

Координационная химия, 2022, T. 48, № 7, стр. 428-444
Синтез, строение и свойства комплексов редкоземельных металлов (Pr(III), Тb(III), Ho(III) и Er(III)) c 2-меркаптопиридин-N-оксидом
В. П. Штефанец 1, Г. В. Шилов 2, Е. И. Куницына 2, Р. Б. Моргунов 2, Н. А. Санина 2, *, С. М. Алдошин 2
1 Московский физико-технический институт (национальный исследовательский университет)
Долгопрудный, Россия
2 Институт проблем химической физики РАН
Черноголовка, Россия
* E-mail: sanina@icp.ac.ru
Поступила в редакцию 18.02.2022
После доработки 24.02.2022
Принята к публикации 25.02.2022
- EDN: EFMBJT
- DOI: 10.31857/S0132344X22070088
Аннотация
Представлен синтез и структурные исследования методом РСА четырех соединений, полученных взаимодействием нитратов редкоземельных металлов(III) с 2-меркапто-пиридин-N-оксидом (HSC5H4NO) в водно-щелочных растворах: [Pr2(SC5H4NO)6(H2O)2] (I), [Tb2(SC5H4NO)6(H2O)2] (II), [Ho2(SC5H4NO)6(H2O)2] (III), [Er2(SC5H4NO)6(H2O)2] (IV). Каждый ион металла в биядерных комплексах формирует восьмикоординационную структуру. Она включает в себя два лиганда SC5H4NO, хелатным способом (через S и O) координирующие каждый ион металла (3+), два мостиковых атома кислорода, принадлежащие двум SC5H4NO и две молекулы координационной воды. Методом СКВИД-магнитометрии установлено, что в синтезированных комплексах при температурах от 300 до 150 K произведение χT и эффективный момент (μэфф) не изменяются. При температурах T < 150 K в I (Т < 30 K) и II (T < 50 K) наблюдается спад значения μэфф с понижением температуры, что может свидетельствовать о наличии магнитного перехода и об антиферромагнитном взаимодействии между ионами Pr3+ и Tb3+. В комплексе III наблюдается повышение значения μэфф и резкий спад при T < 10 K, а в IV – плавное увеличение μэфф с понижением температуры. Этот факт может свидетельствовать о появлении ферромагнитных корреляций в IV. Для I как в подмагничивающем поле, так и без него отсутствует магнитный релаксационный отклик во всем диапазоне частот переменного поля HAC от 0.2 до 1400 Гц. Зависимости магнитного момента от поля при температурах 2 и 5 K для II–IV имеют вид, характерный для парамагнетиков при данных температурах. При T = 2 K образцы находятся в том же спиновом состоянии, что и при T = 300 K: охлаждение не изменяет спиновое состояние ионов Tb3+, Ho3+ и Er3+ в комплексах. Магнитного гистерезиса при низких температурах (2 и 5 K) в этих комплексах не наблюдалось. Зависимости действительной и мнимой частей магнитной восприимчивости от частоты переменного поля (AC) для II–IV были записаны в отсутствие постоянного поля (DC) и в подмагничивающем поле. Диаграммы Cole-Cole демонстрируют, что в II–IV наблюдается релаксационная мода движения доменной стенки, но отсутствует смещение максимума мнимой части при изменении температуры (туннельный механизм релаксации).
Дизайн новых магнетиков, установление корреляций структура–свойства c последующей оптимизацией свойств перспективных соединений и создание на их основе новейших функциональных материалов являются актуальными направлениями в области современного материаловедения, связанными с непрерывно возрастающей потребностью уменьшения электронной базы вычислительных устройств, увеличения плотности записи информации и пр. Такие материалы могут стать основой для развития молекулярной спинтроники и квантового компьютинга [1]. Создание молекулярных наномагнитов с высокими температурами блокирования намагниченности может привести к технологическим прорывам в области информационных технологий. Идеология использования спинов электронов для хранения и обработки информации представляет значительный практический интерес в связи с возможными применениями получаемых молекулярных систем в молекулярной спинтронике (спиновые переключатели, молекулярные аналоги мультиферроиков и т.д.) и квантовых вычислениях (молекулярные квантовые клеточные автоматы). Существенный вклад в развитие этого направления вносит прогресс в области мономолекулярного магнетизма, а также открытие в недавнее время (2010) моноионного магнетизма. Особый интерес представляет дизайн молекулярных наномагнитов на основе комплексов 3d- и 4f-металлов с органическими лигандами, которые при гелиевых температурах демонстрируют необычные магнитные свойства в масштабе одной молекулы или одной молекулярной цепочки [2].
Ионы лантанидов являются хорошими кандидатами для создания мономолекулярных магнитов (МММ), что в основном обусловлено их значительной магнитной анизотропией, возникающей из-за их большого непогашенного орбитального углового момента. МMM на основе лантанидов демонстрируют превосходство в магнетизме по сравнению с остальными, особенно соединения, содержащие ионы Gd3+, Tb3+ и Dy3+ [3]. Так фталоцианиновые комплексы диспрозия(III) и тербия(III) [DyPc2](TBA) и [TbPc2](TBA) (TBA = = тетрабутиламмоний, Pc = фталоцианин) были первыми МММ, содержащими ионы лантанидов [4]. К настоящему времени синтезирован целый ряд МММ на основе лантанидов [5–14], большинство из которых представляют комплексы с Dy(III) [15–24].
Для синтеза этих МММ, наряду с такими лигандами, как полиоксометалаты, фталоцианины, основания Шиффа, используются β-дикетоны [25], нитронил-нитроксильные лиганды ряда имидазола [26–31], а также комплексы с пиридиновыми спиртами (Pyn(CH2)m(OH)k (n = 1, 2; m = = 1, 2; k = 0, 1, 2)) [32].
Комплексы на основе редкоземельных элементов (РЗЭ) и 2-меркаптопиридин-N-оксида (HSC5H4NO) в качестве лиганда представляют интерес для дизайна новых МММ и материалов на их основе [24]. Их строение и магнитные свойства практически не описаны в литературе. Одним из немногих примеров является описание структуры моноядерного комплекса самария состава Sm(HSC5H4NO)3(ДМСO)2 (ДМСО = диметилсульфоксид), в котором растворитель входит в состав комплекса, но магнитные свойства этого соединения не были исследованы [33]. Позже были получены аналогичные моноядерные комплексы на основе празеодима [34] и диспрозия [35]. Среди “немагнитных“ металлов встречается больше примеров: описано строение комплексов олова [36], висмута [37], платины [38], никеля [39], цинка [40] и меди [41].
В настоящей работе впервые синтезированы биядерные комплексы редкоземельных металлов Pr(III), Тb(III), Ho(III) и Er(III) c меркаптопиридин-N-оксидом (HSC5H4NO) и исследовано влияние нитроксилсодержащего фрагмента на их структурообразование в водных растворах и магнитные характеристики с помощью методов рентгеноструктурного анализа, ИК-спектроскопии и СКВИД-магнитометрии (SQUID, Superconducting Quantum Interference Device).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Все реагенты, использованные в работе – коммерческие реактивы (Aldrich) и применялись без дальнейшей очистки (КОН и Ho(NO)3 ⋅ 5H2O, Pr(NO)3 ⋅ 6H2O, Tb(NO)3 ⋅ 6H2O, Er(NO)3 ⋅ 5H2O и лиганд HSC5H4NO ⋅ H2O). Для приготовления растворов использовали бидистиллированную воду.
Синтез комплексов I–IV (общая методика). Растворяли KOH (1.5 ммоль, 0.0846 г) в 10 мл воды, добавляли сухую навеску HSC5H4NO ⋅ H2O (1.5 ммоль, 0.0191 г), интенсивно перемешивали для получения прозрачного желтого раствора, а затем добавляли в него 40 мл воды и приготовленного раствора нитрата соответствующего металла – M(NO)3 ⋅ nH2O (0.5 ммоль, 0.2270 г) в 50 мл воды. Эту реакционную смесь перемешивали в течение нескольких минут и оставляли на неделю. Продукты реакций однофазные, о чем свидетельствуют данные элементного анализа и ИК-спектроскопии. Образовавшиеся кристаллы комплексов отфильтровывали и сушили на воздухе сутки. Выходы 74–78%. Общая схема синтеза комплексов I–IV представлена на схеме 1 :

Схема 1 .
Анализ на C,H,N,S,O-элементов в комплексах выполняли на CHNS/O-элементном анализаторе Vario El cube в АЦ ИПХФ РАН.
| Найдено, %: | C 33.53; | Н 2.63; | N 7.80; | O 11.92; | S 17.89. |
| Для C30H28N6O8S6Pr2 (I) | |||||
| вычислено, % | C 33.55; | Н 2.61; | N 7.82; | O 11.90; | S 17.90. |
| Найдено, %: | C 32.40; | Н 2.55; | N 7.55; | O 11.50; | S 17.30. |
| Для C30H28N6O8S6Tb2 (II) | |||||
| Вычислено, %: | C 32.44; | Н 2.53; | N 7.57; | O 11.52; | S 17.32. |
| Найдено, %: | C 32.10; | Н 2.52; | N 7.50; | O 11.40; | S 17.10. |
| Для C30H28N6O8S6Ho2 (III) | |||||
| Вычислено, %: | C 32.09; | Н 2.51; | N 7.48; | O 11.41; | S 17.13. |
| Найдено, %: | C 32.00; | Н 2.50; | N 7.43; | O 11.36; | S 17.04. |
| Для C30H28N6O8S6Ho2 (IV) | |||||
| вычислено, %: | C 31.96; | Н 2.51; | N 7.45; | O 11.35; | S 17.06. |
ИК-спектры комплексов I–IV снимали на Фурье спектрометре Bruker ALPHA в интервале частот 400–4000 см–1 в режиме нарушенного полного внутреннего отражения (НВПО).
ИК I (ν, cм–1): 3199 сл, 2192 o.сл, 1592 cp, 1537 cp, 1452 о.с, 1413 о.с, 1214 о.с, 1191 о.с, 1156 о.с, 1139 о.с, 1032 cр, 978 сл, 838 с, 822 с, 752 с, 701 с, 594 с, 579 с, 542 о.с, 469 о.с.
ИК II (ν, cм–1): 3104 о.сл, 1594 о.сл, 1537.2 о.сл, 1450 ср, 1401 о.сл, 1259 о.сл, 1215.8 ср, 1186 о.сл., 1131 о.сл., 1073 о.сл., 1027 о.сл., 838 о.сл., 823.7 о.сл., 763 ср, 752.3 ср, 709 ср, 596.2 ср, 576 о.сл, 547 ср.
ИК III (ν, cм–1): 3609 о.сл, 3047 о.сл, 3022 о.сл, 1595 cp, 1535 ср, 1454 о.с, 1413 о.с, 1267 cp, 1199 о.с, 1148 о.с, 1089 о.с, 1032 ср, 827 о.с, 767 с, 746 о.с, 707 с, 592 с, 551 с, 541 о.с, 451 о.с.
ИК IV (ν, cм–1): 3046 о.сл, 1596 cp, 1533 cp, 1454 о.с, 1414 cp, 1267 cp, 1199 о.с, 1158 о.с, 1149 о.с, 1090 о.с, 1033 ср, 827 ср, 770 ср, 746 с, 708 с, 592 с, 553 с, 521 с, 452 с, 421 ср, 405 ср.
РСА соединений I–IV проведен на монокристальном дифрактометре Agilent XCalibur c детектором EOS (AgilentTechnologies UK Ltd., Yarnton, Oxfordshire, England). Сбор, обработка данных, определение и уточнение параметров элементарной ячейки выполнены по программе CrysAlis PRO [42]. Структуры расшифрованы прямым методом. Позиции и температурные параметры неводородных атомов уточнены в изотропном, а затем в анизотропном приближении полноматричным МНК. Позиции атомов водорода в анионе выявлены из разносных синтезов и уточнены в схеме наездника. Позиции атомов водорода молекулы воды выявлены из разносных синтезов Фурье и уточнены с наложением ограничений на длины связей и тепловые параметры.
Все расчеты выполнены с использованием комплекса программ SHELXTL [43]. Параметры элементарной ячейки и основные кристаллографические данные комплексов I–IV представлены в табл. 1.
Таблица 1.
Кристаллографические данные, параметры эксперимента и уточнения структур I−IV
| Параметры | Значение | |||
|---|---|---|---|---|
| I | II | III | IV | |
| Брутто-формула | C30H28N6O8S6Pr2 | C30H28N6O8S6Tb2 | C30H28N6O8S6Ho2 | C30H28N6O8S6Er2 |
| М | 1074.76 | 1110.78 | 1122.80 | 1127.46 |
| Температура, K | 150(1) | 100(1) | 150(1) | 150.01(10) |
| Размер кристалла, мм | 0.21 × 0.15 × 0.11 | 0.21 × 0.14 × 0.11 | 0.2 × 0.13 × 0.10 | 0.2 × 0.12 × 0.10 |
| λ, Å | 0.7107 | 0.7107 | 0.7107 | 0.7107 |
| Сингония | Moноклинная | Moноклинная | Moноклинная | Moноклинная |
| Пр. группа | P21/n | P21/c | P21/c | P21/c |
| a, Å | 19.6342(2) | 9.99724(15) | 9.9671(2) | 9.9519(3) |
| b, Å | 9.81100(10) | 9.91810(16) | 9.8900(2) | 9.8585(3) |
| c, Å | 20.2743(2) | 19.1807(3) | 19.1711(5) | 19.1517(5) |
| α, град | 90 | 90 | 90 | 90 |
| β, град | 94.1140(10) | 94.4022(14) | 94.392(2) | 94.363(2) |
| γ, град | 90 | 90 | 90 | 90 |
| V, Å3 | 3895.40(7) | 1896.23(5) | 1884.23(7) | 1873.56(9) |
| Z | 4 | 2 | 2 | 2 |
| ρ(выч.), г/cм3 | 1.833 | 1.945 | 1.979 | 1.999 |
| μ, мм–1 | 2.848 | 4.085 | 4.557 | 4.839 |
| F(000) | 2112 | 1080 | 1088 | 1092 |
| Диапазон съемки θ, град | 29.070 | 29.069 | 29.070 | 29.066 |
| Измеренные отражения (Rint) | 22 923 (0.0206) | 10 682 (0.0223) | 1001 (0.0222) | 10 017 (0.0265) |
| Независимые отражения | 10 411 | 5084 | 5051 | 5006 |
| Число уточняемых параметров | 481 | 241 | 241 | 241 |
| R1 (I > 2σ(I)) | 0.0244 | 0.0222 | 0.0252 | 0.0301 |
| wR2 (полные данные) | 0.0524 | 0.0537 | 0.0545 | 0.0691 |
| Tmin/Tmax | 0.74639/1.00000 | 0.81502/1.00000 | 0.81999/1.00000 | 0.69106/1.00000 |
| Остаточная плотность (ρmax/ρmin), e Å–3 | 0.470/–0.766 | 0.540/–0.979 | 0.890/–1.154 | 1.090/–1.885 |
Рентгеновские монокристальные данные структур I–IV депонированы в Кембриджском банке структурных данных (CCDC № 2143309–2143312; deposit@ccdc.cam.ac.uk, http://www.ccdc.cam.ac.uk).
СКВИД-магнитометрия. Магнитные измерения проводили с помощью магнитометра MPMS 5XL Quantum design). Температурную зависимость магнитной восприимчивости (χ(T)) измеряли в интервале температур 2–300 K при значении постоянного магнитного поля HDC = 1 kOe. Зависимость намагниченности от поля измеряли при температурах 2 и 5 K. Частотные зависимости действительной m' и мнимой m'' частей магнитной восприимчивости (χ) в переменном магнитном поле с амплитудой 2 Э записывали в диапазоне температур 1.8–10 К. Частоты переменного магнитного поля составляли 0.2–1400 Гц.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Комплексы I–IV получали взаимодействием соответствующего нитрата редкоземельного металла с лигандом 2-меркаптопиридин-N-оксидом (пиритионом) в водных аэробных растворах в присутствии КОН, в отличиe от работы [39], где используются спиртовые растворы хлоридов редкоземельных металлов и NaOH. Пиритион в протонных средах существует в виде пары таутомеров, основная форма которых представляет собой 1-гидрокси-2(1Н)-пиридинтион, а другая – 2‑меркаптопиридин-тиол-N-оксид [44] (схема 2 ). Пиритион (C5H5NOS) может легко координироваться с ионами РЗЭ (3+) посредством двух донорных атомов – кислорода и серы, а группа NO тиольной формы лиганда может действовать и как отдельный магнитный центр между соседними ионами РЗЭ (3+).

Схема 2 .
Координационная способность 1-гидрокси-2-пиридинтиона с различными ионами 3d-металлов хорошо изучена: были синтезированы и исследованы их спектральные свойства и магнитная восприимчивость в [45]. Установлено, что комплексы образуются путем хелатирования. Сопряженное основание пиритиона представляет собой анион, содержащий два донорных атома, атом серы и атом кислорода, каждый из которых несет формально отрицательный заряд; атом азота остается формально положительно заряженным. Тиолат-анион может образовываться в результате реакции с карбонатом натрия, а при добавлении соли металла(II) образуются пиритионаты. Анион может действовать как монодентатный или бидентатный лиганд и образует комплекс с цинком(II) в стехиометрическом соотношении лиганд : металл = 1 : 2. В своей мономерной форме пиритион цинка имеет два аниона, хелатированных к цинку с тетраэдрической геометрией. В твердом состоянии он образует димер, в котором каждый цинковый центр принимает тригональную бипирамидальную геометрию с двумя анионами, действующими в качестве осветляющих лигандов, координированными через атомы кислорода в осевых положениях [46]. Тиолат-анион координируется как бидентатный лиганд в кластерах Mo [47] и в комплексах хрома(III) [48] или празеодима(III) [34]. При этом в случае редкоземельного металла в его координационной сфере появляются координационные места, занятые молекулами растворителя, и их можно использовать, осуществляя дизайн новых структур, включая МMM.
Соединение I кристаллизуется в моноклинной системе, кристаллическая структура уточнена в пространственной группе P21/n, ассиметричная часть включает два иона Pr3+, шесть анионов (SC5H4NO)– и две молекулы воды в общем положении. На рис. 1 представлена молекулярная структура I, представляющая биядерный комплекс. Каждый ион Pr3+ формирует восьмикоординационную (додекаэдрическую) структуру. Она включает в себя три лиганда (SC5H4NO)–, хелатным способом координирующие каждый ион Pr3+, мостиковые атомы кислорода О(3), O(4), принадлежащие двум лигандам (SC5H4NO)– и молекулы координационной воды. В молекуле на два катиона Pr3+ приходится шесть однозарядных анионов (SC5H4NO)–, исходя из электронейтральности степень окисления Pr равна +3. В табл. 2 представлены длины связей и валентные углы в биядерном комплексе I. Соответствующие длины связей в каждом из восьми координационных комплексов имеют близкие значения, а некоторые углы отличаются. Так, хелатные углы в обоих комплексах одинаковы, тогда как остальные заметно различаются, что связано с разворотами анионов.
Рис. 1.
Молекулярная структура комплекса I. Атомы представлены в виде эллипсоидов с 50%-ной вероятностью.

Таблица 2.
Длины связей и углы в комплексе I
| Связь | d, Å | Связь | d, Å |
|---|---|---|---|
| Pr(2)–O(5) | 2.3625(17) | Pr(1)–O(1) | 2.3638(18) |
| Pr(2)–O(6) | 2.3665(16) | Pr(1)–O(2) | 2.3487(16) |
| Pr(2)–O(w2) | 2.4479(16) | Pr(1)–O(w1) | 2.4797(17) |
| Pr(2)–O(4) | 2.5087(15) | Pr(1)–O(3) | 2.5129(15) |
| Pr(2)–O(3) | 2.5302(15) | Pr(1)–O(4) | 2.5103(15) |
| Pr(2)–S(6) | 2.8742(7) | Pr(1)–S(2) | 2.9253(7) |
| Pr(2)–S(5) | 2.9801(6) | Pr(1)–S(1) | 2.9445(6) |
| Pr(2)–S(4) | 3.0015(6) | Pr(1)–S(3) | 2.9938(6) |
| Угол | ω, град | Угол | ω, град |
| O(5)Pr(2)O(6) | 140.62(6) | O(2)Pr(1)O(1) | 136.78(6) |
| O(5)Pr(2)O(w2) | 98.18(7) | O(1)Pr(1)O(w1) | 89.34(7) |
| O(6)Pr(2)O(w2) | 78.37(6) | O(2)Pr(1)O(w1) | 72.31(6) |
| O(5)Pr(2)O(4) | 77.02(6) | O(1)Pr(1)O(3) | 74.82(6) |
| O(6)Pr(2)O(4) | 138.67(5) | O(2)Pr(1)O(3) | 134.65(5) |
| O(w2)Pr(2)O(4) | 79.76(5) | O(w1)Pr(1)O(3) | 78.16(5) |
| O(5)Pr(2)O(3) | 140.11(6) | O(1)Pr(1)O(4) | 138.08(6) |
| O(6)Pr(2)O(3) | 77.74(5) | O(2)Pr(1)O(4) | 78.33(5) |
| O(w2)Pr(2)O(3) | 75.90(5) | O(w1)Pr(1)O(4) | 80.63(5) |
| O(4)Pr(2)O(3) | 63.09(5) | O(4)Pr(1)O(3) | 63.31(5) |
| O(5)Pr(2)S(6) | 104.31(5) | O(1)Pr(1)S(2) | 122.12(6) |
| O(6)Pr(2)S(6) | 66.54(4) | O(2)Pr(1)S(2) | 66.17(4) |
| O(w2)Pr(2)S(6) | 144.20(5) | O(w1)Pr(1)S(2) | 138.45(4) |
| O(4)Pr(2)S(6) | 132.10(4) | O(3)Pr(1)S(2) | 132.38(4) |
| O(3)Pr(2)S(6) | 102.25(4) | O(4)Pr(1)S(2) | 89.70(4) |
| O(5)Pr(2)S(5) | 63.46(4) | O(1)Pr(1)S(1) | 64.05(4) |
| O(6)Pr(2)S(5) | 78.06(4) | O(2)Pr(1)S(1) | 74.72(4) |
| O(w2)Pr(2)S(5) | 74.72(4) | O(w1)Pr(1)S(1) | 82.38(4) |
| O(4)Pr(2)S(5) | 128.34(4) | O(3)Pr(1)S(1) | 134.39(4) |
| O(3)Pr(2)S(5) | 144.99(4) | O(4)Pr(1)S(1) | 151.46(4) |
| S(6)Pr(2)S(5) | 90.79(2) | S(2)Pr(1)S(1) | 87.965(19) |
| O(5)Pr(2)S(4) | 71.86(4) | O(1)Pr(1)S(3) | 74.21(5) |
| O(6)Pr(2)S(4) | 132.09(4) | O(2)Pr(1)S(3) | 141.76(4) |
| O(w2)Pr(2)S(4) | 142.65(4) | O(w1)Pr(1)S(3) | 140.96(4) |
| O(4)Pr(2)S(4) | 63.00(4) | O(3)Pr(1)S(3) | 63.55(4) |
| O(3)Pr(2)S(4) | 89.01(4) | O(4)Pr(1)S(3) | 88.40(4) |
| S(6)Pr(2)S(4) | 71.920(18) | S(2)Pr(1)S(3) | 78.20(2) |
| S(5)Pr(2)S(4) | 125.988(17) | S(1)Pr(1)S(3) | 118.824(18) |
В структуре I наблюдаются сокращенные межмолекулярные контакты (табл. 3). На рис. 2 представлен фрагмент кристаллической структуры I. Из рис. 2 видно, что в биядерном комплексе фрагменты, которые могли бы быть связанными центром инверсии, по-разному участвуют в межмолекулярных контактах. Влияние этих сокращенных контактов и приводит к нарушению центра инверсии. В кристаллической структуре I биядерные комплексы объединяются в пары за счет π-стэкинг-взаимодействий (C(32)…C(33) 3.384, C(32)…C(34) 3.291 Å). Расстояния между атомами металлов внутри молекул – 4.29 Å, между соседними – 10.61 Å.
Таблица 3.
Межмолекулярные контакты в комплексе I
| Атом1 | Атом2 | Симметрические операции 1 | Симметрические операции 2 | d, Å |
|---|---|---|---|---|
| O(w2) | C(34) | x, y, z | x, –1 + y, z | 3.208 |
| S(3) | C(16) | x, y, z | 1.5 – x, –1/2 + y, 1.5 – z | 3.496 |
| S(1) | O(w1) | x, y, z | 1 – x, –y, 1 – z | 3.309 |
| S(5) | O(w2) | x, y, z | 1 – x, –y, 2 – z | 3.195 |
| C(32) | C(33) | x, y, z | 1 – x, 1 – y, 2 – z | 3.384 |
| C(32) | C(34) | x, y, z | 1 – x, 1 – y, 2 – z | 3.291 |
Рис. 2.
Фрагмент кристаллической структуры комплекса I. Штриховыми линиями показаны сокращенные межмолекулярные контакты.

В кристаллической структуре I также наблюдаются как внутри, так и межмолекулярные водородные связи (O(w)…S 3.10, 3.23 Å) (рис. 3).
Рис. 3.
Фрагмент кристаллической структуры комплекса I. Штриховыми линиями показаны водородные связи.

Cоединения II–IV полностью изоструктурны, поэтому приведено описание лишь структуры комплекса Ho(III).
Соединение III кристаллизуется в моноклинной системе, ассиметричная часть включает ион Ho3+, три аниона (SC5H4NO)– и молекулу воды в общем положении. На рис. 4 представлена молекулярная структура III. Молекула представляет центросимметричный биядерный комплекс. Каждый ион Ho3+ формирует восьмикоординационную структуру, Она включает в себя три лиганда 2‑меркапто-пиридин-N-оксида, хелатным способом координирующие каждый ион Ho3+, мостиковый атом кислорода О(3) и молекулу координационной воды. В молекуле на два катиона Ho3+ приходится шесть однозарядных анионов (SC5H4NO)–, исходя из электронейтральности степень окисления Ho равна +3. В табл. 4 представлены длины связей и валентные углы в биядерном комплексе III.
Рис. 4.
Молекулярная структура комплекса III. Атомы представлены в виде эллипсоидов с 50%-ной вероятностю.
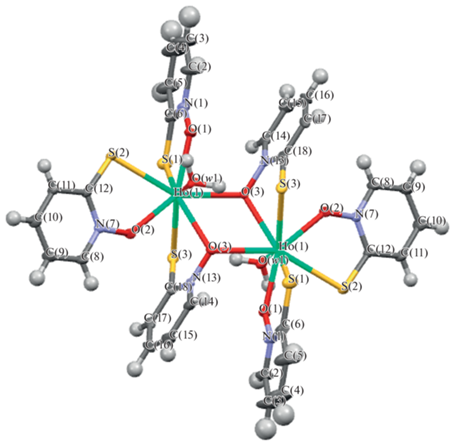
Таблица 4.
Длины связей и валентные углы в комплексе III
| Связь | d, Å | Связь | d, Å |
|---|---|---|---|
| Ho(1)–O(1) | 2.257(2) | Ho(1)–S(1) | 2.7906(8) |
| Ho(1)–O(2) | 2.263(2) | Ho(1)–S(2) | 2.8775(7) |
| Ho(1)–O(w1) | 2.346(2) | Ho(1)–S(3)#1 | 2.9027(8) |
| Ho(1)–O(3) | 2.4105(19) | S(2)–C(12) | 1.705(3) |
| Ho(1)–O(3)#1 | 2.4145(19) | S(3)–C(18) | 1.734(3) |
| Угол | ω, град | Угол | ω, град |
| O(1)Ho(1)O(2) | 138.04(7) | O(3)#1Ho(1)S(1) | 131.44(5) |
| O(1)Ho(1)O(w1) | 75.53(7) | O(1)Ho(1)S(2) | 72.35(5) |
| O(2)Ho(1)O(w1) | 90.25(9) | O(2)Ho(1)S(2) | 66.01(5) |
| O(1)Ho(1)O(3) | 77.90(7) | O(w1)Ho(1)S(2) | 77.33(5) |
| O(2)Ho(1)O(3) | 140.17(7) | O(3)Ho(1)S(2) | 147.50(5) |
| O(w1)Ho(1)O(3) | 82.92(7) | O(3)#1Ho(1)S(2) | 134.35(5) |
| O(1)Ho(1)O(3)#1 | 133.34(7) | S(1)Ho(1)S(2) | 89.85(2) |
| O(2)Ho(1)O(3)#1 | 77.74(7) | O(1)Ho(1)S(3)#1 | 140.29(6) |
| O(w1)Ho(1)O(3)#1 | 76.00(7) | O(2)Ho(1)S(3)#1 | 72.46(6) |
| O(3)Ho(1)O(3)#1 | 62.52(8) | O(w1)Ho(1)S(3)#1 | 139.60(6) |
| O(1)Ho(1)S(1) | 69.55(6) | O(3)Ho(1)S(3)#1 | 87.73(5) |
| O(2)Ho(1)S(1) | 114.27(7) | O(3)#1Ho(1)S(3)#1 | 64.90(5) |
| O(w1)Ho(1)S(1) | 145.00(6) | S(1)Ho(1)S(3)#1 | 74.20(2) |
| O(3)Ho(1)S(1) | 91.84(5) | S(2)Ho(1)S(3)#1 | 123.77(2) |
В кристаллах соединений II–IV (рис. 5) комплексы связаны в трехмерные структуры за счет слабых ван-дер-вальсовых взаимодействий в отличие от кристаллов, включающих комплексы Pr(III) (комплекс I). Во всех трех комплексах наблюдаются внутримолекулярные водородные связи типа O(w)…S 3.152 Å (рис. 6). Однако в кристаллах I, как было отмечено ранее, наблюдаются подобные водородные связи и между соседними комплексами.

Таким образом, кристаллическая структура соединения I стабилизируется за счет водородных связей и ван-дер-ваальсовых взаимодействий, тогда как кристаллическая структура соединений II–IV – только за счет ван-дер-ваальсовых взаимодействий. Другой особенностью кристаллической структуры соединения I является то, что в ней возникают пары из биядерных комплексов за счет π‑стэкинг-взаимодействий.
Температурная зависимость магнитной восприимчивости χ(T) образца I была записана в подмагничивающем поле DC при HDC = 1000 Э (врезка на рис. 7а). При температурах от 300 до 30 K эффективный момент (μэфф = (8χT)1/2) не зависит от температуры (рис. 7а), а при температурах T < < 30 K наблюдается его снижение с понижением температуры. Расчетное значение для ионов редкоземельных элементов в парамагнитном состоянии определяется выражением:
Рис. 7.
Температурные зависимости магнитной восприимчивости (врезка) и эффективного магнитного момента образца комплекса I (пунктиром показано расчетное значение μэфф для двух ионов Pr3+ (a); зависимости магнитного момента образца комплекса I от поля при температурах 2 и 5 K (б).

Зависимости действительной и мнимой частей магнитной восприимчивости от частоты переменного поля (AC) были измерены без подмагничивающего поля (DC) и в постоянном магнитном поле HDC = 2500 Э (рис. 8a и 8б соответственно). В обоих случаях изменений действительной части магнитной восприимчивости с частотой поля AC не наблюдалось. Поскольку величина мнимой части магнитной восприимчивости составляла не более 1% от величины действительной части (как в подмагничивающем поле, так и без него), можно судить об отсутствии магнитного релаксационного отклика в комплексе I в диапазоне частот от 0.2 до 1400 Гц.
Рис. 8.
Зависимости действительной и мнимой частей магнитной восприимчивости образца I от частоты, измеренные без подмагничивающего поля DC (a) и в подмагничивающем поле (HDC = 2500 Э) (б).

Отсутствие релаксационного отклика комплекса I можно объяснить различием окружения Pr3+, когда все атомы находятся в додекаэдрическом окружении, но только комплекс I нецентросимметричный в отличие от комплексов II–IV.
Температурная зависимость магнитной восприимчивости образца II в подмагничивающем поле DC (HDC = 1000 Э) показана на врезке рис. 9a. Эффективный момент μэфф в диапазоне температур от 300 до 50 K не изменяется (рис. 9a), а при температурах T < 50 K наблюдается спад значения μэфф с понижением температуры. Параметры для расчета μэф иона Tb3+ следующие: gJ = 3/2; J = 6, S = 3, L = 3, терм основного состояния – 7F6, электронная конфигурация – 4f 8. Расчетное значение для двух ионов Tb3+ (μэфф = 13.70 μB) оказывается ниже, чем экспериментально полученное значение (μэфф = 16.00 μB). Экспериментальное значение μэфф может быть описано следующими параметрами: gJ = 7/4 (завышенное значение); J = 6. Уменьшение μэфф при температуре ниже 50 K, так же как и в случае комплекса I, может свидетельствовать об антиферромагнитном взаимодействии между ионами тербия. Зависимости магнитного момента комплекса II от поля при температурах 2 и 5 K (рис. 9б) имеют вид, характерный для парамагнитика при данных температурах. Зависимости описываются функцией Бриллюэна, но значения спина, как и ожидалось, выше, чем для ионов Tb3+ (J = 6.7). Магнитного гистерезиса при температурах 2 и 5 K также не наблюдалось (рис. 9б).
Рис. 9.
Температурные зависимости магнитной восприимчивости (врезка) и эффективного магнитного момента образца комплекса II (пунктиром показано расчетное значение μэфф для двух ионов Tb3+ и экспериментальное значение 16.00 μB, описанное в тексте) (a); зависимости магнитного момента образца комплекса II от поля при температурах 2 и 5 K (б).

На рис. 10 показаны зависимости действительной и мнимой частей AC магнитной восприимчивости II от частоты в отсутствие магнитного поля. Мнимая часть магнитной восприимчивости χ" обращается в ноль при температуре выше 3 K. В диапазоне температур от 1.8 до 3.0 K мнимая часть восприимчивости χ" демонстрирует максимум при частоте ~50 Гц, который не зависит от температуры, что указывает на нулевое значение энергии активации, извлекаемой из выражения Аррениуса:
Рис. 10.
Зависимости действительной (a) и мнимой (б) частей магнитной восприимчивости II от частоты, измеренные без подмагничивающего поля DC.

Рис. 11.
Диаграммы Cole-Cole для мнимой и действительной частей магнитной восприимчивости II при температурах от 1.8 до 3.0 K. Внешнее поле DC HDC = 0 Э.

Температурная зависимость эффективного магнитного момента образца III показана на рис. 12a. При температурах от 300 до 150 K эффективный момент не изменяется, при T < 150 K наблюдается увеличение μэфф с понижением температуры и резкий спад при T < 10 K. Параметры для расчета эффективного момента иона Но3+ следующие: gJ = 5/4; J = 8, S = 2, L = 6, терм основного состояния – 5I8, электронная конфигурация 4f 10. Расчетное значение μэфф для двух ионов Ho3+ (15.00 μB) совпадает с экспериментальным значением, полученным при комнатной температуре (14.98 μB). Возрастание магнитной восприимчивости при T < 150 K может свидетельствовать о появлении ферромагнитных корреляций в образце при данных температурах, а дальнейшее уменьшение магнитного момента при T < 10 K отвечает антиферромагнитному взаимодействию между ионами гольмия при данной температуре. Зависимости магнитного момента комплекса III от поля (рис. 12б) имеют вид, характерный для парамагнитика и описываются функцией Бриллюэна со значениями спина и g‑фактора, определенными ранее из температурной зависимости μэфф при комнатной температуре (gJ = 5/4; J = 8). Таким образом, при T = 2 K образец находится в том же спиновом состоянии, что и при T = 300 K (охлаждение не изменяет спиновое состояние ионов гольмия в комплексе). Магнитного гистерезиса при низких температурах (2 и 5 K) также не наблюдалось (рис. 12б).
Рис. 12.
Температурные зависимости магнитной восприимчивости (врезка) и эффективного магнитного момента образца комплекса II (пунктиром показано расчетное значение μэфф для двух ионов Ho3+ (a); зависимости магнитного момента образца комплекса II от поля при температурах 2 и 5 K (б).

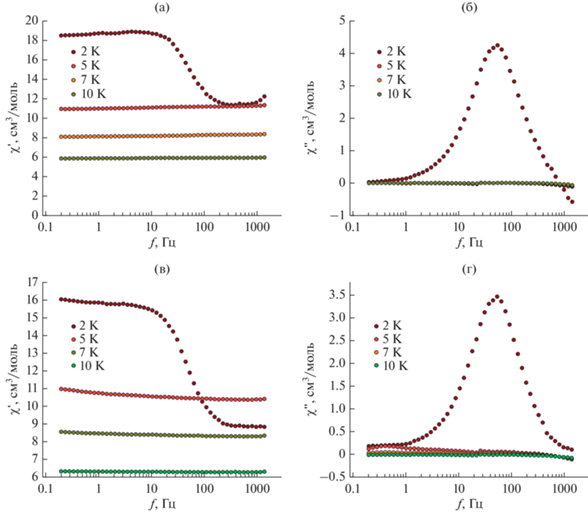
На рис. 13 показаны зависимости действительной и мнимой частей AC магнитной восприимчивости от поля для комплекса III при разных температурах и полях. При температуре T = 2 K максимум мнимой части в обоих случаях наблюдался при одинаковой частоте ~50 Гц.
Рис. 13.
Зависимости действительной (a, в) и мнимой (б, г) частей магнитной восприимчивости от частоты, измеренные без подмагничивающего поля DC (a, б) и в подмагничивающем поле HDC = 2500 Э.
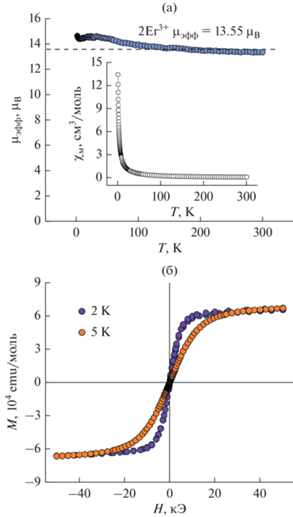
Температурная зависимость эффективного магнитного момента образца IV показана на рис. 14a. При температурах от 300 до 150 K μэфф не изменяется с температурой, при T < 150 K наблюдается увеличение эффективного момента. Параметры для расчета эффективного момента иона Er3+ следующие: gJ = 6/5; J = 15/2, S = 3/2, L = 6, терм основного состояния – 4I15/2, электронная конфигурация – 4f 11 и расчетное значение μэфф для двух ионов Er3+ равно 13.55 μB. Расчетное значение совпадает с экспериментом при T = 300 K (13.32 μB). Возрастание магнитной восприимчивости при T < 150 K может свидетельствовать о появлении ферромагнитных корреляций в образце. Зависимости магнитного момента комплекса III от поля при температурах 2 и 5 K (рис. 14б) имеют вид, характерный для парамагнетика и описываются функцией Бриллюэна со значениями спина и g-фактора, определенными ранее из температурной зависимости μэфф при комнатной температуре (gJ = 5/4; J = 8). Таким образом, охлаждение не изменяет спиновое состояние ионов эрбия в комплексе. Магнитного гистерезиса при низких температурах (2 и 5 K) также не наблюдалось (рис. 14б).
Рис. 14.
Температурные зависимости магнитной восприимчивости (врезка) и эффективного магнитного момента образца комплекса II (пунктиром показано расчетное значение μэфф для двух ионов Er3+ (a); зависимости магнитного момента образца комплекса II от поля при температурах 2 и 5 K (б).
На рис. 15 показаны зависимости действительной и мнимой частей магнитной восприимчивости от частоты переменного поля AC без подмагничивающего поля DC (рис. 15a, 15б). Мнимая часть магнитной восприимчивости χ" обращается в ноль при температуре выше 2.5 K, а в диапазоне температур от 2.0 до 2.5 K мнимая часть восприимчивости χ" демонстрирует максимум при частоте 50 Гц, который не зависит от температуры. Приложение внешнего поля DC (HDC = 500 Э, значение определено по максимуму полевой зависимости мнимой части магнитной восприимчивости) к образцу IV не изменяет частотное поведение действительной и мнимой частей магнитной восприимчивости (рис. 15в, 15г). Мнимая часть восприимчивости χ" в обоих случаях (без внешнего поля и в присутствии внешнего поля) демонстрирует максимум при частоте ~50 Гц, который не зависит от температуры, что аналогично поведению комплекса II.
Рис. 15.
Зависимости действительной (a, в) и мнимой (б, г) частей магнитной восприимчивости IV от частоты, измеренные без подмагничивающего поля DC (a, б) и в подмагничивающем поле HDC = 500 Э.

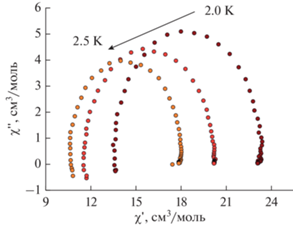
Диаграмма Cole-Cole (рис. 16) показывает, что в образце наблюдается релаксационная мода движения доменной стенки, но отсутствует смещение максимума мнимой части при изменении температуры, как и в случае с образцом II (туннельный механизм релаксации).
Рис. 16.
Диаграммы Cole-Cole для мнимой и действительной частей магнитной восприимчивости IV при температурах 2.00, 2.25 и 2.50 K. Внешнее DC поле HDC = 500 Э.
В заключение с помощью РСА установлено, что лиганд 2-меркапто-пиридин-N-оксид обладает несколькими электронодонорными центрами в реакциях комплексообразования с редкоземельными металлами Pr(III), Тb(III), Ho(III) и Er(III) и проявляет би- и тридентатные свойства с образованием биядерных комплексов I–IV в щелочной среде при соотношении лиганд : РЗЭ = 3 : 1.
Результаты РCА показали, что в комплексообразовании наряду с атомами кислорода и серы лиганда участвуют молекулы воды. В кристаллической структуре I также наблюдаются как внутри-, так и межмолекулярные водородные связи (O(w)…S). В комплексах II–IV наблюдаются только внутримолекулярные водородные связи (O(w)…S).
Магнитные свойства биядерных комплексов I–IV изучены методом СКВИД-магнитометрии в широком диапазоне температур. Комплексы II–IV демонстрируют магнитное поведение, характерное для комплексов с редкоземельными металлами. Температурные зависимости магнитного момента предполагают наличие значительной магнитной анизотропии. Частотные зависимости демонстрируют наличие медленной релаксации намагниченности и могут быть описаны обобщенной моделью Дебая, при этом задействовано одно время релаксации независимо от температуры. Такое поведение характерно для мономолекулярных магнитов с доминированием процесса квантового туннелирования. Этот факт подтверждается отсутствием эффектов гистерезиса на полевых зависимостях магнитного момента.
Авторы заявляют, что у них нет конфликта интересов.
Список литературы
Montgomery C.P., Murray B.S., New E.J. et al. // Acc. Chem. Res. 2009. V. 42. № 7. P. 925.
Stamatatos T.C., Teat S. J., Wernsdorfer W. et al. // Angew. Chem. Int. Ed. 2009. V. 48. № 3. P. 521.
Kühne I.A., Magnani N., Mereacre V. et al. // Chem. Commun. 2014. V. 50. P. 1882.
Ishikawa N., Sugita M., Ishikawa T. et al. // J. Am. Chem. Soc. 2003. V. 125. № 29. P. 8694.
Zhang Y.Z., Duan G.P., Sato O. et al. // J. Mater. Chem. 2006. V. 16. № 26. P. 2625.
Gao S., Su G., Yi T. et al. // Phys. Rev. B. 2001. V. 63. № 5. P. 054431.
Sugita M., Ishikawa N., Ishikawa T. et al. // Inorg. Chem. 2006. V. 45. № 3. P. 1299.
Benelli C., Gatteschi D. // Chem. Rev. 2002. V. 102. № 6. P. 2369.
Ishikawa N., Sugita M., Ishikawa, T. et al. // J. Am. Chem. Soc. 2003. V. 125. № 29. P. 8694.
Ishikawa N., Sugita M., Ishikawa T. et al. // J. Phys. Chem. B. 2004. V. 108. № 31. P. 11265.
Ishikawa N., Sugita M., Wernsdorfer W. // Angew. Chem. Int. Ed. 2005. V. 44. № 19. P. 2931.
Ishikawa N., Sugita M., Wernsdorfer W. // J. Am. Chem. Soc. 2005. V. 127. № 11. P. 3650.
AlDamen M.A., Clemente-Juan J.M., Coronado E. et al. // J. Am. Chem. Soc. 2008. V. 130. № 28. P. 8874.
AlDamen M.A., Cardona-Serra S., Clemente-Juan J.M. et al. // Inorg. Chem. 2009. V. 48. № 8. P. 3467.
Layfield R.A., McDouall J.J., Sulway S.A. et al. // Chem. Eur. J. 2010. V. 16. № 15. P. 4442.
Tang J., Hewitt I., Madhu N. et al. // Angew. Chem. 2006. V. 118. № 11. P. 1761.
Zheng Y.Z., Lan Y., Anson C.E. et al. // Inorg. Chem. 2008. V. 47. № 23. P. 10813.
Gamer M.T., Lan Y., Roesky P.W. et al. // Inorg. Chem. 2008. V. 47. № 15. P. 6581.
Wang Y., Li X.L., Wang T.W. et al. // Inorg. Chem. 2010. V. 49. № 3. P. 969.
Sessoli R., Powell A.K. // Coord. Chem. Rev. 2009. V. 253. № 19–20. P. 2328.
Jiang S.D., Wang B. W., Su G. et al. // Angew. Chem. 2010. V. 122. № 41. P. 7610.
Guettas D., Gendron F., Fernandez Garcia G. et al. // Chem. Eur. J. 2020. V. 26. № 19. P. 4389.
Yi X., Bernot K., Pointillart F. et al. // Chem. Eur. J. 2012. V. 18. № 36. P. 11379.
Yi X., Pointillart F., Le Guennic B. et al. // Polyhedron. 2019. V. 164. P. 41.
Wang W.M., Zhang H.X., Wang S.Y. et al. // Inorg. Chem. 2015. V. 54. № 22. P. 10610.
Bernot K., Luzon J., Bogani L. et al. // J. Am. Chem. Soc. 2009. V. 131. № 15. P. 5573.
Zhou N., Ma Y., Wang C. et al. // Dalton Trans. 2009. № 40. P. 8489.
Bernot K., Pointillart F., Rosa P. et al. // Chem. Commun. 2010. V. 46. № 35. P. 6458.
Wang X. L., Li L. C., Liao D. Z. // Inorg. Chem. 2010. V. 49. № 11. P. 4735.
Mei X.L., Ma Y., Li L. C. et al. // Dalton Trans. 2012. V. 41. № 2. P. 505.
Liu R. N., Zhang C. M., Li L.C. et al. // Dalton Trans. 2012. V. 41. № 39. P. 12139.
Bao D.X., Xiang S., Wang J. et al. // J. Coord. Chem. 2016. V. 69. № 21. P. 3131.
Xiong R.G. Zuo J.L., You X.Z. et al. // Polyhedron. 1996. V. 15. № 19. P. 3321.
Niu D.Z., Mu L.L., Lu Z. S. et al. // J. Chem. Crystallogr. 2004. V. 34. № 3. P. 195.
Niu D.Z., Chen J.T. // Chin. J. Struct. Chem. 2002. V. 21. № 5. P. 520.
Karwasara S., Jha C.K., Sinhababu S. et al. // Dalton Trans. 2016. V. 45. № 17. P. 7200.
Niu D.Z., Mu L.L., Yu S.Z. et al. // J. Chem. Crystallogr. 2003. V. 33. № 1. P. 27.
Ravindran Durai Nayagam B., Jebas S.R., Schollmeyer D. // Acta Crystallogr. E. 2008. V. 64. № 2. P. m425.
Chen X., Hu Y., Wu D. et al. // Polyhedron. 1991. V. 10. № 23–24. P. 2651.
Liu R., Liu S., Zhu H. // J. Nanjing Univ. Technol. (Nat. Sci. Ed.). 2007. V. 12. № 3. P. 48.
Niu D.Z., Yao L., Min X. et al. // Z. Kristallogr. NCS. 2011. V. 226. № 4. P. 527.
Agilent CrysAlis PRO. AgilentTechnologies UK Ltd., Yarnton, Oxfordshire, England, 2011.
Sheldrick G.M. SHELXTL. Version 6.14. Structure Determination Software Suite. Madison (WI, USA): Bruker AXS., 2000.
Bond A., Jones W. // Acta Crystallog. C. 1999. V. 55. № 9. P. 1536.
Das A., Han Z., Brennessel W.W. et al. // ACS Catal. 2015. V. 5. № 3. P. 1397.
Barnett B.L., Kretschmar H.C., Hartman F.A. // Inorg. Chem. 1977. V. 16. № 8. P. 1834.
Liang H., Chen Z.F., Hu R.X. et al. // Transition Met. Chem. 2002. V. 27. № 1. P. 102.
Wen T.B., Shi J.C., Liu Q.T. et al. // Acta Crystallogr. C. 1996. V. 52. № 5. P. 1204.
Kleemann W. // Annu. Rev. Mater. Res. 2007. V. 37. P. 415.
Guo Y.N., Xu G.F., Guo Y. et al. // Dalton Trans. 2011. V. 40. № 39. P. 9953.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия