = = 111.0930(10)°, γ = 90°, V = 6546.25(14) Å3, Z = 4, ρ(расч.) = 1.212 г/см3, μ = 0.903 мм–1). Измерено 35698 отражений, 6819 независимых отражений (Rint = 0.0238) использованы для решения структуры и последующего уточнения 397 параметров полноматричным МНК в анизотропном приближении для неводородных атомов. Водородные атомы в II · Py помещены в геометрически рассчитанные положения и уточнены изотропно с фиксированными тепловыми параметрами U(H)изо = 1.2U(C)экв (U(H)изо = 1.5U(C)экв для метильных групп). После финального уточнения wR2 = 0.1244 и S(F 2) = = 1.054 для всех отражений (R1 = 0.0402 для всех отражений, удовлетворяющих условию I > 2σ(I)). Остаточные максимум и минимум электронной плотности составили 0.34/–0.25 e/Å–3.
Структура аддукта II · Py депонирована в Кембриджский банк структурных данных (CCDC № 2113818; ccdc.cam.ac.uk/structures/).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Основная методика получения бис-хелатных комплексов ZnL2 (I), CuL2 (II), CoL2 (III), NiL2 (IV) с 2,4,9,11-тетра-трет-бутил-бензо[5,6][1,4]-оксазино[2,3-b]феноксазин-1-олом (LH) представляет собой взаимодействие перхлоратных солей соответствующих металлов с лигандом в соотношении 1 : 2 в среде ТГФ. В качестве депротонирующего агента использован Et3N. Поскольку представленный гетеропентаценовый лиганд является потенциально редокс-активным, синтез проводился с использованием вакуумной линии с целью исключения влияния кислорода воздуха на механизм и продукты реакции. Кипячение реакционных смесей в течение часа приводит к изменению окраски растворов с красной на сине-фиолетовую. Добавление небольшого количества этанола приводит к выпадению осадка. Комплексы I–IV выделены фильтрацией на воздухе в виде тeмно-фиолетовых порошков с высоким выходом (~80%). Полученные соединения устойчивы к действию кислорода и влаги воздуха (схема 1 ).
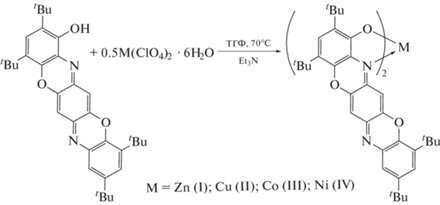
Схема 1 .
В качестве альтернативной методики получения комплексов рассмотрен синтез с использованием ацетатов металлов с LH в соотношении 1 : 2 в среде изопропилового спирта на воздухе. Реакционную смесь кипятили на воздухе с обратным холодильником в течение 1 ч. Окраска изменялась с красной на сине-фиолетовую. Данный подход приводит к получению идентичных соединений I–IV, однако характеризуется меньшими выходами конечных продуктов (~65%).
Спектр ЯМР 1Н, измененный в ТГФ-d8, характеризуется одним набором сигналов координированного лиганда, что указывает на эквивалентность хелатных лигандов в полученном комплексе. Сигналы от пяти протонов при ароматических кольцах находятся в диапазоне 7.33–6.59 м.д., в то время как сигналы от протонов четырех трет-бутильных заместителей проявляются в характерной области 1.46–1.34 м.д.
Все исследуемые комплексы показали слабую тенденцию к образованию монокристаллов. Однако при медленном упаривании раствора комплекса II в смеси толуол–пиридин (1 : 1) в течение 1 месяцa были выделены игольчатые монокристаллы, пригодные для РСА. На рис. 1 приведена молекулярная структура аддукта II · Py. Согласно рентгеноструктурным данным, комплекс II взаимодействует с молекулой пиридина с образованием пентакоординированного аддукта II · Py. Координационное окружение иона меди представляет собой тригональную бипирамиду, в основании которой лежат атомы азота N двух O,N-хелатных лиганов и молекулы пиридина, в то время как в апикальных позициях находятся атомы кислорода. Для анализа координационной сферы пентакоординированных комплексов используется геометрический параметр τ5; в случае идеальной квадратной пирамиды τ5 = 0, а в случае идеальной тригональной бипирамиды τ5 = 1 [48]. В соединении II · Py вычисленная величина τ5 = 0.71 соответствует тетрагонально-искаженному тригонально-бипирамидальному окружению иона меди.
Рис. 1.
Молекулярная структура аддукта II · Py (a); фрагмент координационного окружения иона меди(II) (б); межатомные расстояния в пентациклическом лиганде (Å) (в). Для ключевых атомов тепловые эллипсоиды приведены с 50%-ной вероятностью. Атомы водорода не показаны для ясности.
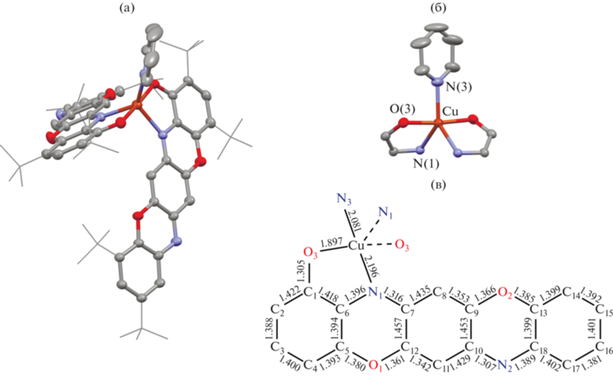
В кристаллической упаковке аддукта II · Py наблюдаются межмолекулярные взаимодействия различной природы: CH···арен (~2.8 Å) и π–π-стэкинг-взаимодействия (3.3–3.9 Å) между сопряженными системами гетеропентаценовых лигандов соседних молекул.
Длины связей Cu–O(3) 1.897 и Cu–N(1) 2.196 Å входят в диапазон, характерный для близких по структуре O,N-хелатных комплексов меди [48–51]. Расстояние между атомом меди и атомом азота (N(3)) координированной молекулы пиридина составляет 2.081 Å. Оба пентациклических лиганда имеют идентичное строение и структурные параметры. Длины связей С(1)–С(6) и С(13)–С(18) в шестичленных кольцах, содержащих трет-бутильные заместители, близки к ароматическим (1.40 Å), в то время как в центральной части пентациклического лиганда наблюдается значительное хиноидное искажение, характеризующееся чередованием длинных и коротких связей С–С (рис. 1). Расстояния С(6)–N(1) 1.396(2) и С(18)–N(2) 1.389(2) Å характерны для одинарных связей углерод–азот, в то время как С(7)–N(1) 1.316(2) и С(10)–N(2) 1.307(2) Å заметно короче, что указывает на их кратную (C=N) природу. Таким образом, на основании имеющихся структурных данных можно заключить, что в аддукте II · Py пентациклические лиганды находятся в моноанионной диамагнитной форме.
Данное заключение хорошо согласуется с результатами квантово-химических расчетов. Оптимизированная геометрия (DFT/B3LYP/6-311++g(d,p)) аддукта II · Py характеризуется слегка увеличенными длинами связей Cu–O и Cu–N и демонстрирует заметное тетрагонально-пирамидальное искажение координационного полиэдра. Это обусловлено тем фактом, что при оптимизации геометрии единичной молекулы не учитываются эффекты кристаллической упаковки. Отсутствие спиновой плотности (qs) на атомах сопряженной системы лиганда подтверждает заключение о его моноанионной природе, в то время как один неспаренный электрон делокализован между ионом двухвалентной меди ($q_{s}^{{{\text{Cu}}}}$ = 0.60) и донорными атомами N (Σqs = 0.21) и O (Σqs = 0.19).
С целью установления молекулярного и электронного строения выполнена оптимизация геометрии тетракоординированных комплексов I–IV (DFT/B3LYP/6-311++g(d,p)). Согласно расчетам, координационное окружение центрального иона металла в I–IV, составленное из атомов O и N хелатирующего лиганда, занимает промежуточное положение между плоско-квадратным и тетраэдрическим. Для количественной характеристики искажения полиэдра был использован геометрический параметр τ4, равный нулю в случае плоско-квадратной и единице в случае тетраэдрической геометрий [51]. В качестве примера на рис. 2 приведено строение комплекса II. Вычисленная величина τ4 для II составила 0.35, что позволяет описать геометрию комплекса как тетраэдрически-искаженную плоско-квадратную. В табл. 1 приведены избранные длины связей металл–донорный атом для серии I–IV. Длины связей C–C и C–N в пентациклических лигандах близки к полученным ранее для аддукта II · Py, что указывает на их моноанионную форму. На основании полученных данных, несмотря на потенциальную редокс-активность пентациклического лиганда, электронное строение всей серии тетракоординированных комплексов I–IV можно описать следующим образом: ион двухвалентного металла (M2+), координированный двумя O,N-хелатными моноанионными лигандами (L–). Спиновая плотность в комплексе II, как и в аддукте II · Py, делокализована между атомом меди ($q_{s}^{{{\text{Cu}}}}$ = 0.58) и донорными атомами хелатного узла (Σqs = 0.41).
Рис. 2.
Оптимизированная геометрия комплекса II (DFT/B3LYP/6-311++g(d,p)). Длины связей приведены в ангстремах. Атомы водорода и метильные заместители трет-бутильных групп не показаны для ясности.
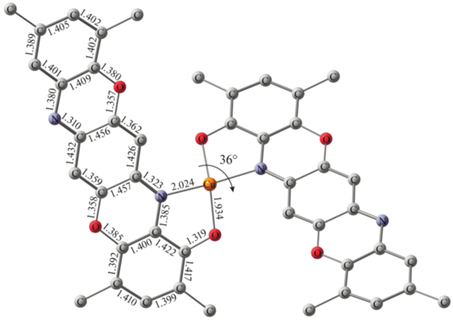
Таблица 1.
Избранные длины связей (Å) исследуемых комплексов I–IV согласно DFT B3LYP/6-311++g(d,p).
| Комплекс | Связь (d, Å) | τ4 | |
|---|---|---|---|
| M–O | M–N | ||
| I | 1.943 | 2.082 | 0.76 |
| II | 1.934 | 2.024 | 0.35 |
| III | 1.936 (hs) | 2.094 (hs) | 0.72 |
| 1.883 (ls) | 1.988 (ls) | 0.31 | |
| IV | 1.923 (hs) | 2.045 (hs) | 0.71 |
| 1.875 (ls) | 1.926 (ls) | 0.15 | |
Вследствие высокой электронной лабильности ионов кобальта(II) и никеля(II), способных существовать как в низко- (ls), так и в высокоспиновых (hs) состояниях, квантово-химически исследованы возможные изомеры комплексов III и IV на соответствующих поверхностях потенциальной энергии (ППЭ). Согласно расчетам, псевдотетраэдрическое высокоспиновое (S = 3/2) состояние комплекса III, hs-Co(II) является основным, а низкоспиновый изомер (S = 1/2), содержащий ls-Co(II), дестабилизирован относительно него на 12.5 ккал/моль. Последний характеризуется значительно искаженной (близкой к плоско-квадратной) геометрией координационного узла. Рассчитанное значение qs на ионе кобальта в hs-Co(II) изомере комплекса III составляет 2.705, что несколько ниже ожидаемой для свободного иона высокоспинового кобальта(II) ($q_{s}^{{{\text{Co}}}}$ = 3). Данный факт обусловлен частичной делокализацией спиновой плотности между донорными атомами кислорода и азота координированных лигандов.
В случае комплекса NiL2 (IV) предсказана потенциальная возможность конфигурационной изомерии (КИ), заключающейся в обратимом взаимопревращении плоско-квадратного изомера с тетраэдрическим [52, 53]. Согласно расчетам, основным состоянием соединения IV является парамагнитный псевдотетраэдрический изомер, содержащий ион двухвалентного никеля в высокоспиновом состоянии (S = 1). С другой стороны, диамагнитный плоскоквадратный изомер (S = 0) дестабилизирован относительно основного состояния всего на 3 ккал/моль, что указывает на высокую вероятность реализации КИ. Анализ распределения спиновой плотности в высокоспиновом изомере показал слегка заниженное значение ($q_{s}^{{{\text{Ni}}}}$ = 1.618) на ионе металла (hs-Ni(II)), что, как и в соединении III, обусловлено ее частичной делокализацией в хелатных фрагментах.
Для соединения II (S = 1/2) зарегистрирован изотропный спектр ЭПР (рис. 3а) в растворе толуола при 330 K, демонстрирующий анизотропно уширенный квартет, обусловленный сверхтонким взаимодействием (СТВ) неспаренного электрона с магнитными изотопами меди (63, 65Cu, I = 3/2). Данная форма спектра является характерной для подобного типа тетракоординированных комплексов меди(II) [49, 50, 54]. Необходимо отметить, что наиболее интенсивная и высокопольная компонента спектра демонстрирует сверхтонкую структуру (СТС), обусловленную СТВ неспаренного электрона с двумя эквивалентными магнитными ядрами атома азота (14N, I = 1) координированных хелатных лигандов. Экспериментально определенные величины констант СТВ и g-фактора составляют: ai(63, 65Cu) = 66.5 Э, ai(14N) = 10 Э и gi = 2.14.
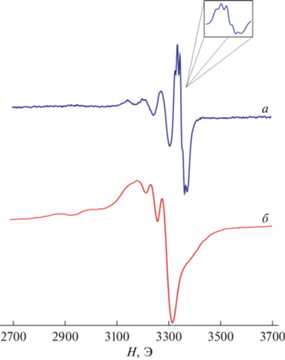
Замена растворителя на пиридин приводит к значительным изменениям в спектре ЭПР комплекса II (рис. 3б), обусловленным дополнительной координацией молекулы пиридина с образованием аддукта II · Py. При этом происходит значительное изменение геометрии координационного полиэдра c искаженной плоско-квадратной на тригонально-пирамидальную. Величина константы СТВ с 63, 65Cu составила 140 Э (gi = 2.24). На основании имеющихся данных можно заключить, что координация молекулы пиридина сопровождается изменением электронной структуры центрального иона меди, что находит отражение в значительном изменении формы и параметров спектра ЭПР.
С целью изучения влияния природы металла на спектральные свойства координационных соединений I–IV проведено сравнительное исследование оптических и фотолюминесцентных свойств в растворе дихлорметана при комнатной температуре. На рис. 4 приведены ЭСП растворов лиганда LH и полученных на его основе комплексов I–IV. Экспериментальные данные систематизированы в табл. 2.
Рис. 4.
Электронные спектры поглощения LH и I–IV в дихлорметане при T = 293 K (с = 2 × 10–5 моль/л, l = 1 см).
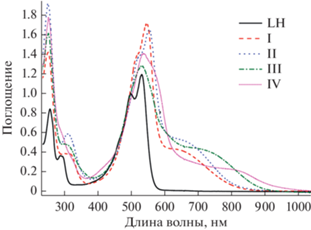
Таблица 2.
Спектральные данные для LH и комплексов I–IV в дихлорметане при 293 K
| Соединение | Поглощение λmax, нм (ε, 103 M–1 см–1) |
|---|---|
| LH | 254 (41.9), 286 (18.1), 496 (49.9), 529 (59.7) |
| I | 249 (71.0), 309 (19.2), 545 (85.8), 625 пл (21.6) |
| II | 249 (96.3), 309 (29.2), 550 (82.5), 630 пл (26.9) |
| III | 250 (80.8), 305 пл (23.7), 530 (64.2), 700 пл (21.6) |
| IV | 250 (89.3), 287 пл (30.6), 536 (70.6), 800 пл (11.0) |
В спектральном диапазоне 400–1000 нм ЭСП лиганда LH характеризуется интенсивной структурированной полосой поглощения с двумя максимумами при 496 и 529 нм и молярными коэффициентами экстинкции – 49 900 и 59 700 M–1 см–1 соответственно (табл. 2, рис. 4.). Для установления природы наблюдаемых электронных переходов выполнены TD-DFT (Time Dependent DFT) расчеты теоретических ЭСП, которые показали хорошее согласие с экспериментальными данными. Вычисленные интенсивные полосы в области 490–530 нм отвечают π–π-переходам между ВЗМО (высшая занятая молекулярная орбиталь) и НСМО (низшая свободная молекулярная орбиталь). НСМО в основном локализована на центральном пара-хиноновом фрагменте гетеропентацена, в то время как ВЗМО на ароматических кольцах аминофенолятных частей лиганда (рис. 5).
Рис. 5.
Формы граничных орбиталей LH и комплекса I, участвующих в переходах при 500–530 и 625 нм соответственно.
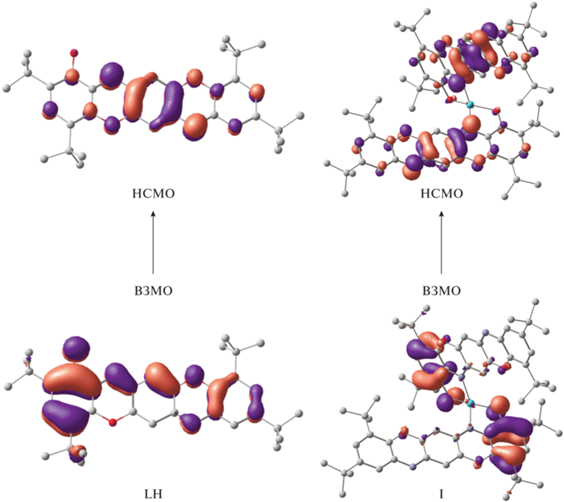
В отличие от лиганда LH, для комплексов I–IV в том же спектральном диапазоне, наряду с полосой при 530–550 нм (ε = 64 200–85 800 M–1 см–1), наблюдается новая полоса поглощения с максимумами в районе 625–800 нм (ε = 11 000–26 900 M–1 см–1) (табл. 2, рис. 4). Комплексообразование приводит к значительному батохромному смещению максимумов полос длинноволнового поглощения координационных соединений I–IV относительно лиганда на 96–271 нм, что визуально проявляется в углублении окраски растворов комплексов в сравнении с лигандом от оранжевой до темно-фиолетовой. Согласно результатам TD-DFT расчетов, полоса поглощения в спектрах соединений I–IV в области 530–550 нм имеет ту же природу, что и наблюдаемая в свободном лиганде LH, в то время как новая полоса в диапазоне 625–800 нм обусловлена переносом заряда с O,N-хелатных металлоциклов молекулы (ВЗМО) на центральные пара-хиноновые (НСМО) фрагменты лигандов. На рис. 5 приведена форма граничных орбиталей LH и комплекса I, участвующих в обсуждаемых переходах.
Анализ спектральных характеристик исследуемых соединений показывает, что комплексы цинка и меди обладают практически одинаковыми оптическими свойствами, что подтверждается близкими по форме, положению и интенсивности полосами поглощения (табл. 2, рис. 4). Батохромное смещение максимумов полос длинноволнового поглощения комплексов I и II по сравнению с лигандом LH составляет 96 и 101 нм соответственно. В то же время комплексы кобальта III и никеля IV демонстрируют значительное смещение поглощения (Δλ = 75–175 нм) в ближнюю ИК-область спектра в сравнении с аналогичными по составу комплексами меди и цинка.
Из литературных данных известно, что близкие к использованному в работе по структуре гетеропентаценовые лиганды демонстрируют интенсивную флуоресценцию и характеризуются высокой фотостабильностью [33]. В то же время исследование фотофизических свойств LH и комплексов I–IV на его основе показало, что как лиганд, так полученные на его основе комплексы не проявляют достаточно заметных фотолюминесцентных свойств в растворах при комнатной температуре.
Таким образом, синтезирована серия бис-хелатных комплексов цинка, меди, кобальта и никеля с 2,4,9,11-тетра-трет-бутилбензо[5,6][1,4]-оксазино[2,3-b]феноксазин-1-олом. Геометрические и электронные характеристики полученных соединений исследованы методом теории функционала плотности. Тетракоординированные комплексы характеризуются искаженной геометрией координационного полиэдра, промежуточной между псевдотетраэдрической и плоско-квадратной. Молекулярное строение пентакоординированного аддукта комплекса меди с пиридином установлено методом РСА. Анализ структурных параметров указывает на диамагнитную моноанионную форму гетеропентаценового лиганда во всех полученных соединениях. Изучены оптические свойства лиганда и синтезированных комплексов.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Chen C.H., Shi J. // Coord. Chem. Rev. 1998. V. 171. P. 161.
Mitschke U., Bauerle P. // J. Mater. Chem. 2000. V. 10. P. 1471.
Organic Light-Emitting Diodes (OLEDs): Materials, Devices and Applications. Buckley A. Woodhead Publishing Limited, Philadelphia, PA, 2013.
OLED Fundamentals: Materials, Devices, and Processing of Organic Light-Emitting Diodes / Eds Gaspar D.J., Polikarpov E. Boca Raton (FL, USA): Taylor & Francis Group, 2015.
Bunzli J.-C.G., Piguet C. // Chem. Soc. Rev. 2005. V. 34. P. 1048.
Tsuboi T., Nakai Y., Torii Y. // Centr. Eur. J. Phys. 2012. V. 10. P. 524.
Shen L., Li F.Y., Sha Y.W. et al. // Tetrahedron Lett. 2004. V. 45. P. 3961.
Monzon L.M.A., Burke F., Coey J.M.D. // J. Phys. Chem. C. 2011. V. 115. P. 9182.
Hamada Y., Sano T., Fujita M. et al. // Jpn. J. Appl. Phys. Pt 2. 1993. V. 32. P. L514.
Sapochk L.S., Benincasa F.E., Schofield R.S. et al. // J. Am. Chem. Soc. 2002. V. 124. P. 6119.
Dumur F. // Synth. Met. 2014. V. 195. P. 241.
Hopkins T.A., Meerholz K., Shaheen S. et al. // Chem. Mater. 1996. V. 8. P. 344.
Mishra A., Nayak P.K., Periasamy N. // Tetrahedron Lett. 2004. V. 45. P. 6265.
Mastropietro T.F., Szerb E.I., Deda M.L. et al. // Eur. J. Inorg. Chem. 2013. V. 2013. P. 2188.
Balashova T.V., Polyakova S.K., Arsenyev M.V. et al. // Eur. J. Inorg. Chem. 2021. P. 1441.
Balashova T.V., Arsenyev M.V., Polyakova S.K. et al. // J. Mol. Struct. 2021. V. 1229. P. 129798.
Miozzo L., Papagni A., Casalbore-Miceli G., Del Buttero P. et al. // Chem. Mater. 2004. V. 16. P. 5124.
Aiello I., Aiello D., Ghedini M. // J. Coord. Chem. 2009. V. 62. P. 3351.
Ukwitegetse N., Femia D., Sylvinson Muthiah Ravinson D. et al. // Inorg. Chem. 2021. V. 60. P. 866.
Stegmann H.B., Scheffler K. // Chem. Ber. 1968. V. 101. P. 262.
Whalen A.M., Bhattacharya S., Pierpont C.G. // Inorg. Chem. 1994. V. 33. P. 347.
Speier G., Whalen A.M., Csihony J., Pierpont C.G. // Inorg. Chem. 1995. V. 34. P. 1355.
Ivakhnenko E.P., Starikov A.G., Minkin V.I. et al. // Inorg. Chem. 2011. V. 50. P. 7022.
Антипин М.Ю., Ивахненко Е.П., Кощиенко Ю.В. и др. // Изв. АН. Сер. хим. 2013. № 8. С. 1744 (Antipin M.Yu., Ivakhnenko E.P., Koshchienko Yu.V. et al. // Russ. Chem. Bull. 2013. V. 62. P. 1744).
Romanenko G.V., Ivakhnenko E.P., Minkin V.I. et al. // Inorg. Chim. Acta. 2014. V. 418. P. 66.
Ivakhnenko E.P., Starikov A.G., Lyssenko K.A. et al. // Inorg. Chim. Acta. 2014. V. 410. P. 144.
Ranis L.G., Werellapatha K., Pietrini N.J. et al. // Inorg. Chem. 2014. V. 53. P. 10203.
Ивахненко Е.П., Кощиенко Ю.В., Князев П.А. и др. // Коорд. химия. 2016. Т. 42. № 4. С. 221 (Ivakhnenko E.P., Koshchienko Yu.V., Knyazev P.A. et al // Russ. J. Coord. Chem. 2016. V. 42. P. 509). https://doi.org/10.1134/S1070328416040011
Ivakhnenko E.P., Romanenko G.V., Simakov V.I. et al. // Inorg. Chim. Acta. 2017. V. 458. P. 116.
Galley S.S., Pattenaude S.A., Gaggioli C.A. et al. // J. Amer. Chem. Soc. 2019. V. 141. P. 2356.
Чегерев М.Г., Арсеньева К.В., Черкасов А.В., Пискунов А.В. // Коорд. химия. 2020. Т. 46. № 11. С. 672 (Chegerev M.G., Arsenyeva K.V., Cherkasov A.V. Piskunov A.V. // Russ. J. Coord. Chem. 2020. V. 46. P. 746). https://doi.org/10.1134/S1070328420110019
Seidel P. // Chem. Ber. 1890. V. 23. P. 182.
Ivakhnenko E.P., Romanenko G.V., Makarova N.I. et al. // Dyes Pigments. 2020. V. 176. P. 108174.
Chong-an D., Li J., Yu G. et al. // Org. Lett. 2008. V. 10. P. 3025.
Nicolas Y., Castet F., Devynck M. et al. // Org. Electron. 2012. V. 13. P. 1392.
Gruntz G., Lee H., Hirsch L. et al. // Adv. Electron. Mater. 2015. P. 1500072.
Tanaka T, Ashida T, Matsumoto S. // Chem. Lett. 2011. V. 40. P. 573.
Allama N.Y., Lepeltier F., Massin M. et al. // Chem. Eur. J. 2014. V. 20. P. 3678.
Gong X., Han P., Wen H. et al. // Eur. J. Org. Chem. 2017. P. 3689.
Miao Q. // Synlett. 2012. V. 23. P. 326.
Stoll S., Schweiger A.J. // J. Magn. Reson. 2006. V. 178. P. 42.
Frisch M.J., Trucks G.W., Schlegel H.B. et al. Gaussian 16. Revision C. 01. Wallingford: Gaussian, 2019.
Becke A.D. // J. Chem. Phys. 1993. V. 98. P. 5648.
Barone V., Cossi M. // J. Phys. Chem. A. 1998. V. 102. P. 1995.
CrysAlisPro. Version 171.41.93a. Rigaku Oxford Diffraction. 2015. https://www.rigaku.com/products/crystallography/crysalis
Sheldrick G.M. // Acta Crystallogr. A. 2015. V. 71. P. 3.
Sheldrick, G.M. // Acta Crystallogr. C. 2015. V. 71. P. 3.
Addison A.W., Rao T.N., Reedijk J. et al. // Dalton Trans. 1984. P. 1349.
Lada Z.G., Sanakis Y., Raptopoulou C.P. et al. // Dalton Trans. 2017. V. 46. P. 8458.
Ivakhnenko E.P., Knyazev P.A., Vitkovskaya Y.G. et al. // Eur. J. Inorg. Chem. 2021. V. 2021. P. 2055.
Yang L., Powell D.R., Houser R.P. // Dalton Trans. 2007. Is. 9. P. 955.
Ernst R.E., O’Connor M.J., Holm R.H. // J. Am. Chem. Soc. 1967. V. 89. P. 6104.
Garnovskii A.D., Nivorozhkin A.L., Minkin V.I. // C-oord. Chem. Rev. 1993. V. 126. P. 1.
Antholine W.E., Bennett B., Hanson G.R. // Multifrequency Electron Paramagnetic Resonance / Ed. Misra S.K. Wiley-VCH Verlag, 2011. P. 647.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия