

Кристаллография, 2020, T. 65, № 2, стр. 255-259
Синтез, структура и электрохимические свойства нового пространственно-экранированного о-бензохинона – 6,9-ди-трет-бутил-1,2,3a,4-тетрагидробензо[b]оксазоло[3,2-d] [1, 4]-оксазин-7,8-диона
М. В. Арсеньев 1, *, Е. А. Челноков 1, Е. В. Баранов 1, С. А. Чесноков 1
1 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
* E-mail: mars@iomc.ras.ru
Поступила в редакцию 14.05.2019
После доработки 03.07.2019
Принята к публикации 08.07.2019
Аннотация
Синтезирован новый представитель пространственно-экранированного 3,6-ди-трет-бутил-о-бензохинона с аннелированным тетрагидрооксазоло[3,2-d][1,4]оксазиновым фрагментом – 6,9-ди-трет-бутил-1,2,3a,4-тетрагидробензо[b]оксазоло[3,2-d][1,4]-оксазин-7,8-дион. Его строение подтверждено методами ИК-, УФ-, ЯМР-спектроскопий, а также методом рентгеноструктурного анализа. По данным метода цикловольтамперометрии первый потенциал восстановления хинона в ацетонитриле равен –0.83 В (отн. Ag/AgCl/KCl).
ВВЕДЕНИЕ
Пространственно-экранированные о-бензохиноны, производные 3,6-ди-трет-бутил-о-бензохинона, представляют большой интерес в химии высокомолекулярных соединений как в качестве ингибиторов свободнорадикальных процессов [1], так и в качестве компонентов фотоинициирующих полимеризацию олигоэфирметакрилатов систем, работающих в видимом диапазоне [2, 3]. Данные соединения интересны и в качестве редокс-активных лигандов в координационной химии [4]. Особый интерес представляют N-замещенные о-бензохиноны, свойства которых существенно зависят от возможности участия в сопряжении электронной пары атома азота с π-системой о-хинона [5]. Ранее по реакции нуклеофильного присоединения и дальнейшего окисления продуктов присоединения из диэтаноламина и 3,6-ди-трет-бутил-о-бензохинона был получен о-хинон 1 с аннелированным морфолиновым фрагментом [6]. В настоящей работе представлен новый O,N-замещенный 3,6-ди-трет-бутил-о-бензохинон, содержащий тетраоксазоло[3,2-d] [1, 4]оксазиновый фрагмент, полученный при дальнейшем окислении хинона 1, и наглядно продемонстрировано изменение структурных характеристик при нарушении сопряжения неподеленной электронной пары атома азота с π-системой о-хинона.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Тетрагидрооксазоло[3,2-d] [1, 4]оксазиновый фрагмент ранее формировали путем конденсации аминоспиртов с дикарбонильными соединениями [7, 8]. В настоящей работе этот фрагмент формировался при окислении морфолинового фрагмента хинона 1. По-видимому, при окислении образуется иминиевый интермедиат 2, который подвергается внутримолекулярной атаке гидроксильной группы (схема 1). Данная внутримолекулярная циклизация приводит к образованию о-хинона 3 с тетрагидрооксазолооксазиновым фрагментом. Строение о-хинона подтверждено данными 1H-, 13С ЯМР, УФ-, ИК-спектроскопий. Из смеси ацетон–гексан были выращены монокристаллы о-хинона 3, и молекулярное строение было подтверждено с помощью метода рентгеноструктурного анализа (РСА).
ИК-спектры регистрировали на ИК-фурье-спектрометре “ФСМ 1201” (суспензии в вазелиновом масле) в области 400–4000 см–1. ЯМР-спектры регистрировали на спектрометре Bruker Avance III 400 МГц. Химические сдвиги приведены в шкале δ (м. д.) относительно дейтерированного растворителя в качестве внутреннего стандарта. Потенциалы окисления измеряли методом циклической вольтамперометрии (ЦВА) в трехэлектродной ячейке с помощью потенциостата “Elins P-45X” в среде аргона. Рабочий электрод – стационарный стеклоуглеродный (СУ) электрод с диаметром 2 мм, вспомогательный электрод – платиновая проволока. Электрод сравнения – Ag/AgCl/KCl с водонепроницаемой диафрагмой. Скорость развертки потенциала 0.1 В с–1. Фоновый электролит – 0.1 М Bu4NClO4 (99%, “Acros”), дважды перекристаллизованный из водного EtOH и высушенный в вакууме (48 ч) при 50°C. Концентрация о-бензохинона составляла 5 ммоль л–1.
Рентгеноструктурное исследование соединения 1 проводили на автоматическом дифрактометре “Bruker Smart APEX” при температуре 150(2) К (ω-сканирование, МоKα-излучение, λ = = 0.71073 Å). Экспериментальные наборы интенсивностей были собраны и интегрированы с помощью программ SMART [9] и SAINTPlus [10] соответственно. Кристалл соединения 1 – немероэдрический двойник, соотношение монокристальных компонентов (доменов) составляет ∼53 : 47%. Для учета поглощения использовали программу TWINABS [11]. Структура решена прямым методом и уточнена полноматричным МНК по $F_{{hkl}}^{2}$ в анизотропном приближении для всех неводородных атомов. Водородные атомы были помещены в геометрически рассчитанные положения и уточнены в модели “наездника”. Уточнение структуры 1 проводили по двум доменам двойника с использованием инструкции HKLF5. Расчеты по расшифровке и уточнению структуры выполнены в программном пакете SHELXTL [12]. Рисунок молекулярной структуры получен при помощи программы Olex2 [13]. Кристаллографические данные и параметры рентгеноструктурных экспериментов приведены в табл. 1. Основные длины связей и углы в 1 даны в табл. 2. Кристаллографическая информация для соединения 1 депонирована в Кембриджском банке структурных данных (CCDC № 1905483; deposit@ccdc.cam.ac.uk или https://www.ccdc.cam.ac.uk/structures/).
Таблица 1.
Кристаллографические данные, параметры рентгеноструктурных экспериментов и уточнения для кристалла о-хинона 3
| Брутто-Формула | C18H25NO4 |
|---|---|
| Молекулярная масса | 319.39 |
| Т, K | 150(2) |
| Сингония | Моноклинная |
| Пр. гр. | P21/n |
| a, b, c, Å | 10.3054(9), 10.1731(9), 16.7316(15) |
| α, β, γ, град | 90, 105.930(3), 90 |
| V, Å | 1686.7(3) |
| Z | 4 |
| ρ, г см–3 | 1.258 |
| µ, мм–1 | 0.088 |
| F(000) | 688 |
| Размер кристалла, мм | 0.36 × 0.15 × 0.07 |
| Область измерений, θ/град | 2.097–25.998 |
| Пределы h, k, l | –12 ≤ h ≤ 12 0 ≤ k ≤ 12 0 ≤ l ≤ 20 |
| Число независимых отражений | 6398 |
| Число отражений I > 2σ(I) | 3516 |
| Добротность (F2) | 1.041 |
| R1/wR2 (I > 2σ(I)) | 0.0488/0.0821 |
| R1/wR2 (по всем параметрам) | 0.0995/0.0883 |
| Остаточная электронная плотность ρmax/ρmin, эÅ–3 | 0.264/–0.253 |
Таблица 2.
Некоторые длины связей и валентные углы в хиноне 3
| Связь | d, Å | Связь | d, Å |
|---|---|---|---|
| O(1)–C(1) | 1.222(2) | C(1)–C(6) | 1.455(3) |
| O(2)–C(2) | 1.222(2) | C(1)–C(2) | 1.552(3) |
| O(3)–C(5) | 1.358(2) | C(2)–C(3) | 1.453(3) |
| O(3)–C(15) | 1.430(2) | C(3)–C(4) | 1.378(3) |
| O(4)–C(16) | 1.415(2) | C(4)–C(5) | 1.504(3) |
| O(4)–C(17) | 1.419(3) | C(5)–C(6) | 1.353(3) |
| N(1)–C(4) | 1.386(2) | C(15)–C(16) | 1.490(3) |
| N(1)–C(16) | 1.446(2) | C(17)–C(18) | 1.518(3) |
| N(1)–C(18) | 1.471(3) | ||
| Угол, град | Угол, град | ||
| C(5)O(3)C(15) | 117.3(2) | C(3)C(4)N(1) | 122.2(2) |
| C(16)O(4)C(17) | 105.7(2) | C(3)C(4)C(5) | 121.3(2) |
| C(4)N(1)C(16) | 122.2(2) | N(1)C(4)C(5) | 116.6(2) |
| C(4)N(1)C(18) | 120.1(2) | C(6)C(5)O(3) | 117.7(2) |
| C(16)N(1)C(18) | 104.3(2) | C(6)C(5)C(4) | 124.8(2) |
| O(1)C(1)C(6) | 124.7(2) | O(3)C(5)C(4) | 117.3(2) |
| O(1)C(1)C(2) | 116.3(2) | C(5)C(6)C(1) | 113.2(2) |
| C(6)C(1)C(2) | 119.1(2) | O(3)C(15)C(16) | 110.5(2) |
| O(2)C(2)C(3) | 124.6(2) | O(4)C(16)N(1) | 106.3(2) |
| O(2)C(2)C(1) | 115.5(2) | O(4)C(16)C(15) | 111.3(2) |
| C(3)C(2)C(1) | 119.8(2) | N(1)C(16)C(15) | 111.1(2) |
| C(4)C(3)C(2) | 114.3(2) | O(4)C(17)C(18) | 107.9(2) |
| N(1)C(18)C(17) | 102.9(2) |
о-Хинон 1 был получен по методике [6].
Получение 6,9-ди-трет-бутил-1,2,3a,4-тетрагидробензо[b]оксазоло[3,2-d] [1, 4]-оксазин-7,8-диона (3) проведено следующим образом. К суспензии 1.28 г (4 ммоль) о-хинона 1 в 50 мл ацетонитрила добавили 2.39 г (10 ммоль) PbO2, реакционная смесь нагревалась при 60°С в течение 5 ч. Далее смесь фильтровали через вату от механических примесей, промывали водой и экстрагировали эфиром. Полученный экстракт упарили. о-Хинон 3 выделен с помощью колоночной хроматографии (элюент бензол:ацетон 20:1). Выделено 0.45 г (35%) в виде рубиновых кристаллов. C18H25NO4. Найдено: C, 67.70; H, 7.85; N, 4.43. Вычислено: C, 67.69; H, 7.89; N, 4.39. 1H ЯМР (400 MГц, CDCl3, δ, м.д., J, Гц): 1.29 и 1.34 (с, по 9Н, t-Bu), 3.37 (дт, 2JHH = 10.4, 3JHH = 6.7, 4.9, 1H, C(18)H2), 3.71 (дт, 2JHH = 10.4 Гц, 3JHH = 7.5 Гц, 1H, C(18)H2), 3.86–3.97 (м, 2H, C(17)H2), 4.01 (д, 1H, C(15)–H, 2JHCH = 12.8 Гц), 4.52 (д, 1H, C(15)–H, 2JHCH = = 12.8 Гц), 4.84 (с, 1H, C(16)–H). Спектр ЯМР 13C (CDCl3, 100 МГц, δ, м.д.): 181.78, 181.77 (2 C=O), 161.11, 146.65, 133.68, 126.11, 83.66 (C(16)H), 65.23 (C(17)H2), 63.25 (C(15)H2), 58.93 (C(18)H2), 35.99, 35.05, 30.34, 30.19. ИК-спектр (вазелиновое масло, ν/см–1): 474 (ср), 509 (ср), 546 (сл), 592 (сл), 641 (ср), 711 (ср), 778 (ср), 812 (ср), 827 (ср), 850 (ср), 866 (сл), 898 (ср), 932 (сл), 946 (сл), 982 (сл), 1005 (ср), 1025 (ср), 1045 (ср), 1080 (ср), 1163 (ср), 1188 (ср), 1210 (ср), 1242 (ср), 1287 (с), 1315 (с), 1346 (с), 1365 (с), 1394 (с), 1423 (с), 1481 (с), 1522 (с), 1584 (с), 1628 (с), 1660v(с).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В ИК-спектре соединения 3 наблюдаются характерные для о-хинонов интенсивные полосы поглощения при 1627 и 1660 см–1.
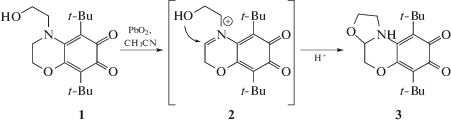
Электронный спектр поглощения соединения 3 является типичным для о-хинонов. В видимом диапазоне наблюдаются полосы, относящиеся к ππ*- (λmax = 435 нм, lgε = 3.24) и nπ*-переходам карбонильных групп о-хинона. Последняя полоса не разрешается и регистрируется в виде плеча (рис. 1).
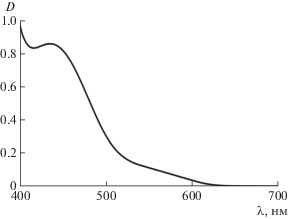
Образование оксазолидинового цикла фиксирует геометрию молекулы, что приводит к существенному усложнению спектра 1H ЯМР для соединения 2 по сравнению с хиноном 1. В соединении 1 возможна конформационная динамика, атомы водорода в каждом из метиленовых фрагментов являются эквивалентными и в 1H ЯМР-спектре наблюдаются четыре уширенных сигнала, относящиеся к четырем CH2-группам. В соединении 2 за счет фиксации геометрии дополнительным гетероциклическим фрагментом невозможна конформационная динамика, и метиленовые протоны при атомах C(15), C(17), C(18) становятся неэквивалентными (нумерация атомов представлена на рис. 3). Это отражается в существенном усложнении 1H ЯМР-спектра соединения 2. На рис. 2 приведен фрагмент 1H ЯМР-спектра в области 3.2–5.0 м.д. Наличие хинонового фрагмента также подтверждают данные 13C ЯМР-спектроскопии – характеристичные сигналы карбонильных групп наблюдаются при 181.78, 181.77 м.д.
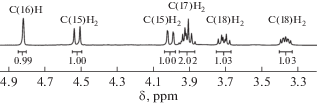
Рис. 3.
Молекулярная структура R-изомера о-хинона 3. Тепловые эллипсоиды приведены с 30%-ной вероятностью. Me-группы t-Bu заместителей показаны “каркасными” линиями.
Молекулярное строение о-хинона 3 определено методом РСА (рис. 3). Кристаллографические характеристики и основные длины связей в о-хиноне 3 представлены в табл. 1 и 2. Оксазолидиновый цикл формируется за счет образования одинарной связи C(16)–O(4) (1.415(2) Å). Атом углерода C(16) в соединении 3 является хиральным. В кристалле о-хинон 3 присутствует в виде рацемической смеси R- и L-изомеров. На рис. 3 представлена молекулярная структура R-изомера о-хинона 3.
Молекула соединения 3 имеет полигетероциклическое строение. Все три цикла заметно неплоские. Угол между средними плоскостями хинонового (С(1)–С(6)) и морфолинового (C(4), C(5), O(3), C(15), C(16), N(1)) циклов составляет 15°, а угол между средними плоскостями морфолинового и оксазолидинового (C(16)–C(18), O(4), N(1)) фрагментов равен 66°. Торсионный угол C(18)N(1)C(4)C(3) равен –58.7(3)°. Атом азота N(1) имеет пирамидальное строение. Выход атома N(1) из плоскости C(4)C(16)C(18) составляет ∼0.3 Å, сумма углов при атоме азота 346.6°. В соединении 1 атом азота находится практически в плоскости основания тетраэдра (отклонение от плоскости ∼0.14 Å), сумма углов при N(1) близка к 360° (357.3°). Формирование оксалидинового цикла приводит к уменьшению донирования электронной плотности p-орбитали азота на π-систему хинона, что отражается в перераспределении электронной плотности в хиноновом фрагмента и изменении всех длин связей хинонового кольца. Наблюдается удлинение связи C(4)–N(1) на 0.026 Å, что свидетельствует об уменьшении двоесвязанности данного фрагмента, однако значение связи 1.360(6) Å меньше, чем в N,N-замещенном о-хиноне с пиперазиновым заместителем (1.438(2) Å), в котором сопряжение с неподеленной электронной парой атома азота невозможно [14]. Наблюдается выравнивание длин связей карбонильных фрагментов O(1)–C(1) и O(2)–C(2) (1.222(2) Å в 3, 1.218(6) и 1.251(6) Å в 1). При переходе от 1 к 3 некоторые связи хинонового цикла заметно удлиняются: C(1)–C(2) (с 1.517(7) до 1.552(3) Å), C(2)–C(3) (с 1.417(6) до 1.453(3) Å); с одновременным укорочением связи C(3)–C(4) (с 1.396(3) до 1.378(3) Å). В целом можно заключить, что в соединении 3 наблюдается более выраженное альтернирование связей по сравнению с 1. Торсионный угол O(1)C(1)C(2)O(2) в молекуле 3 составляет 15.7(3)°.
Методом ЦВА изучены редокс-превращения хинона 3. На рис. 4 приведены кривые ЦВА соединения 3. Электрохимическое восстановление хинона 3 в анаэробных условиях протекает в две последовательные стадии. Первая стадия – одноэлектронная и квазиобратимая (кривая 1, E1/2 = = ‒0.83 В) – приводит к образованию стабильного (Ip.a/Ip.k = 1.0) в условиях ЦВА-эксперимента о-семихинонового анион-радикала, вторая – необратимая – к образованию катехолат-дианиона, который из-за высокой основности подвергается протонированию, и на обратной ветви наблюдается появление пика окисления при –0.45 В. В анодной области наблюдается необратимое окисление аминофрагмента о-хинона 3 при 1.3 В.
Рис. 4.
Кривые ЦВА восстановления соединения 3 в ацетонитриле в анаэробных условиях (Ar) в интервалах: 1 – от –2.0 до +1.0 В; 2 – от –1.3 до –0.5 В. C = 5 × × 10–3 М, 0.1 M NBu4ClO4, 100 мВ/с, отн. Ag/AgCl/KCl.
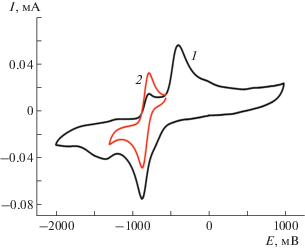
Обращает на себя внимание смещение первого потенциала восстановления хинона 3 в катодную область по сравнению с описанными ранее O,O- и N,N-димезамещенными производными 3,6-ди-трет-бутил-о-бензохинона [15], которые нашли применение при создании соединений поглощающих молекулярный кислород [16, 17] и редокс-изомерных систем [18].
ЗАКЛЮЧЕНИЕ
Синтезирован новый представитель 4,5-(N,O)-дизамещенного 3,6-ди-трет-бутил-о-бензохинона – о-хинона c аннелированным тетрагидрооксазоло[3,2-d] [1,4]оксазиновым фрагментом. Молекулярная структура соединения подтверждена данными ЯМР, ИК-, УФ-спектроскопий, а также данными рентгеноструктурного анализа. Показано, что данный хинон является слабым окислителем в ряду о-бензохинонов.
Работа выполнена с использованием оборудования центра коллективного пользования “Аналитический центр ИМХ РАН” в рамках государственного задания.
Список литературы
Shushunova N.Y., Chesnokov S.A. // Polym. Sci.B. 2009. V. 51. P. 427.
Chesnokov S.A., Arsenyev M.V., Kovylin R.S. et al. // Appl. Organomet. Chem. 2017. V. 31. P. e3553.
Chesnokov S.A., Cherkasov V.K., Abakumov G.A. et al. // Polym. Sci. B. 2014. V. 56. P. 11.
Konovalov A.I., Antipin I.S., Burilov V.A. et al. // Russ. J. Org. Chem. 2018. V. 54. P. 157.
Abakumov G.A., Cherkasov V.K., Kocherova T.N. et al. // Russ. Chem. Bull. 2006. V. 55. P. 1195.
Prokof’eva T.I., Vol’eva V.B., Prokof’ev A.I. et al. // Chem. Heterocycl. Compd. 2000. V. 36. P. 923.
Kicsak M., Mandi A., Varga S. et al. // Org. Biomol. Chem. 2018. V. 16. P. 393.
Alcaide B., Garcia-Blanco S., Garcia-Gonzalez M.T. et al. // Tetrahedron Lett. 1986. V. 27. P. 4217.
Bruker SMART-Bruker Molecular Analysis Research Tool. V. 5.632. Madison: Bruker AXS, 2005.
Bruker SAINTPlus Data Reduction and Correction Program. V. 6.45a. Madison: Bruker AXS, 2003.
Sheldrick G.M. TWINABS. V. 2012/1. Bruker/Siemens Area Detector Absorption Correction Program for Twinned Crystals. Madison: Bruker AXS, 2012.
Sheldrick G.M. SHELXTL. V. 6.14. Structure Determination Software Suite. Madison: Bruker AXS, 2003.
Dolomanov O.V., Bourhis L.J., Gildea R.J. et al. // J. Appl. Cryst. 2009. V. 42. P. 339.
Abakumov G.A., Cherkasov V.K., Kocherova T.N. et al. // Russ. Chem. Bull. 2007. V. 56. P. 1849.
Smolyaninov I.V., Kuzmin V.V., Arsenyev M.V. et al. // Russ. Chem. Bull. 2017. V. 66. P. 1217.
Cherkasov V.K., Abakumov G.A., Grunova E.V. et al. // Chem. Eur. J. 2006. V. 12. P. 3916.
Poddel’sky A.I., Smolyaninov I.V., Kurskii Y.A. et al. // J. Organomet. Chem. 2010. V. 695. P. 1215.
Bubnov M.P., Skorodumova N.A., Baranov E.V. et al. // Inorg. Chim. Acta. 2012. V. 392. P. 84.
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография